Cation-reinforced donor-acceptor pseudorotaxanes
Sofia I.
Pascu
,
Thibaut
Jarrosson
,
Christoph
Naumann
,
Sijbren
Otto
,
Guido
Kaiser
and
Jeremy K. M.
Sanders
*
Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge, UK CB2 1EW. E-mail: jkms@cam.ac.uk
First published on 15th December 2004
Abstract
Formation of a series of pseudorotaxanes from an electron-rich crown ether (host 1), pyromellitic diimide (guest 2) and alkali salt templates MX, where M = Li+ and Na+, and X− = Br−, I−, CF3SO3−, [B(C6F5)4]− and [B{3,5-(CF3)2(C6H3)}4]−, is reported. Mixing of 1 and 2 in CH2Cl2 or a mixture of CHCl3 and MeOH (98∶2) gave a pale yellow solution indicative of a very weak charge-transfer interaction. Upon addition of MX, brightly coloured solutions were obtained, resulting from a red shift and an increase in the intensity of the charge-transfer band. Structural and kinetic studies of the pseudorotaxanes were performed by NMR. The solution-phase structures of [M2·1·2]2+ are in good agreement with the solid-phase structure determined by X-ray crystallography. The remarkable templating properties of Li+ for the 1·2 donor-acceptor complex are due to the almost perfect coincidence of coordinative geometries in [Li2·1]2+ and [Li2·1·2]2+.
Introduction
Recent years have seen increased attention to the development of functional supramolecular architectures.1–10 Whilst research into supramolecular chemistry continues to focus on understanding and using non-covalent forces to construct multi-component assemblies, significant effort is now directed on applying this fundamental knowledge to develop molecular assemblies that can perform tasks analogous to the machines of everyday life.5,11–14 Examples include the cation-crown ether complexes developed into highly sensitive and selective fluoroionophores and simple viologen donor-acceptor interactions employed for switchable rotaxanes and catenanes.15–18 The number of publications in the area of functional rotaxane and catenane structures has risen dramatically since the early reports on synthesis by metal–ligand coordination and statistical threading.19–21 The functionality of hydrogen-bonding catenanes and rotaxanes has been explored with particular emphasis on the study of the relative motion of the components, that is in the synthesis and study of a molecular muscle where an interlocked rotaxane can be contracted or extended by changing the metal cation.22,23 Leigh et al. have shown that photoinduced electron transfer to a naphthalimide unit alters the hydrogen-bond acceptor ability of the carbonyl groups.24 Stoddart and co-workers have reported a series of rotaxanes and catenanes that demonstrate a diverse array of properties, such as photochemically driven molecular switches, redox switchable catenanes and slow shuttling rotaxanes.18,25–47 Analogous neutral donor-acceptor interactions have been used in our work: the acceptor units are electron-deficient aromatic diimide units, such as pyromellitic or naphthalene diimide, and a range of symmetric and asymmetric donor-acceptor catenanes have been synthesised using either reversible alkene metathesis or irreversible Glaser coupling of terminal acetylenes.48–52 In general additional non-covalent interactions (C–H⋯O bonding in particular) are required to assemble the donor and acceptor components.52We have recently described a switching experiment between naphthalene diimide and pyromellitic diimide pseudorotaxanes induced by lithium cations and have incorporated this switch into a neutral rotaxane.53,54 In an attempt to understand and generalise this cation-induced effect we have now carried out a detailed structural and kinetic study of the formation of pseudorotaxanes from donor (crown ether 1) and acceptor (2) units (Fig. 1), in the presence or absence of a series of alkali salts MX such as LiBr, LiI, Li(CF3SO3), Li[B(C6F5)4], NaBr, NaI, Na(CF3SO3) and Na[B{3,5-(CF3)2(C6H3)}4]. Solid-state structures of new pseudorotaxanes (determined by X-ray diffraction) and solution structures [determined by NMR in CD2Cl2 or a mixture of CHCl3 and MeOH (98∶2)] are discussed.
Results and discussion
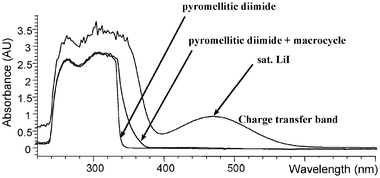
We were surprised by the way alkali metal ions affected the association of host 1 and guest 2. When we first added alkali metal salts to solutions of 1 and 2 we intended to use the well-established interaction between crown ethers and alkali metal cations to dissociate the 1·2 pseudorotaxane complex. As it turned out, the envisaged cation-crown interaction did occur but had exactly the opposite effect to that expected: the cation reinforced the complexation between 1 and 2. Evidence for this unexpected behaviour comes from visual inspection of the solutions and UV-Vis analysis. Whereas a 10.0 mM solution of guest 2 is essentially colourless, addition of 5.0 mM of the colourless host 1 gives rise to a faint yellow colour resulting from relatively inefficient complexation and charge transfer between 1 and 2. Subsequent addition of excess alkali salt had a dramatic effect, giving coloured solutions ranging from orange to dark-red, depending on the salt. The UV-Vis spectrum in Fig. 2 shows the appearance of a charge transfer band at 468 nm upon addition of LiI, suggesting that the donor and acceptor systems are able to interact much more efficiently in the presence of LiI. The variation of λmax in lithium complexes ranges from 432 nm (LiBr) to 468 nm (LiI). In Na complexes λmax ranges from 430 nm to 482 nm (NaB[{3,5-(CF3)2(C6H3)}4]) (Table 1). These intriguing observations prompted us to investigate the effect of a series of alkali-metal salts on the formation of 1·2 pseudorotaxanes in solution [in CH2Cl2 or a mixture of CHCl3 and MeOH (98∶2], using NMR and ITC, and in the solid state by single crystal X-ray diffraction. Crystal structures of the resulting complexes revealed that the Li and Na salts insert into two different remarkably well pre-organised binding sites.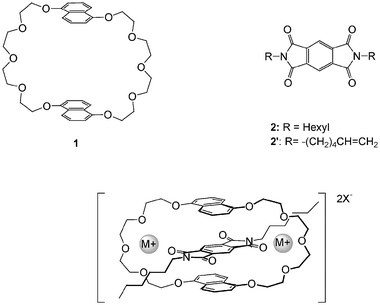 | ||
| Fig. 1 Representations of the donor host 1 and acceptor guest 2, templates and the pseudorotaxane assembly. | ||
| MX | λ max/nm | ΔE/eV | E max/L mol−1 cm−1 |
|---|---|---|---|
| LiBr | 432 | 2.87 | 187.8 |
| LiI | 468 | 2.65 | 610.6 |
| Li(CF3SO3) | 466 | 2.66 | 453.0 |
| LiB(C6F5)4 | 462 | 2.68 | — |
| NaBr | 430 | 2.88 | 151.2 |
| NaI | 452 | 2.77 | 763.6 |
| Na(CF3SO3) | 454 | 2.73 | 578.0 |
| NaB[{3,5-(CF3)2(C6H3)}4] | 482 | 2.65 | — |
Pseudorotaxane structures in solution: NMR studies
We observed that after addition of 1 equiv. of LiI, 50% of the organic material remains unchanged, while 50% is present in a new species in slow exchange on the chemical shift timescale. Integration confirmed unambiguously the M∶1∶2 stoichiometry of 2∶1∶1, implying a dramatic cooperativity between binding of the first and second cations. The same is true for all LiX investigated. The 1H NMR spectra allowed the elucidation of the structure of the donor-acceptor complexes of MX, where for M+ = Li+, X− = I−, Br−, CF3SO3−, [B(C6F5)4]− and for M+ = Na+, X− = I−, CF3SO3−, [B{3,5-(CF3)2(C6H3)}4]−. For M+ = Li+ or Na+, the 1H NMR spectrum of the 2∶1∶1 complexes [M2·1·2]X2 show a remarkable degree of symmetry. Based on the number of signals observed and discussed below, we believe that the new species exhibit in solution the general structure shown in Fig. 1.The NMR evidence includes ring-current induced shift in the aromatic protons of guest 2, ring-current induced shift in the naphthyl protons of the crown ether 1, shifts in tetraethylene glycol signals and determination of effective symmetry. While the aromatic protons of the unbound pyromellitic diimide (2) in the presence of 1 resonate at δ 8.19 in CD2Cl2 at 280 K, this resonance shifts to 6.55 ppm upon addition of 10 equiv. of LiI, indicating that they are held in close proximity to the shielding region of the naphthyl rings. Separate peaks are observed for bound and unbound guest 2, indicating that dissociation of 2 is slow on the NMR chemical shift timescale. Similar shifts were observed for a range of alkali salts (Table 2).
| MX | T/K | H1 | H2 | H3 | H4 | H6a |
|---|---|---|---|---|---|---|
| a H6: for metal complexes only the downfield-shifted diastereotopic protons are reported. b 19F: δ − 131.6 (br, ortho-F), −161.8 (td, 3JFF = 18.8 Hz, meta-F), −165.8 (td, 3JFF = 18.8 Hz, para-F); 11B{1H}: δ − 3.7; 7Li: δ 0.02. c 19F: δ − 63.65; 11B{1H}: δ − 3.79. | ||||||
| None | 280 | 7.70 | 7.17 | 6.48 | 8.19 | 4.04 |
| LiI | 280 | 6.98 | 6.81 | 6.58 | 6.55 | 5.06 |
| LiI | 200 | 6.90 | 6.77 | 6.54 | 6.18 | 4.66 |
| LiBr | 280 | 6.98 | 6.78 | 6.56 | 6.75 | 5.17 |
| Li(CF3SO3) | 280 | 6.98 | 6.80 | 6.52 | 6.43 | 4.50 |
| LiB(C6F5)4b | 280 | 7.07 | 6.84 | 6.48 | 6.64 | 3.64 |
| NaI | 200 | 6.88 | 6.75 | 6.54 | 7.13 | 5.28 |
| Na(CF3SO3) | 240 | 7.06 | 6.81 | 6.36 | 6.76 | — |
| NaB[{3,5-(CF3)2(C6H3)}4]c | 280 | 7.17 | 6.86 | 6.30 | 7.07 | 3.56 |
The four (shifted) aromatic peaks are not split, showing that the high degree of symmetry is retained when the alkali metal is incorporated into the complex. The large upfield shifts of the signals of the naphthalene proton (H1, H2, H3 labelled as in Fig. 3) confirm the presence of the pyromellitic diimide guest in a sandwich structure, as in our related catenane structure.52 Chemical shifts of the aromatic proton H4 of the bound guest differ in LiI vs. NaI complexes by ca. 1 ppm (in CD2Cl2 at 200 K, Fig. 4 and Table 2). Almost identical chemical shifts for the aromatic bound crown ether resonances H1–H3 were observed regardless of the nature of the cation. For all complexes, COSY experiments confirm the expected proton couplings of the naphthalene rings. The free macrocycles contain four distinct carbon environments for the tetraethylene glycol moiety. The number of different environments does not change in the [M2·1·2]2+ complex (according to the 13C NMR spectrum), consistent with the retention of the high degree of symmetry after complexation. Binding of two lithium cations results in the geminal splitting of the tetraethylene glycol protons, as indicated by HMQC. The proposed structure [M2·1·2]X2 (Fig. 1) satisfies all steric, electrostatic, charge transfer and symmetry requirements for all complexes. Since no significant binding was observed by NMR for NaBr, further investigations were not performed. A lower ion-pair dissociation constant for NaBr combined with low solubility in CH2Cl2 probably explains its lack of complexation.55
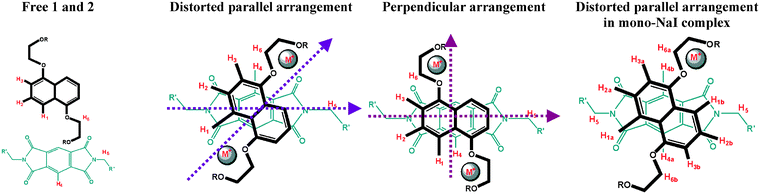 | ||
| Fig. 3 Schematic drawing of main geometry arrangements for pseudorotaxane complexes. | ||
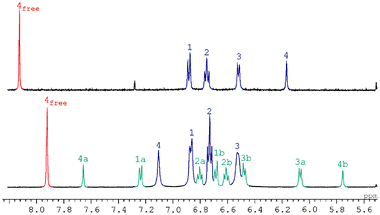 | ||
| Fig. 4 1H NMR spectrum of (a) LiI and (b) mono-NaI and bis-NaI complexes. | ||
A closer inspection of chemical shift differences in the series suggested that use of sodium instead of lithium salts, combined with differing size of the anions, results in subtly different geometries of the resulting complexes. The orientation of the guest with respect to the host (Table 1, Fig. 3) was derived from variable temperature 1D and 2D NOESY experiments in CD2Cl2. Fig. 3 shows the labelling for free and bound host and guest molecules. The donor-acceptor arrangement varies in the series from parallel (with varying degrees of distortion from the ideal geometry) to perpendicular and will be discussed below.
For all lithium complexes, the guest aromatic protons H4 show NOEs with the naphthalene rings protons H1–H3, as well as with the tetraethylene glycol protons. H3 also shows NOEs to glycol loops OCH2CH2 and H4 shows NOE to the most downfield shifted OCH2CH2 proton H6. In addition, the NOESY spectrum reveals NOEs between the aromatic protons of the naphthalene rings and the alkyl chains of the pyromellitic diimide as follows: H1 shows NOEs to NCH2CH2 > NCH2 > N(CH2)2CH2, H2 ‘sees’ NCH2CH2 > N(CH2)2CH2. These results suggest a distorted parallel host–guest arrangement in solution for all lithium-containing pseudorotaxanes.
No satisfactory NOE results were obtained for the Na complexes due to a combination of precipitation and low intensities. The study of the NaI complex was complicated by the formation of a mono-NaI complex, in ca. 36% yield as estimated by integration. Since the bis-NaI complex showed similar chemical shifts for the proton resonances of the bound crown (at 200 K in CH2Cl2) to those found for the bis-LiI complex, a distorted parallel host–guest arrangement is proposed (Table 1 and Fig. 3).
At 200 K and in CD2Cl2 or CD3OD∶MeOD 98∶2 solutions, a slow exchange was observed between the bis-NaI complex and the mono-NaI complex.56 Saturation transfer experiments show that this exchange takes place stepwise: free guest 2 is in exchange with the mono-NaI complex and the mono-NaI complex is in exchange with the free crown ether 1 as well as the bis-NaI pseudorotaxane. This behaviour was not observed (on the NMR timescale and above 200 K) for the complexes of Na(CF3SO3) or Na[B{3,5-(CF3)2(C6H3)}4]. Interestingly, the diagnostic resonances (H4) for the guest molecule in the mono-NaI complex were found at δ 5.75 and 7.75, consistent with the lower symmetry of the system caused by shielding from only one side of the molecule (Table 3). Assignments of the mono-NaI complex were aided by a COSY spectrum. Correlations were observed between H1a and H1b, H4a and H4b, H1a, H2a and H3a, H1b, H2b and H3b. There is little exchange between the asymmetric sides of both the crown and the pyromellitic diimide, but H1a and H1b can be ‘linked’ via the bis-NaI complex H1. Similarly H2a and H2b are ‘linked’ via H2 of the bis-NaI complex and H3a and H3b are linked via H3 of the bis-NaI complex. The structure of the mono-NaI complex has not yet been determined in the solid state and exchange with the bis-NaI complex in solution made uncertain the assignment of the relative donor-acceptor orientation by NMR. Since no formation of mono-MX complexes was detected in any other cases, cooperative binding of the cation is occurring. The absence of a 1∶1∶1 complex is significant with respect to the mechanism of formation of the [Li2·1·2]2+ complex. The stability of the 2∶1∶1 complex [Li2·1·2]2+ is clearly orders of magnitude higher than that of [Li·1·2]2+.
| NMR shift δ | Assignment |
|---|---|
| 7.26 | H1a |
| 6.82 | H2a |
| 6.08 | H3a |
| 7.69 | H4a |
| 4.75 | H6a |
| 6.70 | H1b |
| 6.62 | H2b |
| 6.49 | H3b |
| 5.76 | H4b |
| 4.17 | H6b |
It appears that the choice of cation is limited by the size of the binding cavity, since attempted template reactions with K, Rb and Cs ions had no effect on the neutral donor-acceptor complex. However, the low solubility of these salts in CH2Cl2 may also explain this observation.
Solid-state pseudorotaxane structures
A search of the Cambridge Structural Database revealed that only 16 supramolecular complexes using the donor crown ether 1 as the host have been reported so far,30,52,57–67 in addition to the structure of the free 1.30 Amongst these, only two are derivatives of the guest 2 and they are both catenanes.52,66 Crystallisation of single crystals from either CH2Cl2 (complexes [Li2·1·2][B(C6F5)4]2 and [Na2·1·2][B{3,5-(CF3)2(C6H3)}4]2) or a mixture of CHCl3∶MeOH 98∶2 was successful {complexes [Li2·1·2′]I2,53 [Li2·1·2]Br2, [Li2·1]I2 and [Na2·1·2]I2} and the solid-phase structures of these complexes have been determined by X-ray crystallography (Figs. 5–10, Tables 4 and 5).![Two views (ORTEP plot at 30% probability) of the X-ray molecular structure of complex [Li2·1·2]Br2. Hydrogens are omitted for clarity. (Br: brown, N: light blue, Li: dark grey, O: red, C: light grey)](/image/article/2005/NJ/b415418e/b415418e-f5.gif) | ||
| Fig. 5 Two views (ORTEP plot at 30% probability) of the X-ray molecular structure of complex [Li2·1·2]Br2. Hydrogens are omitted for clarity. (Br: brown, N: light blue, Li: dark grey, O: red, C: light grey) | ||
![Two views (ORTEP plot at 30% probability) of the X-ray molecular structure of [Li2·1·2][B(C6F5)4]2. Counter-ions and hydrogen atoms are omitted for clarity. (N: light blue, Li: dark grey, O: red, C: light grey)](/image/article/2005/NJ/b415418e/b415418e-f6.gif) | ||
| Fig. 6 Two views (ORTEP plot at 30% probability) of the X-ray molecular structure of [Li2·1·2][B(C6F5)4]2. Counter-ions and hydrogen atoms are omitted for clarity. (N: light blue, Li: dark grey, O: red, C: light grey) | ||
![Packing diagram of the [Li2·1·2][B(C6F5)4]2 complex, views over the a and c axes, respectively. (B: pink, F: green, N: light blue, Li: dark grey, O: red, C: light grey)](/image/article/2005/NJ/b415418e/b415418e-f7.gif) | ||
| Fig. 7 Packing diagram of the [Li2·1·2][B(C6F5)4]2 complex, views over the a and c axes, respectively. (B: pink, F: green, N: light blue, Li: dark grey, O: red, C: light grey) | ||
![Two views (ORTEP plot at 30% probability) of the X-ray molecular structure of [Li2·1]I2. Hydrogens are omitted for clarity. (N: light blue, Li: dark grey, O: red, C: light grey, I: purple)](/image/article/2005/NJ/b415418e/b415418e-f8.gif) | ||
| Fig. 8 Two views (ORTEP plot at 30% probability) of the X-ray molecular structure of [Li2·1]I2. Hydrogens are omitted for clarity. (N: light blue, Li: dark grey, O: red, C: light grey, I: purple) | ||
![Two views (ORTEP plot at 30% probability) of the X-ray molecular structure of [Na2·1·2]I2. Hydrogens are omitted for clarity. (N: light blue, Li: dark grey, O: red, C: light grey, I: purple, Na: dark blue)](/image/article/2005/NJ/b415418e/b415418e-f9.gif) | ||
| Fig. 9 Two views (ORTEP plot at 30% probability) of the X-ray molecular structure of [Na2·1·2]I2. Hydrogens are omitted for clarity. (N: light blue, Li: dark grey, O: red, C: light grey, I: purple, Na: dark blue) | ||
![Two views (ORTEP plot at 30% probability) of the X-ray molecular structure of [Na2·1·2][B{3,5-(CF3)2(C6H3)}4]. Hydrogens are omitted for clarity. (N: light blue, Li: dark grey, O: red, C: light grey, I: purple, Na: dark blue)](/image/article/2005/NJ/b415418e/b415418e-f10.gif) | ||
| Fig. 10 Two views (ORTEP plot at 30% probability) of the X-ray molecular structure of [Na2·1·2][B{3,5-(CF3)2(C6H3)}4]. Hydrogens are omitted for clarity. (N: light blue, Li: dark grey, O: red, C: light grey, I: purple, Na: dark blue) | ||
| Compound | [Li2·1·2′]I2a | [Li2·1·2]Br2 | [Li2·1·2]B[(C6F5)4]2 | [Li2·1]I2 | [Na2·1]I2 | [Na2·1·2][B{3,5-(CF3)2(C6H3)}]4 |
|---|---|---|---|---|---|---|
| a Ref. 53. b Ligand = H2O. c Ligand = I. d Ligands = two fluorines (disordered CF3). e Ligand = MeOH. f 2 × (distance D1–D1) = 6.42 Å. | ||||||
| M–O (A1)/Å | 1.8783 (x) | 1.8874 | 1.89 | — | 2.2716 | 2.33 |
| M–O (D1)/Å | 2.2488 (x) | 2.2401 | 2.3 | 2.1425 | 2.4871 | 2.35 |
| 1.9668 (x) | 1.954 | 1.97 | 1.978 | 2.3181 | 2.34 | |
| 2.2903 (x) | 2.234 | 2.19 | 2.5513 | 2.4226 | 2.35 | |
| M–Ligand/Å | 1.8823 (y)b | 1.9251b | 1.95b | 1.9096b | 3.0308c | 2.93d |
| — | — | — | 1.9246e | — | — | |
| M–M/Å | 11.02 | 11.06 | 10.83 | 10.89 | 10.08 | 10.63 |
| D1–A1/Å | 3.27 | 3.28 | 3.34 | —f | 3.41 | 3.48 |
| Torsion angle D1–A1/° | 38.3 | 37.1 | 41.5 | — | 57.6 | 89.05 |
| Compound | [Li2·1·2]Br2·5CH2Cl2·2H2O | [Li2·1·2][B(C6F5)4]2·2CH2Cl2·2H2O | [Li2·1]I2·2MeOH·2H2O | [Na2·1·2]I2·6CH2Cl | [Na2·1·2][B{3,5-(CF3)2(C6H3)}4]2 |
|---|---|---|---|---|---|
| a Synchrotron radiation, Station 9.8, Daresbury SRS. | |||||
| Formula | C63H81Br2Cl15N2O16 | C108H76B2Cl4F40Li2N2O16 | C38H56I2Li2O14 | C64H78Cl18I2N2Na2O14 | C122H96B2F48N2Na2O14 |
| M | 1827.73 | 2595.03 | 1004.55 | 2037.16 | 2793.36 |
| λ/Å | 0.71073 | 0.71073 | 0.6923a | 0.71073 | 0.71073 |
| T/K | 180 (2) | 180 (2) | 150 (2) | 180 (2) | 180 (2) |
| Crystal system | Monoclinic | Triclinic | Triclinic | Triclinic | Monoclinic |
| Space group | P21/c | P -1 | P -1 | P -1 | C2/c |
| a/Å | 15.8820(3) | 10.7147(2) | 7.566(1) | 10.8071(2) | 38.5110(1) |
| b/Å | 10.2424(2) | 13.4449(2) | 10.3074(14) | 11.2503(2) | 15.7246(2) |
| c/Å | 26.2432(6) | 19.5512(3) | 15.598(2) | 18.3062(3) | 22.8791(3) |
| α/° | 103.2331(7) | 72.355(2) | 78.4706(8) | ||
| β/° | 94.5550(7) | 90.1735(7) | 88.102(2) | 81.8524(7) | 93.6400(5) |
| γ/° | 94.5532(7) | 81.429(2) | 86.3758(12) | ||
| U/Å3 | 4255.49(15) | 2732.42(8) | 1146.2(3) | 2157.41(7) | 13826.9(3) |
| Z | 2 | 1 | 1 | 1 | 4 |
| ρ calc/g cm−3 | 1.426 | 1.577 | 1.455 | 1.568 | 1.342 |
| μ/mm−1 | 1.481 | 0.243 | 1.430 | 1.352 | 0.135 |
| Total data | 24872 | 20750 | 8794 | 21614 | 87214 |
| Unique data | 7714 | 12240 | 6117 | 9407 | 15601 |
| R int | 0.057 | 0.045 | 0.03 | 0.033 | 0.078 |
| R [I > 3σ(I)] | 0.1000 | 0.0452 | 0.0342 | 0.0344 | 0.1580 |
| wR | 0.1184 | 0.0562 | 0.0367 | 0.0444 | 0.1728 |
The two related geometries found in solution, distorted parallel and perpendicular, are retained in the solid state. Host–guest torsion angles (defined as in Fig. 3) are around 40° for the Li complexes, 57° for NaI and almost 90° for NaB[{3,5-(CF3)2(C6H3)}4]. The distortion from the ideal geometry accompanies the increase in the bulkiness of the anion. For Li complexes, the variation in host–guest torsion angle is not large (up to ca. 3.4°), whereas the corresponding difference between [Na2·1·2][B{3,5-(CF3)2(C6H3)}4]2 and [Na2·1·2]I2 is larger than 30°.
In the complex [Na2·1·2]I2, NaI remains associated (Na–I separation ca. 3.03 Å) with the sodium centre in a distorted trigonal bipyramidal geometry rendering a distorted parallel donor-acceptor arrangement. In [Na2·1·2][B{3,5-(CF3)2(C6H3)}4]2, coordination of counter-ions to Na+ occurs through the CF3 groups of the bulky anion. As expected, each of the CF3 groups of [B{3,5-(CF3)2(C6H3)}4]− is disordered over two positions and a difference electron density map showed six clear positions for the fluorine atoms in each of the CF3 groups, with almost equal peak size. It was possible to observe that two fluorine atoms on the same CF3 group occupy the 5th and 6th coordination sites of the sodium centre (the two Na–F separations are 2.45 and 2.87 Å). The geometry of the Na atoms is therefore octahedral (but greatly distorted), in which two of the equatorial sites are occupied by two of the fluorine atoms belonging to one of the CF3 groups of B[3,5-(CF3)2(C6H3)]4−.
The lithium cations are all five-coordinate with a greatly distorted coordination geometry that most closely resembles a trigonal bipyramidal arrangement. The fifth coordination site is filled by an additional donor molecule, for example a water molecule for [Li2·1·2′]I2, [Li2·1·2]Br2, [Li2·1·2][B(C6F5)4]2 and [Li2·1]I2. For [Li2·1·2′]I253 and [Li2·1·2]Br2 the preference for water over methanol to fill the fifth coordination site is remarkable considering the large excess of methanol in solutions of [Li2·1]I2, [Li2·1·2′]I2 and [Li2·1·2]Br2 (2% solution ≡ 90 equiv. of methanol). In the case of [Li2·1]I2, in the absence of a guest, this site is occupied by one molecule of methanol for each of the lithium atoms, therefore maintaining the five-coordinate geometry.
Intermolecular stacking between pseudorotaxane units in the unit cell has not been observed for any of the complexes. For the lithium halide complexes, the anion is not directly coordinated to the complex and no ion-pairing was observed. This observation is also consistent with NMR experiments that suggested that the solution structures of the lithium complexes are not significantly influenced by the nature of the anion when X− = I− or Br−. Our observations are consistent with those by Huang et al. regarding the direct correlation between ion-pairing in solid state and in solution.55
By contrast, for complexes [Li2·1·2][B(C6F5)4]2 and [Na2·1·2][B{3,5-(CF3)2(C6H3)}4]2 the large counter-ion is involved in a series of intermolecular hydrogen bonds, which in the case of [Li2·1·2][B(C6F5)4]2 give rise to a supramolecular array. In the 3D network of [Li2·1·2][B(C6F5)4]2 hydrogen bonds link pseudorotaxane units and the counter-ions: the lithium-coordinated molecule of water is strongly hydrogen-bonded to F(6) from a neighbouring [B(C6F5)4]− unit (and the same is true for the symmetry-generated pair). The separation O(8)–F(6A) [where F(6A) belongs to the asymmetric unit generated by the symmetry operator x, y, 1 + z] is 2.983(1) Å. Furthermore, the acceptor molecule within the pseudorotaxane shows intermolecular hydrogen bonds between the N(CH2) units and the fluorine atoms of adjacent counter-ions. These interactions are directed perpendicular with respect to the acceptor’s plane (below and above this plane) with a separation between C(6) and F(13A) of 2.991(1) Å (where the latter atom belongs to the asymmetric unit generated by the symmetry operator x, 1 + y, z). In addition, it appears that all fluorine atoms of [B(C6F5)4]− are within short distances to neighbouring hydrogen of the glycol chain of the crown ether, with C(glycol)–F separations ranging between 2.9 and 3.7 Å. These short distances seem to be maintained in CD2Cl2 solutions, since 1H NMR showed reduced chemical shift differences between the diastereotopic O–CH2 (glycol) chain in [Li2·1·2][B(C6F5)4]2 (Δδ = 0.1) with respect to those found in the LiI or LiBr (Δδ ca. 1.2) and Li(CF3SO3) complexes (Δδ ca. 0.5).
The Na–Na distance in [Na2·1·2][B{3,5-(CF3)2(C6H3)}4]2 is 10.63 Å, similar to the 10.08 Å measured for [Na2·1·2]I2. The Li–Li distance does not vary significantly in the series, being 11.02 Å in [Li2·1·2′]I2, 10.8 Å in [Li2·1·2]B(C6F5)4]2 and 10.89 Å in [Li2·1]I2. In general, the inter-planar separations between the host and guest rings in the series range from 3.27 to 3.48 Å with higher values for sodium complexes. The cavity size of the guest-free complex [Li2·1]I2 is 6.42 Å, that is the mid-distance between naphthyl groups of the crown ether is 3.21 Å. The two donor units are essentially coplanar with a deviation smaller than 0.5°. These values are consistent with the inter-planar separations of published catenanes52,66,67 and confirm the donor-acceptor interaction between the host and the guest, as well as the pre-organised nature of the host. The crystal structures indicate that the size of the cation that can be accommodated is limited by the size of the binding cavity (Fig. 11).
![Binding of cations inside the pseudorotaxane cavity: (a) [Li2·1·2]Br2, (b) [Na2·1·2]I2, (c) [Na2·1·2][B{3,5-(CF3)2(C6H3)}4].](/image/article/2005/NJ/b415418e/b415418e-f11.gif) | ||
| Fig. 11 Binding of cations inside the pseudorotaxane cavity: (a) [Li2·1·2]Br2, (b) [Na2·1·2]I2, (c) [Na2·1·2][B{3,5-(CF3)2(C6H3)}4]. | ||
NMR studies on the kinetic stability of complexes
In an attempt to estimate the influence of different MX salts on the kinetic stability of the pseudorotaxanes [M2·1·2]X2, we studied the rate constants for complex dissociation in CD2Cl2 solution (k−1) using 1D NOESY NMR experiments.68–71 Complexes were obtained using a twofold excess of guest 2 and a tenfold excess of the MX salt. The 1D NOESY experiments showed that the free and bound guest 2 species are in exchange, for each of the different MX salts.The temperature for each experiment was chosen such that the exchange process was slow on the NMR chemical shift timescale. The chemical shifts of the exchanging resonances were not significantly temperature dependent. For each complex, the resonances corresponding to the methyl and the H4 protons of the bound guest 2 were irradiated in turn and the responses of the corresponding resonances of the free guest were measured. The rates of complex dissociation were then extracted using the initial rate approximation method.69–71
For [Li2·1·2]I2, rate constants k−1 in CD2Cl2 were obtained for temperatures ranging from 290 to 315 K and the activation parameters for complex dissociation were determined from an Eyring plot (Fig. 12).72
![Estimation of dissociation barrier for the LiI complex by Eyring plot [Eyring plot: ln(k/T) = −ΔH#/RT + ΔS#/R + 23.76; estimated uncertainty on k
±20%]. (a) In CD2 Cl2: ΔH# = 54.6 kJ mol−1, ΔS# = −72.0 × 10−3 kJ mol−1 K−1, ΔG# (273 K) = 74.3 kJ mol−1. (b) In a CHCl3∶MeOD mixture (98∶2): ΔH# = 47.9 kJ mol−1, ΔS# = −39.6 × 10−3 kJ mol−1 K−1, ΔG# (273 K) = 58.7 kJ mol−1.](/image/article/2005/NJ/b415418e/b415418e-f12.gif) | ||
| Fig. 12 Estimation of dissociation barrier for the LiI complex by Eyring plot [Eyring plot: ln(k/T) = −ΔH#/RT + ΔS#/R + 23.76; estimated uncertainty on k ±20%]. (a) In CD2 Cl2: ΔH# = 54.6 kJ mol−1, ΔS# = −72.0 × 10−3 kJ mol−1 K−1, ΔG# (273 K) = 74.3 kJ mol−1. (b) In a CHCl3∶MeOD mixture (98∶2): ΔH# = 47.9 kJ mol−1, ΔS# = −39.6 × 10−3 kJ mol−1 K−1, ΔG# (273 K) = 58.7 kJ mol−1. | ||
We use relative rates to illustrate the kinetic stability differences in the [M2·1·2]X2 series: the relative rates were obtained by extrapolating each k−1 to 315 K while setting the slowest exchange at 315 K {for the [Li2·1·2]I2 complex in CD2Cl2} to unity (Table 6). This assumes that the free energy of activation does not change within that temperature range. Note that the equilibrium constants K could not be calculated, due to the absence of free host 1 from the system.
| MX | T/K | k −1/s−1 | Rel. rate |
|---|---|---|---|
| a Ref. 72. b Exchange between the mono-NaI complex and the bis-NaI complex. c Estimated based on broadening of resonances at different temperatures. | |||
| LiIa | 315 | 1.0 | 1 |
| LiBr | 280 | 1.7 | 50 |
| Li(CF3SO3) | 280 | 4 | 100 |
| LiB(C6F5)4 | 280 | 3 | 80 |
| NaI | 200 | 2.0b | 106 |
| Na(CF3SO3) | — | — | >104c |
| NaB[{3,5-(CF3)2(C6H3)}4] | 280 | 1.7 | 50 |
A kinetically more stable complex requires a higher temperature to achieve a given rate of exchange. Thus, for the [Li2·1·2]X2 series (in CD2Cl2) LiI forms the most kinetically stable complex, followed by LiBr, LiB(C6F5)4 and Li(CF3SO3) (Table 6).
The relative kinetic stabilities of the [Na2·1·2]X2 complexes could not be determined for a variety of reasons. For example, the NaI complex shows a more complex exchange behavior, involving a fast interchange between the mono-NaI and bis-NaI complexes, as well as a much slower dissociation to the free guest 2 species. Reliable data (from 1D NOESY experiments) could only be obtained for the exchange between the two NaI complexes and not for the dissociation. Although the spectrum of [Na2·1·2]X2 at 200 K is sharp, no dissociation is observed at this temperature: only slow exchange between the mono-NaI and bis-NaI species. At higher temperatures, at which dissociation should occur, the signals for the bound guest 2 resonances are broad: this is due to the fast exchange between the mono- and bis-NaI complexes at these temperatures.
ITC studies on the thermodynamics of pseudorotaxane formation
We have determined the apparent association constants for binding of 1 to 2 in the presence and absence of LiI in a mixture of CHCl3∶MeOH 98∶2, using isothermal titration microcalorimetry (ITC). This technique allows direct determination of the binding constant (and, hence, ΔG°) and enthalpy of binding and therefore allows calculation of the entropy of binding. The results are shown in Table 7. Without any alkali metal ion the association is based solely on stacking interactions between the aromatic systems of the donor and acceptor and is relatively weak (K = 21 M−1 in a mixture of CHCl3∶MeOH 98∶2). In the presence of LiI, analysis of the binding process is complex since a termolecular complex is formed. However, we have chosen to titrate 2 into a solution of 1 containing a large excess of LiI and under these conditions it is reasonable to assume that binding of Li+ to 1 is essentially complete, since this could not be independently measurable due to method limitations. Furthermore, the use of a large excess of LiI ensures that the concentration of I− counterions in the solution is relatively constant independent of the presence of the complex, thus eliminating complications arising in the determination of the dissociation constant for the ion pair.73 Data analysis is now simplified since we only need to consider 1∶1 binding of the acceptor to the pre-formed [Li2·1]I2 complex. Fig. 13 shows the results of the titration of a 200 mM solution of acceptor into a solution containing 5.0 mM crown ether and 50 mM LiI and the fit to a 1∶1 binding model. Also shown is the control experiment in which the acceptor solution is diluted into solvent, which shows nearly constant heat effects, indicating that the acceptor does not self-associate to any significant extent, despite the high concentration that had to be used. The results indicate that in the presence of 50 mM LiI the effective affinity for the acceptor increases 10-fold.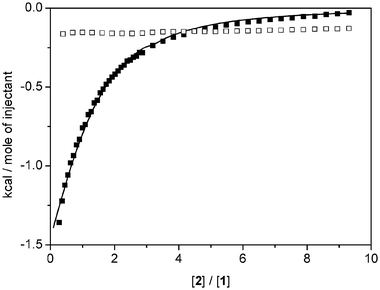 | ||
| Fig. 13 ITC titrations of a 200 mM solution of 2 into solvent CDCl3∶MeOD (98∶2) (□) and into a solution containing 5.0 mM 1 and 50 mM LiI (■) and the fit to a 1∶1 binding model (solid line). | ||
Inspection of the thermodynamic parameters in Table 7 indicates that, in the absence as well as in the presence of LiI, binding is completely enthalpy-driven and involves essentially no change in entropy. Apparently the loss of translational and rotational entropy of the donor and acceptor parts of the complex that has to occur upon binding is exactly compensated for by the gain in entropy caused by liberation of solvent molecules upon binding.
Conclusions
These pseudorotaxanes are selective towards small, singly charged cations with geometrically undemanding s orbitals (i.e., Li+ and Na+). The arrangement of the aromatic stacking is parallel for lithium complexes and perpendicular (or highly distorted parallel) for sodium complexes. Use of extremely bulky anions induces steric constraints and modifies the relative donor-acceptor geometry. Lithium cations fit precisely into the pre-organised tetraethylene glycol cavities, whereas the larger sodium cation sits outside the plane formed by the oxygen atoms of the donor and acceptor centres. In addition, solubility of salts also influences the selectivity of the pseudorotaxane. In the case of NaI, formation of a mono-NaI complex was observed as an intermediate.From a synthetic viewpoint, lithium cations represent ideal templates that could be employed in the formation of rotaxanes and catenanes. The lithium templated pseudorotaxane has ideal properties for the incorporation of this unit into supramolecular devices.
Experimental
Methods
1H and 13C NMR spectra were recorded on a Bruker Avance 500 MHz spectrometer using the residual 1H of the deuterated solvent as a reference. The pulse sequence used for 1D NOESY experiments was selnogp.2.68 UV-Vis spectra were recorded on Hewlett-Packard 8452A diode array spectrometer. All UV-Vis samples were prepared in freshly distilled CH2Cl2 and recorded at 25 °C. Isothermal titration calorimetry experiments were performed using a MCS Isothermal Titration Microcalorimeter (Microcal Inc. Northampton, MA, USA). Host–guest titrations were corrected for heat of dilution of the syringe contents by subtracting blank titrations into solvent (CHCl3∶MeOH 98∶2).General procedure for the formation of the cation templated pseudorotaxanes
To the 5.0 mM solutions of crown ether 1 (3.2 mg, 5.5 µmol, in CH2Cl2 or CHCl3∶MeOH 98∶2), the solid aromatic diimide 2 (1.92 mg, 5.5 µmol) was added. In each case, alkali metal halides (anhydrous) were added in excess (as ca. 20 mM solutions in CH2Cl2 or CHCl3∶MeOH 98∶2). The suspensions were sonicated for 30–60 s. The time the metal halides were exposed to the atmosphere was kept to the absolute minimum since the salts are extremely hygroscopic. When the salts LiB(C6F5)4 and NaB[3,5-(CF3)2(C6H3)]4 were used, manipulations were carried out under an atmosphere of N2 prior to product formation; however, handling of the final product and the NMR experiments were carried out under air and using wet CD2Cl2. Excess salts were separated by filtration and products isolated in quantitative yield after removal of solvent under reduced pressure.Crystal structure determination
Crystals were isolated by filtration and a specimen crystal selected under an inert atmosphere, covered with polyfluoroether, and mounted on the end of a nylon loop. Crystal data are summarised in Table 3.†Data for complexes [Li2·1·2]Br2, [Li2·1·2][B(C6F5)4]2, [Na2·1·2]I2 and [Na2·1·2][B{3,5-(CF3)2(C6H3)}4]2 were collected at 180 K on a Nonius KappaCCD with graphite-monochromated Mo-Kα radiation (λ = 0.710 73 Å), as summarised in Table 5. The images were processed with the DENZO and SCALEPACK programs.74 Crystals of [Li2·1·2]I2 were small and weakly diffracting, so a synchrotron radiation source was used to collect diffraction data for this compound (at 150 K). Data was collected at Station 9.8, Daresbury SRS, UK, using a Bruker SMART CCD diffractometer. The structures were solved by direct methods using the program SIR92.75 The refinement and graphical calculations were performed using the CRYSTALS76,77 and CAMERON78 software packages. The structures were refined by full-matrix least-squares procedure on F. All non-hydrogen atoms were refined with anisotropic displacement parameters. Hydrogen atoms were located in Fourier maps and their positions adjusted geometrically (after each cycle of refinement) with isotropic thermal parameters. Chebychev weighting schemes and empirical absorption corrections were applied.79
For complex [Na2·1·2][B{3,5-(CF3)2(C6H3)}4]2, treatment of 3.5 molecules of H2O (disordered) per asymmetric unit was performed using the procedure described by Spek80 implemented in PLATON.81 Structure contains solvent accessible voids of 265.00 A3, equivalent to ca. 3.5 molecules of H2O per asymmetric unit. Identification of the crystallising solvent as water is based upon additional chemical evidence from 1H NMR. Each of the eight CF3 groups has been modelled as disordered over two sites with refined occupancies. In view of the severe shortage of data their temperature factors have been refined isotropically. One hexyl chain has been modelled as disordered over two sites with refined occupancy and isotropic temperature factors.
Acknowledgements
We thank the Royal Society for a University Research Fellowship (SO) and BBSRC, EPSRC, AstraZeneca and GSK for financial support.References
- J. M. Lehn, Supramolecular Chemistry Concepts and Perspectives, VCH, New York, 1995 Search PubMed.
- C. Dietrich-Buchecker, G. Rapenne and J.-P. Sauvage, Molecular Catenanes, Rotaxanes, and Knots: a Journey through the World of Molecular Topology, Wiley-VCH, New York, 1999 Search PubMed.
- T. J. Hubin and D. H. Busch, Coord. Chem. Rev., 2000, 200, 5 CrossRef.
- C. Reuter, R. Schmieder and F. Vogtle, Pure Appl. Chem., 2000, 72, 2233 CrossRef CAS.
- A. R. Pease, J. O. Jeppesen, J. F. Stoddart, Y. Luo, C. P. Collier and J. R. Heath, Acc. Chem. Res., 2001, 34, 433 CrossRef CAS.
- K. Kim, Chem. Soc. Rev., 2002, 31, 96 RSC.
- L. Raehm, D. G. Hamilton and J. K. M. Sanders, Synlett., 2002, 1743 CAS.
- S. J. Rowan, S. J. Cantrill, G. R. L. Cousins, J. K. M. Sanders and J. F. Stoddart, Angew. Chem., Int. Ed., 2002, 41, 898 CrossRef.
- V. Balzani, Photochem. Photobiol. Sci., 2003, 2, 459 RSC.
- M. J. Gunter, Eur. J. Org. Chem., 2004, 1655 CrossRef CAS.
- J. O. Jeppesen, K. A. Nielsen, J. Perkins, S. A. Vignon, A. Di Fabio, R. Ballardini, M. T. Gandolfi, M. Venturi, V. Balzani, J. Becher and J. F. Stoddart, Chem.-Eur. J., 2003, 9, 2982 CrossRef CAS.
- H. R. Tseng, S. A. Vignon and J. F. Stoddart, Angew. Chem., Int. Ed., 2003, 42, 1491 CrossRef CAS.
- M. Asakawa, G. Brancato, M. Fanti, D. A. Leigh, T. Shimizu, A. M. Z. Slawin, J. K. Y. Wong, F. Zerbetto and S. W. Zhang, J. Am. Chem. Soc., 2002, 124, 2939 CrossRef CAS.
- A. Harada, Acc. Chem. Res., 2001, 34, 456 CrossRef CAS.
- V. Balzani, A. Credi, G. Mattersteig, O. A. Matthews, F. M. Raymo, J. F. Stoddart, M. Venturi, A. J. P. White and D. J. Williams, J. Org. Chem., 2000, 65, 1924 CrossRef CAS.
- C. P. Collier, J. O. Jeppesen, Y. Luo, J. Perkins, E. W. Wong, J. R. Heath and J. F. Stoddart, J. Am. Chem. Soc., 2001, 123, 12632 CrossRef CAS.
- Y. Luo, C. P. Collier, J. O. Jeppesen, K. A. Nielsen, E. Delonno, G. Ho, J. Perkins, H. R. Tseng, T. Yamamoto, J. F. Stoddart and J. R. Heath, ChemPhysChem, 2002, 3, 519 CrossRef CAS.
- M. R. Diehl, D. W. Steuerman, H. R. Tseng, S. A. Vignon, A. Star, P. C. Celestre, J. F. Stoddart and J. R. Heath, ChemPhysChem, 2003, 4, 1335 CrossRef CAS.
- C. Wu, M. C. Bheda, C. Lim, X. S. Ya, J. Sze and H. W. Gibson, Polym. Commun., 1991, 32, 204 CAS.
- J. P. Sauvage and M. Ward, Inorg. Chem., 1991, 30, 3869 CrossRef CAS.
- H. W. Gibson, M. Bheda, P. T. Engen, Y. X. Shen, J. Sze, C. Wu, S. Joardar, T. C. Ward and P. R. Lecavalier, Makromol. Chem., Macromol. Symp., 1991, 42–43, 395 Search PubMed.
- J.-P. Collin, C. Dietrich-Buchecker, P. Gavina, M. C. Jimenez-Molero and J.-P. Sauvage, Acc. Chem. Res., 2001, 34, 477 CrossRef CAS.
- M. C. Jimenez, C. O. Dietrich-Buchecker and J.-P. Sauvage, Angew. Chem., Int. Ed., 2000, 39, 3284 CrossRef CAS.
- A. M. Brouwer, C. Frochot, F. G. Gatti, D. A. Leigh, L. Mottier, F. Paolucci and G. W. H. Wurpel, Science, 2001, 291, 2124 CrossRef CAS.
- D. B. Amabilino, P. R. Ashton, L. Perezgarcia and J. F. Stoddart, Angew. Chem., Int. Ed. Engl., 1995, 34, 2378 CAS.
- F. M. Raymo and J. F. Stoddart, Pure Appl. Chem., 1997, 69, 1987 CAS.
- F. M. Raymo, K. N. Houk and J. F. Stoddart, J. Am. Chem. Soc., 1998, 120, 9318 CrossRef CAS.
- M. Asakawa, P. R. Ashton, V. Balzani, A. Credi, C. Hamers, G. Mattersteig, M. Montalti, A. N. Shipway, N. Spencer, J. F. Stoddart, M. S. Tolley, M. Venturi, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 1998, 37, 333 CrossRef CAS.
- S. J. Rowan and J. F. Stoddart, Org. Lett., 1999, 1, 1913 CrossRef CAS.
- P. R. Ashton, V. Balzani, J. Becher, A. Credi, M. C. T. Fyfe, G. Mattersteig, S. Menzer, B. N. Mogens, F. M. Raymo, J. F. Stoddart, M. Venturi and D. J. Williams, J. Am. Chem. Soc., 1999, 121, 3951 CrossRef CAS.
- P. R. Ashton, R. Ballardini, V. Balzani, A. Credi, K. R. Dress, E. Ishow, C. J. Kleverlaan, O. Kocian, J. A. Preece, N. Spencer, J. F. Stoddart and S. Wenger, Chem.-Eur. J., 2000, 6, 3558 CrossRef.
- V. Balzani, A. Credi, G. Mattersteig, O. A. Matthews, F. M. Raymo, J. F. Stoddart, M. Venturi, A. J. P. White and D. J. Williams, J. Org. Chem., 2000, 65, 1924 CrossRef CAS.
- A. R. Pease, J. O. Jeppesen, J. F. Stoddart, Y. Luo, C. P. Collier and J. R. Heath, Acc. Chem. Res., 2001, 34, 433 CrossRef CAS.
- J. O. Jeppesen, J. Perkins, J. Becher and J. F. Stoddart, Angew. Chem., Int. Ed., 2001, 40, 1216 CrossRef CAS.
- H. B. Yu, Y. Luo, K. Beverly, J. F. Stoddart, H. R. Tseng and J. R. Heath, Angew. Chem., Int. Ed., 2003, 42, 5706 CrossRef CAS.
- V. Balzani, M. Clemente-Leon, A. Credi, J. N. Lowe, J. D. Badjic, J. F. Stoddart and D. J. Williams, Chem.-Eur. J., 2003, 9, 5348 CrossRef CAS.
- M. Alvaro, B. Ferrer, H. Garcia, E. J. Palomares, V. Balzani, A. Credi, M. Venturi, J. F. Stoddart and S. Wenger, J. Phys. Chem. B, 2003, 107, 14319 CrossRef CAS.
- J. D. Badjic, V. Balzani, A. Credi, S. Silvi and J. F. Stoddart, Science, 2004, 303, 1845 CrossRef CAS.
- K. S. Chichak, S. J. Cantrill, A. R. Pease, S. H. Chiu, G. W. V. Cave, J. L. Atwood and J. F. Stoddart, Science, 2004, 304, 1308 CrossRef CAS.
- R. Hernandez, H. R. Tseng, J. W. Wong, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2004, 126, 3370 CrossRef CAS.
- J. O. Jeppesen, C. P. Collier, J. R. Heath, Y. Luo, K. A. Nielsen, J. Perkins, J. F. Stoddart and E. Wong, J. Phys. IV, 2004, 114, 511 Search PubMed.
- S. S. Kang, S. A. Vignon, H. R. Tseng and J. F. Stoddart, Chem.-Eur. J., 2004, 10, 2555 CrossRef CAS.
- Y. Liu, A. H. Flood and J. F. Stoddart, J. Am. Chem. Soc., 2004, 126, 9150 CrossRef CAS.
- H. R. Tseng, D. M. Wu, N. X. L. Fang, X. Zhang and J. F. Stoddart, ChemPhysChem, 2004, 5, 111 CrossRef CAS.
- S. A. Vignon, J. Wong, H. R. Tseng and J. F. Stoddart, Org. Lett., 2004, 6, 1095 CrossRef CAS.
- E. Baranoff, K. Griffiths, J.-P. Collin, J.-P. Sauvage, B. Ventura and L. Flamigni, New J. Chem., 2004, 28, 1091 RSC.
- M. Venturi, S. Dumas, V. Balzani, J. Cao and J. F. Stoddart, New J. Chem., 2004, 28, 1032 RSC.
- D. G. Hamilton, N. Feeder, L. Prodi, S. J. Teat, W. Clegg and J. K. M. Sanders, J. Am. Chem. Soc., 1998, 120, 1096 CrossRef CAS.
- D. G. Hamilton, N. Feeder, S. J. Teat and J. K. M. Sanders, New J. Chem., 1998, 22, 1019 RSC.
- D. G. Hamilton, J. E. Davies, L. Prodi and J. K. M. Sanders, Chem.-Eur. J., 1998, 4, 608 CrossRef CAS.
- Q. Zhang, D. G. Hamilton, N. Feeder, S. J. Teat and J. K. M. Sanders, New J. Chem., 1999, 23, 897 RSC.
- J. G. Hansen, N. Feeder, D. G. Hamilton, M. J. Gunter, J. Becher and J. K. M. Sanders, Org. Lett., 2000, 2, 449 CrossRef CAS.
- G. Kaiser, T. Jarrosson, S. Otto, Y.-F. Ng, A. D. Bond and J. K. M. Sanders, Angew. Chem., Int. Ed., 2004, 43, 1959 CrossRef CAS.
- T. Iijima, S. A. Vignon, H.-R. Tseng, T. Jarrosson, J. K. M. Sanders, F. Marchioni, M. Venturi, E. Apostoli and J. F. Stoddart, Chem.-Eur. J., 2004, 10, 6375 CrossRef.
- F. H. Huang, J. W. Jones, C. Slebodnick and H. W. Gibson, J. Am. Chem. Soc., 2003, 125, 14458 CrossRef CAS.
- For the bis-NaI complex, H1 and H2 have NOEs to NCH2CH2 > NCH2 > N(CH2)2CH2, H1 has small NOE to upfield glycol protons, whereas H2 sees the most downfield-shifted OCH2CH2 proton. H3 has strong NOEs to glycol loops OCH2CH2 and H4 has three small NOEs to OCH2CH2 protons. However, the small NOEs could also be interpreted as transfer NOEs of the mono-NaI complex, introduced via exchange between the mono- and bis-NaI complexes.
- B. Cabezon, J. Cao, F. M. Raymo, J. F. Stoddart, A. J. P. White and D. J. Williams, Angew. Chem., Int. Ed., 2000, 39, 148 CrossRef CAS.
- P. R. Ashton, E. J. T. Chrystal, J. P. Mathias, K. P. Parry, A. M. Z. Slawin, N. Spencer, J. F. Stoddart and D. J. Williams, Tetrahedron Lett., 1987, 28, 6367 CrossRef.
- P. R. Ashton, S. E. Boyd, A. Brindle, S. J. Langford, S. Menzer, L. Perez-Garcia, J. A. Preece, F. M. Raymo, N. Spencer, J. F. Stoddart, A. J. P. White and D. J. Williams, New J. Chem., 1999, 23, 587 RSC.
- P. R. Ashton, R. Ballardini, V. Balzani, M. Gomez-Lopez, S. E. Lawrence, M. V. Martinez-Diaz, M. Montalti, A. Piersanti, L. Prodi, J. F. Stoddart and D. J. Williams, J. Am. Chem. Soc., 1997, 119, 10641 CrossRef.
- P. R. Ashton, O. A. Matthews, S. Menzer, F. M. Raymo, N. Spencer, J. F. Stoddart and D. J. Williams, Liebigs Ann. Recl., 1997, 2485 Search PubMed.
- P. R. Ashton, S. E. Boyd, C. G. Claessens, R. E. Gillard, S. Menzer, J. F. Stoddart, M. S. Tolley, A. J. P. White and D. J. Williams, Chem.-Eur. J., 1997, 3, 788 CAS.
- H. R. Tseng, S. A. Vignon, P. C. Celestre, J. F. Stoddart, A. J. P. White and D. J. Williams, Chem.-Eur. J., 2003, 9, 543 CrossRef CAS.
- P. R. Ashton, S. E. Boyd, S. Menzer, D. Pasini, F. M. Raymo, N. Spencer, J. F. Stoddart, A. J. P. White, D. J. Williams and P. G. Wyatt, Chem.-Eur. J., 1998, 4, 299 CrossRef CAS.
- P. R. Ashton, V. Balzani, A. Credi, O. Kocian, D. Pasini, L. Prodi, N. Spencer, J. F. Stoddart, M. S. Tolley, M. Venturi, A. J. P. White and D. J. Williams, Chem.-Eur. J., 1998, 4, 590 CrossRef CAS.
- D. G. Hamilton, J. K. M. Sanders, J. E. Davies, W. Clegg and S. J. Teat, Chem. Commun., 1997, 897 RSC.
- P. R. Ashton, R. Ballardini, V. Balzani, A. Credi, M. T. Gandolfi, S. Menzer, L. Perez-Garcia, L. Prodi, J. F. Stoddart, M. Venturi, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 1995, 117, 11171 CrossRef.
- 1D NOESY using selective excitation with a shaped pulse; dipolar coupling may be due to NOE or chemical exchange. Pulse sequence used: selnogp.2 (Bruker, advanced version 00/02/07).
- A. Kumar, G. Wagner, R. R. Ernst and K. Wuthrich, J. Am. Chem. Soc., 1981, 103, 3654 CrossRef CAS.
- K. Stott, J. Stonehouse, J. Keeler, T.-L. Hwang and A. J. Shaka, J. Am. Chem. Soc., 1995, 117, 4199 CrossRef CAS.
- C. Naumann, B. O. Patrick and J. C. Sherman, Tetrahedron, 2002, 58, 787 CrossRef CAS.
- For [Li2·1·2]I2 the rate of dissociation was also determined in a mixture of CDCl3 and MeOD (98∶2). At 250 K, k−1 was 2.3 s−1. An Eyring plot was obtained to calculate activation parameters for the formation of the [Li2·1·2]I2 complex in CDCl3∶MeOD (98∶2) within the temperature range 230–265 K. The free activation energy is 15.6 kJ·mmol−1 lower than that of [Li2·1·2]I2 formation in CH2Cl2. Therefore [Li2·1·2]I2 is less stable kinetically in the more polar medium. Using relative rates is an illustrative way to describe kinetic differences. For example, in the case of the LiI complex, the dissociation/association process takes place 24
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times faster in CDCl3∶MeOD (98∶2) than in CD2Cl2!.
000 times faster in CDCl3∶MeOD (98∶2) than in CD2Cl2!. - J. W. Jones and H. W. Gibson, J. Am. Chem. Soc., 2003, 125, 7001 CrossRef CAS.
- Z. Otwinowski and W. Minor, in Methods in Enzymology, eds. C. N. Carter, Jr. and R. M. Sweet, Academic Press, London, 1996 Search PubMed.
- A. Altomare, G. Carascano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343 CrossRef.
- D. J. Watkin, C. K. Prout, J. R. Carruthers and P. W. Betteridge, CRYSTALS, Oxford, UK, 1996 Search PubMed.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
- D. J. Watkin, C. K. Prout and L. J. Pearce, CAMERON, Oxford, UK, 1996 Search PubMed.
- N. Walker and D. Stuart, Acta Crystallogr., Sect. A, 1983, 39, 158 CrossRef.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht, The Netherlands, 1998 Search PubMed.
Footnote |
| † CCDC reference numbers 254110–254114. See http://www.rsc.org/suppdata/nj/b4/b415418e/ for crystallographic data in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2005 |