Tuning the synthesis of a dinitroaromatic towards a new trinitroaromatic stabilized energetic material
Olivier
Siri
* and
Pierre
Braunstein
*
Laboratoire de Chimie de Coordination (UMR 7513 CNRS), Université Louis Pasteur, 4, rue Blaise Pascal, F-67070, Strasbourg cedex, France. E-mail: braunst@chimie.u-strasbg.fr
First published on 8th December 2004
Abstract
A new isomer of diaminotrinitrobenzene, 3, has been prepared from 1,2-diaminobenzene under high dilution conditions in which the two NH2 electron-donating groups increase the thermal stability of the 1,2,3-trinitrobenzene backbone. Its thermal decomposition only begins around 230 °C and deflagration occurs at 298 °C, which is of interest for heat-resistant explosives. The crystal structure of the known 1,2-diamino-4,5-dinitrobenzene, 4, which has been obtained in place of 3 under different dilution conditions, is also reported and reveals interesting van der Waals and intermolecular N–H⋯O–N hydrogen-bonding interactions.
Introduction
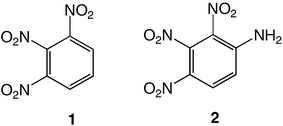
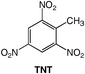
Energetic materials are metastable molecules endowed with high reactivity and numerous studies have been performed to develop capabilities to predict the sensitivity of explosives,1 that is, the ease with which these molecules undergo explosive decomposition by application of stimuli, producing smaller molecules and large amounts of energy.2 Aromatic compounds with nitro groups are known to be potential explosives,3 particularly when the number of nitro groups per ring is equal or superior to 3. In general, the power of an explosive molecule and its sensitivity increase with the number of NO2 groups.4 Explosives with high-temperature properties, usually called “heat-resistant explosives”, or secondary explosives, are the subject of intensive research and the relationship between their high thermal stability and their molecular and crystal structures are not yet fully understood.3 Previous works reported that the best way to generate much safer nitroaromatics, as for example, to desensitize a trinitro explosive while still keeping its explosive power, is to increase the number of electron-donating groups (e.g., NH2).2 This has allowed the reduction of trinitrotoluene (TNT)5 sensitivity by factors of 3 or 4.2 Furthermore, the amino substituents in TNT bring about an added benefit since the hydrogen bonding interactions between NO2 and NH2 groups increase the bulk density and thus the power, while providing desensitization.2 Trinitroaromatic molecules bearing an amino group represent a class of energetic materials classified as secondary explosives.3 For example, 1,2,3-trinitrobenzene, 1,6,7 is the parent molecule to secondary explosives such as 2, 6–8 which is stabilized due to the presence of an amino group.Therefore, we anticipated that further addition of an electron-donating substituent (e.g., −NH2) could give access to an even more stabilized form of 1,2,3-trinitrobenzene. Polynitroaromatic molecules can be prepared by direct oxidation of amines, which represents a convenient methodology owing to the availability of amines and the resulting high yields of the reactions.7,9 Polynitroaromatics bearing amino groups may be synthesized by nucleophilic aromatic substitution10,11 or by direct amination of nitrobenzene derivatives, proceeding by vicarious nucleophilic substitution of hydrogen.12,13 However, some polynitroaromatic compounds are still not accessible. This was the case until now for the new isomer 1,2-diamino-3,4,5-trinitrobenzene 3, for which we report a three-step synthesis from 1,2-diaminobenzene under high dilution conditions (Scheme 1). Under different dilution conditions,14 1,2-diamino-4,5-dinitrobenzene, 4, is isolated instead. Since this molecule has previously given rise to numerous theoretical calculations of its hyperpolarizability and electronic properties in relation to nonlinear optical properties,15–17 we decided to determine its precise structure by X-ray diffraction analysis.
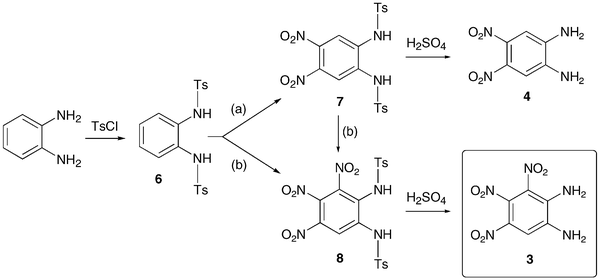 | ||
| Scheme 1 Synthesis of 3. (a) HNO3/glacial AcOH, low dilution conditions; (b) HNO3/glacial AcOH, high dilution conditions. | ||
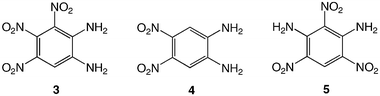
To the best of our knowledge, only one isomer of 3 appears to have been reported so far, 1,3-diamino-2,4,6-trinitrobenzene, 5, in which the nitro groups are not in sequential positions.18 Depending on the nature of the stimulus that triggers explosive decomposition, different types of sensitivity can be defined, such as heat, impact or shock, but also friction and electrostatic sensitivity.2 In the present study, only thermal behaviour will be discussed.
Results and discussion
Nitration of 1,2-diaminobenzene itself is not feasible and N,N′-protected derivatives have been generally used for the preparation of nitro compounds bearing two amino groups.14 In the course of the preparation of 1,2-diamino-4,5-dinitrobenzene, 4, an intermediate in the synthesis of functional ligands, we observed that, following the described procedure (route a, Scheme 1),14 intermediate 7 remained partly soluble although it was reported to precipitate in AcOH. Therefore, we anticipated that if the same reaction was carried out under high dilution conditions, precipitation of 7 could be prevented and further nitration facilitated. Whereas treatment of 6 with concentrated fuming HNO3/glacial AcOH under low dilution conditions afforded 7 in high yield, we have now found that the same reaction carried out under high dilution conditions leads instead to 8, which can be subsequently deprotected into 3 (route b, Scheme 1).Compound 8 was fully characterized except by X-ray diffraction and its 1H NMR data were consistent with the presence of three nitro groups that lower the molecular symmetry present in 7. Thus, the methyl groups of the tosyl moieties in 8 give rise to two singlets at δ 2.45 and 2.48, in contrast to 7 for which the tosyl groups appear as a singlet at δ 2.41. Detosylation was carried out in concentrated H2SO4 at 90 °C and recrystallization from a mixture of acetone–H2O afforded 3 as orange crystals. 3 revealed, as expected, a high thermal stability since the product begins to decompose , with a disappearance of the orange colour, only around 230 °C (gradient of 5 °C min−1). The measurements were made with specific safety precautions on a ca. 0.5 mg sample. For comparison, thermal data of related molecules are reported in Table 1.
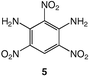
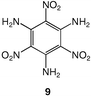
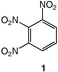
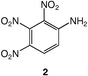
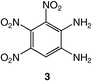
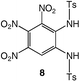
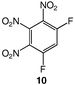
These data show the very significant thermal stabilization brought by the introduction of electron-donating groups on the trinitrobenzene skeleton.19 In the case of substituted 1,3,5-trinitrobenzene derivatives, the thermal stability of the material increases with the number of NH2 groups as indicated by the values of the melting (or decomposition) point of TNT, 5 and 9, the former being among the most stable trinitrobenzene derivatives20 (Table 1). Similarly, when 1,2,3-trinitrobenzene, 1,7 is stabilized by the presence of one NH2 group, as in molecule 2,7 the melting (or decomposition) temperature increases from 121–122 to 208–210 °C. Introduction of a second amino substituent, as in 3, leads to an even higher stability, with a decomposition temperature of 230 °C. Compared to 3, N-substitution by electron-withdrawing groups such as tosyls in 8 destabilizes the trinitroaromatic molecule (m.p. = 185 °C). Consistently, introduction of fluorine atoms in 10 destabilizes the parent molecule 1 (m.p. = 88–90 vs. 121–122 °C).7 Note however, that TNT is less stable than 1,3,5-trinitrobenzene, even though the methyl group is more electron-donating than hydrogen.21
Compound 3 was found to decompose with deflagration at 298 °C. Recrystallization from MeCN instead of a mixture of acetone–H2O afforded 3 as brown crystals, which decomposed with deflagration at 290 °C. The temperature at which this sample begins to decompose could not be determined with precision due to its darker colour, itself suggesting a different packing arrangement in the solid state relative to that in the orange crystals.22
Although 4 is a long-known compound14 that has given rise to numerous theoretical calculations regarding NLO properties (investigations on the electrostatic first hyperpolarizabilities of different chromophores with various donor-acceptor pairs),15–17 no X-ray structure has been reported so far despite its potential to reveal interesting repulsive intramolecular and attractive intermolecular interactions, depending on the degree of internal rotation of the amino and nitro groups about the C–N bonds.25 Single crystals of 4, suitable for an X-ray diffraction study, were grown from acetone. Crystallographic data and selected bond lengths and angles are given in Tables 2 and 3, respectively.
| Chemical formula | C6H6N4O4 |
| Formula weight/g mol−1 | 198.15 |
| Crystal system | Monoclinic |
| Space group | P21/n |
| a/Å | 8.7780(10) |
| b/Å | 5.6200(10) |
| c/Å | 15.635(2) |
| β/° | 98.504(5) |
| U/Å3 | 762.83(19) |
| Z | 4 |
| ρ calcd/g cm−3 | 1.725 |
| μ(MoKα)/mm−1 | 0.147 |
| Temperature/K | 173(2) |
| Total reflections | 5056 |
| Unique reflections | 2215 |
| R int | 0.0421 |
| Observed data [I > 2σ(I)] | 1670 |
| R 1 | 0.0489 |
| wR 2 | 0.1124 |
| C(1)–N(1) | 1.360(2) | C(5)–C(6)–C(1) | 121.5(1) |
| C(2)–N(2) | 1.372(2) | C(4)–C(3)–C(2) | 121.7(1) |
| C(4)–N(3) | 1.451(2) | C(6)–C(1)–0(1) | 120.2(1) |
| C(5)–N(4) | 1.451(2) | C(3)–C(2)–N(2) | 121.0(1) |
| C(1)–C(2) | 1.423(2) | C(3)–C(4)–N(3) | 117.5(1) |
| C(4)–C(5) | 1.396(2) | C(6)–C(4)–N(4) | 116.9(1) |
| O(1)–N(3) | 1.231(2) | C(5)–C(4)–N(3) | 122.9(1) |
| O(2)–N(3) | 1.230(2) | C(1)–C(2)–N(2) | 120.2(1) |
| O(3)–N(4) | 1.230(2) | C(6)–C(5)–C(4) | 119.9(1) |
| O(4)–N(4) | 1.226(2) | C(4)–C(5)–N(4) | 122.7(1) |
Compound 4 crystallizes in a centrosymmetric space group, which rules out second-order NLO properties in the solid state. Its crystal structure determination revealed that the nitrogen atoms N(3) and N(4) are not coplanar with the aromatic ring, in contrast with the amino nitrogen atoms N(1) and N(2). This results from the steric hindrance between the NO2 groups, which are twisted with respect to the phenyl ring (Fig. 1), as already observed in other o-dinitrobenzene derivatives.26
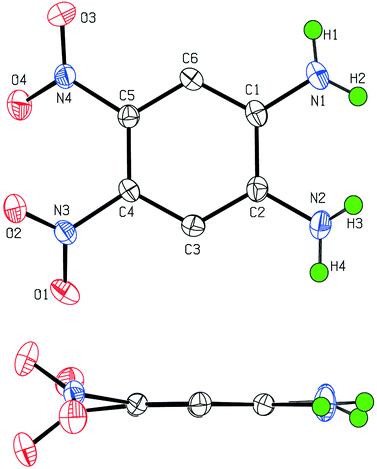 | ||
| Fig. 1 ORTEP representations of the molecular structure of 4 in the crystal. | ||
Examination of the bond distances (Table 3) shows that the two C–NH2 bonds [1.360(2) and 1.372(2) Å] are shorter than the C–NO2 bonds [1.451(2) Å], which is consistent with previous structural data collected on aromatic compounds bearing amino and nitro groups.27 In the solid state, van der Waals interactions lead to the formation of parallel sheets as indicated by the separations between atoms facing each other in two successive layers, with C(3B)⋯N(3E), C(6A)⋯C(6D) and N(1A)⋯N(4D) distances of 3.551(5), 3.218(5) and 3.411(5) Å, respectively (Fig. 2). Within each of these layers, all the chromophores are arranged in alternating orientations along the C(3D)–C(6D) molecular axis and assembled via N–H⋯O–N hydrogen bridges of 2.439(4) and 2.554(4) Å (Table 4).
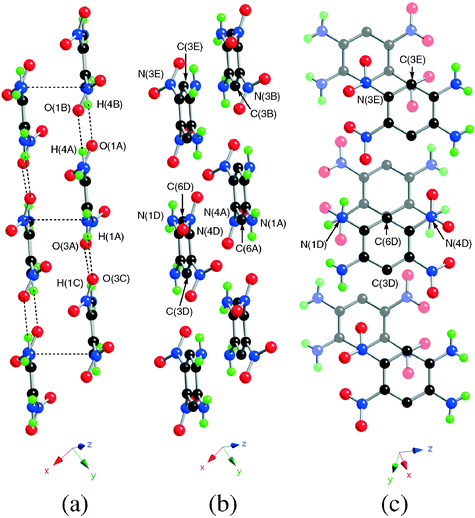 | ||
| Fig. 2 (a) Side view of the stacking arrangement generated by 4 in the solid state. (b) Intermediate view. (c) View of the sheet structure generated by 4 in the solid state. Colour coding: nitrogen, blue; oxygen, red; hydrogen, green. For clarity, the neighbouring molecules have been labelled A–E. | ||
Fig. 3 illustrates the intermolecular packing in which a central layer can be identified where each molecule interacts with four neighbours from the adjacent layers via N–H⋯O–N hydrogen bonds in the range 2.428(4)–2.552(4) Å (Table 4). Inter- and intralayer hydrogen bonding distances are similar and reveal a highly bifurcated system that forms a three-dimensional network.
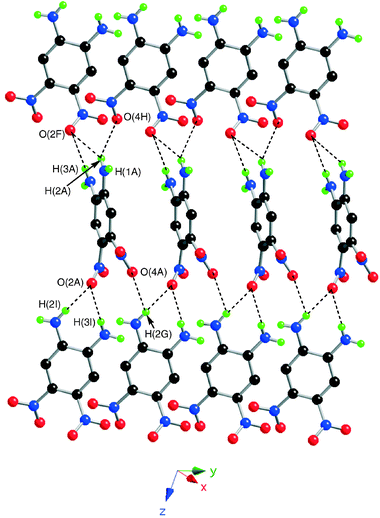 | ||
| Fig. 3 View of the N–H⋯O–N hydrogen-bonding network in the solid state structure of 4. Colour coding: nitrogen, blue; oxygen, red; hydrogen, green. | ||
| Intramolecular interactions (Fig. 1) | |
| H(2)⋯N(2) = 2.525(4) | |
| H(3)⋯N(1) = 2.555(4) | |
| Intermolecular interactions (Fig. 2) | Symmetry transformations used to generate equivalent atoms |
| O(1A)⋯H(4B) = 2.439(4); O(1A)⋯N(2B) = 3.286(5) | −x, 2 − y, −z |
| O(3A)⋯H(1C) = 2.554(4); O(3A)⋯N(1C) = 3.358(5) | 1 − x, −y, −z |
| C(6A)⋯C(6D) = 3.218(5) | 1 − x, 1 − y, −z |
| N(1A)⋯N(4D) = 3.411(5) | 1 − x, 1 − y, −z |
| C(3B)⋯N(3E) = 3.551(5) | −x, 2 − y, −z; x, 1 + y, z |
| Intermolecular interactions (Fig. 3) | |
| H(2A)⋯O(2F) = 2.552(4); N(1A)⋯O(2F) = 3.319(5) | 1/2 + x, 3/2 − y, 1/2 + z |
| H(3A)⋯O(2F) = 2.434(4); N(2A)⋯O(2F) = 3.231(5) | 1/2 + x, 3/2 − y, 1/2 + z |
| O(4A)⋯H(2G) = 2.428(4); O(4A)⋯N(1G) = 2.916(5) | −1/2 + x, 1/2 − y, −1/2 + z |
| H(2A)⋯O(4H) = 2.428(4); N(1A)⋯O(4H) = 2.916(5) | 1/2 + x, 1/2 − y, 1/2 + z |
| O(2A)⋯H(2I) = 2.552(4); O(2A)⋯N(1I) = 3.319(5) | −1/2 + x, 3/2 − y, −1/2 + z |
| O(2A)⋯H(3I) = 2.434(4); O(2A)⋯N(2I) = 3.231(5) | −1/2 + x, 3/2 − y, −1/2 + z |
Conclusion
3 constitutes the second known isomer of diaminotrinitrobenzene and reveals a high thermal stability for an explosive compound. Its synthesis represents a simple and new approach in the preparation of trinitrobenzene derivatives, which proceeds by a nitration reaction under high dilution conditions. Furthermore, such molecules often display unique chemical and electrochemical properties that should attract the interest of chemists.28 The determination of the crystal structure of 4 by X-ray diffraction provides new structural data on packing arrangements in aminonitro aromatic compounds resulting from van der Waals interactions and N–H⋯O–N hydrogen bonds. Such data will also be useful for a better understanding of the factors governing the conformation of the nitro groups in polynitroaromatic compounds bearing amino substituents since molecular structures have been recently established mostly by theoretical calculations.1,8,15–17 It is noteworthy that experimental studies on the polymorphism of 1,3,5-trinitrobenzene29 and on the crystal structures of reduction products of TNT have appeared very recently.30Experimental
General
1H NMR (300 MHz) spectra were recorded on a Bruker AC-300 instrument. FAB mass spectral analyses were recorded on an Autospec HF mass spectrometer and EI mass spectral analyses were recorded on a Finnigan TSQ 700. Elemental analyses were performed by the Service de Microanalyse, Université Louis Pasteur, Strasbourg. Compounds 4, 6 and 7 were prepared as reported in the literature.14Syntheses
X-Ray analysis
A single crystal of 4 suitable for X-ray analysis (obtained by slow evaporation of an acetone–H2O mixture) was mounted on a Nonius Kappa-CCD area detector diffractometer (Mo Kα, λ = 0.710 69 Å). A list of data collection conditions (Denzo software) and structure refinements are given in Table 2. The cell parameters were determined from reflections taken from one set of ten frames (1.0° steps in phi angle), each at 20 s exposure. The structure was solved using direct methods (SIR97) and refined against F2 using the SHELXL97 software. The absorption was not corrected. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms, which were not found by Fourier differences, were generated according to stereochemistry and refined using a riding model in SHELXL97.31†Acknowledgements
We are grateful to the CNRS and the Ministère de la Recherche et des Nouvelles Technologies for support. We thank Prof. R. Welter (ULP, Strasbourg) for the crystal structure determination of 4, and G. Jacob (SNPE, Vert-le-Petit, France) for ATG measurements and fruitful discussions.References
- B. M. Rice and J. J. Hare, J. Phys. Chem. A, 2002, 106, 1770 CrossRef CAS.
- S. Iyer and N. Slagg, in Structure and Reactivity, eds. J. F. Liebman and A. Greenberg, VCH, New York, 1986, ch. 7, p. 255 Search PubMed.
- T. Urbanski, Chemistry and Technology of Explosives, Pergamon Press, New York, 1984, vol. 4, p. 202 Search PubMed.
- A. J. Bell, E. Eadie, R. W. Read, B. W. Skelton and A. H. White, Aust. J. Chem., 1987, 40, 175 CAS.
- S. Iyer, J. Energ. Mater., 1984, 2, 151 Search PubMed.
- C. Chen and J. C. Wu, Comput. Chem., 2001, 25, 117 CrossRef CAS.
- A. T. Nielsen, R. L. Atkins and W. P. Norris, J. Org. Chem., 1980, 45, 2341 CrossRef CAS.
- P. C. Chen, S. C. Tzeng and F. M. Chang, Comput. Chem., 1999, 23, 503 CrossRef CAS.
- H. O. Larson, in The Chemistry of the Nitro and Nitroso Groups, ed. H. Feuer, Interscience, New York, 1969, part 1 Search PubMed.
- J. J. Wolff, A. Zietsch, T. Oeser and I. Bolocan, J. Org. Chem., 1998, 63, 5164 CrossRef CAS.
- J. J. Wolff, F. Gredel, T. Oeser, H. Irngartinger and H. Pritzkow, Chem. Eur. J., 1999, 5, 29 CrossRef CAS.
- M. Makosza and J. Winiarski, Acc. Chem. Res., 1987, 20, 282 CrossRef CAS.
- V. V. Rozhkov, S. A. Shevelev, I. I. Chervin, A. R. Mitchell and R. D. Schmidt, J. Org. Chem., 2003, 68, 2498 CrossRef CAS.
- G. W. H. Cheeseman, J. Chem. Soc., 1962, 1170 RSC.
- Y.-J. Liu, Y. Liu, X. Zhao and C.-B. Liu, Huaxue Xuebao, 2001, 59, 48 CAS.
- P. Wang, P. Zhu, W. Wu, H. Kang and C. Ye, Phys. Chem. Chem. Phys., 1999, 1, 3519 RSC.
- X.-L. Yang, C. Andraud and J. Zyss, Mol. Cryst. Liq. Cryst. Sci. Technol., Sect. B, 2000, 25, 139 CAS.
- J. Meisenheimer and E. Patzig, Ber. Dtsch. Chem. Ges., 1906, 39, 2533.
- We could not find in the open literature deflagration temperatures to be compared to that of 3 , which would be more relevant than melting/decomposition temperatures.
- R. L. Atkins, R. A. Hollins and W. S. Wilson, J. Org. Chem., 1986, 51, 3261 CrossRef CAS.
- We thank a referee for this comment. J. S. Murray and P. Politzer, in Chemistry and Physics of Energetic Materials, ed. S. N. Bulusu, Kluwer Academic Publishers, Dordrecht, Netherlands, 1990, pp. 157–173 Search PubMed.
- A major exothermic peak was observed by TGA at 274 °C (G. Jacob, personal communication). This may be due to differences in the molecular packing and/or crystallinity of the various samples. The onset temperature of 3 was measured at 251 °C, which is lower than that of its positional isomer 5 (ca. 295 °C2).
- Y. Okamoto and S. T. Attarwala, J. Org. Chem., 1979, 44, 3269 CrossRef CAS.
- D. M. O’Keefe, US Pat. 4833265A, 1989 Search PubMed.
- M. R. Manaa, R. H. Gee and L. E. Fried, J. Phys. Chem. A, 2002, 106, 8806 CrossRef CAS.
- M. Kubicki, T. W. Kindopp, M. V. Capparelli and P. W. Codding, Acta Crystallogr., Sect. B, 1996, 52, 487 CrossRef.
- (a) H. H. Cady and A. C. Larson, Acta Crystallogr., 1965, 18, 485 CrossRef CAS; (b) J. R. Holden, Acta Crystallogr., 1967, 22, 545 CrossRef CAS.
- J. J. Wolff, A. Zietsch, B. Nuber, F. Gredel, B. Speiser and M. Würde, J. Org. Chem., 2001, 66, 2769 CrossRef CAS.
- P. K. Thallapally, R. K. R. Jetti, A. K. Katz, H. L. Carrell, K. Singh, K. Lahiri, S. Kotha, R. Boese and G. R. Desiraju, Angew. Chem., Int. Ed., 2004, 43, 1149 CrossRef CAS.
- D. Graham, A. R. Kennedy, C. J. McHugh, W. E. Smith, W. I. F. David, K. Shankland and N. Shankland, New J. Chem., 2004, 28, 161 RSC.
- (a) B. V. Nonius, Kappa CCD Operation Manual, Delft, The Netherlands, 1997 Search PubMed; (b) G. M. Sheldrick, SHELXL-97, Program for refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
Footnote |
| † CCDC reference number 226339. See http://www.rsc.org/suppdata/nj/b4/b412789g/ for crystallographic data in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2005 |