Weak interactions between resorcinarenes and diquaternary alkyl ammonium cations
Heidi
Mansikkamäki
a,
Christoph A.
Schalley
b,
Maija
Nissinen
a and
Kari
Rissanen
*a
aNanoscience Centre, University of Jyväskylä, P.O. Box 35, FIN-40014, Jyväskylä, Finland. E-mail: hemansik@cc.jyu.fi, majoni@cc.jyu.fi, kari.rissanen@jyu.fi; Fax: +358 14 260 2501; Tel: +358 14 260 2672
bKekulé-Institut für Organische Chemie und Biochemie der Universität, Gerhard-Domagk-Str. 1, D-53121, Bonn, Germany. E-mail: c.schalley@uni-bonn.de
First published on 9th December 2004
Abstract
The interactions of resorcin[4]arenes 1 with alkyl ammonium cations bearing a 1,4-diazabicyclo[2.2.2]octane (DABCO) scaffold (32+, 42+ and 52+) were analyzed in the solid state by X-ray crystallography, in solution by 1H NMR spectroscopy, and in the gas phase by ESI-TOF mass spectrometry. The results are complemented with AM1 calculations and compared to previous reports on complexation studies of resorcinarenes with quaternary alkyl ammonium cations. The NMR titration results indicate that there are hardly any differences in the binding of the quaternary tetramethyl ammonium cation 2+ and the diquaternary N,N’-dimethyl DABCO dication 42+. The large N,N’-dibenzyl DABCO dication 52+ has two potential sites for inclusion, that is, the aryl groups and the central cationic part, and the complexation and interactions of both sites with 1 were verified in the NMR studies as well as in the solid state structures.
Introduction
Self-assembling, reversibly hydrogen-bonded capsules1 have contributed a great deal to our current understanding of enzyme action. The size-selective,2 and in some cases even stereoselective,3 inclusion of suitable guest molecules and the possibility to accelerate or catalyze chemical reactions4 within such capsules provide profound and general insights into the particular properties that govern the substrate specificity and the rate enhancement observed in biocatalysts.One family among these capsules is based on calixarenes or resorcinarenes (e.g, 1a–1f, Scheme 1). They have attracted great interest in supramolecular chemistry as models for receptors5 and as building blocks in various frameworks ranging from molecular capsules6 to tubular assemblies.7 The well-known solvent-assisted hexameric capsule of C-methyl resorcinarene, encapsulating neutral solvent molecules, is no doubt one of the most fascinating solid state resorcinarene assemblies.8 Several studies have shown that lipophilic resorcinarenes, (e.g, 1f) assemble into a hexameric capsule in water-saturated benzene and chloroform.9
 | ||
| Scheme 1 | ||
There are several studies on the dimeric analogues of hexamer capsules.6 Dimeric capsules that are solvent-mediated, like the hexamer capsule in the solid state, have been shown to encapsulate both neutral solvent molecules6b and small alkyl ammonium cations.6a,c–f Resorcinarenes have quite a high affinity towards alkyl ammonium cations in alkaline solution due to the deprotonation of the resorcinarene host and the resulting electrostatic interactions between host and guest.10a Under neutral conditions, closer to the biological environment, the binding of quaternary alkyl ammonium cations is weakened by a factor of several hundreds compared to the binding constants observed in alkaline solutions.10a Even though these results point towards rather weak cation-π interactions, these noncovalent forces are sufficiently high to act as a significant driving force in biological processes such as protein folding.11 Therefore, a better understanding of the subtle details related to them is an important research topic.
Besides crystallography and solution-phase studies of the binding interactions of cations to resorcinarenes, mass spectrometry12 is a perfectly suited tool for their examination, because it makes advantageous use of the cation present in the complex and gives access to data from the gas phase.13 Dimeric capsules of resorcinarenes and related compounds encapsulating small cations have been shown to prevail in the gas phase without solvent mediation and have been predicted to be directly hydrogen bonded.14
In this report, interactions of doubly protonated and N,N’-bisalkylated DABCO derivative cations 3+–5+ with resorcinarenes 1b–1f are analyzed, starting with the solid state structures of their corresponding complexes, followed by an NMR investigation of their interactions in solution, and finally, a discussion on the inclusion properties of diquaternary alkyl ammonium cations in the gas phase. The results are compared to earlier reports of resorcinarene complexes with quaternary and diquaternary alkyl ammonium cations.6e,f,10a
Results and discussion
Single-crystal X-ray analysis
In the following section, seven novel crystal structures of ammonium ion complexes of resorcinarenes 1b–1f are reported: one complex with tetramethyl ammonium 2+, four complexes with N,N’-dimethyl DABCO 42+ and two complexes with N,N’-dibenzyl DABCO 52+.We performed similar crystallization experiments with 1d and 1f, but, so far, we have not been able to co-crystallize quaternary alkyl ammonium cations with these two resorcinarenes. Instead of the complexation of the cation, solvent inclusion was observed or there simply were no crystals suitable for X-ray measurement.
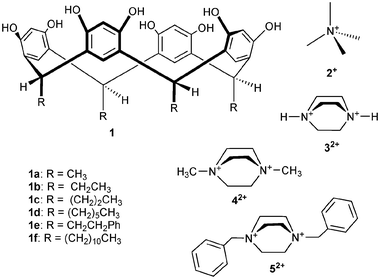
On the contrary, crystallization of 1e with 2+Br− (Me4NBr) from aqueous MeOH produces a dimeric solvent- and bromide-mediated capsule, complex I (1e·0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2+·Br−·2.5MeOH·0.5H2O, Fig. 1). The asymmetric unit comprises half a capsule, that is, one resorcinarene, which is in an almost perfect crown conformation (distances between centroids of opposite aromatic groups are 6.8 and 7.0 Å), stabilized by four intramolecular hydrogen bonds between the phenolic hydroxyl groups. The encapsulated cation 2+ is severely disordered, as are also the co-crystallized methanol molecules and the bromide. Due to the disorder of the capsule-mediating solvents and anions, the hydrogen-bonding scheme and the interactions holding the two halves together are difficult to analyze. However, it is justified to say that this capsule is not fully bound together as were our previous capsules,6e,f since all of the solvents hydrogen bonded to the hydroxy groups of 1d are not linked to both of the capsule halves via H-bonds, i.e. they do not play a mediating role in capsule formation. The capsules pack further to form hydrogen-bonded ribbons where the dimers of 1e are mediated by the bromide anions [Fig. 1(b) and 1(c)].
2+·Br−·2.5MeOH·0.5H2O, Fig. 1). The asymmetric unit comprises half a capsule, that is, one resorcinarene, which is in an almost perfect crown conformation (distances between centroids of opposite aromatic groups are 6.8 and 7.0 Å), stabilized by four intramolecular hydrogen bonds between the phenolic hydroxyl groups. The encapsulated cation 2+ is severely disordered, as are also the co-crystallized methanol molecules and the bromide. Due to the disorder of the capsule-mediating solvents and anions, the hydrogen-bonding scheme and the interactions holding the two halves together are difficult to analyze. However, it is justified to say that this capsule is not fully bound together as were our previous capsules,6e,f since all of the solvents hydrogen bonded to the hydroxy groups of 1d are not linked to both of the capsule halves via H-bonds, i.e. they do not play a mediating role in capsule formation. The capsules pack further to form hydrogen-bonded ribbons where the dimers of 1e are mediated by the bromide anions [Fig. 1(b) and 1(c)].
 | ||
| Fig. 1 X-Ray crystal structure of complex I. (a) A thermal ellipsoid (30% probability level) illustration of the dimeric capsule of 1e. Hydrogen bonds are indicated by dashed lines. Only OH hydrogens are shown. The other hydrogens, as well as the disordered cation and solvent molecules, are omitted for clarity. (b) Packing view showing the hydrogen-bond linkage of adjacent capsules. The cations, hydrogen atoms and solvent molecules are omitted. (c) Top view of the capsule chain. | ||
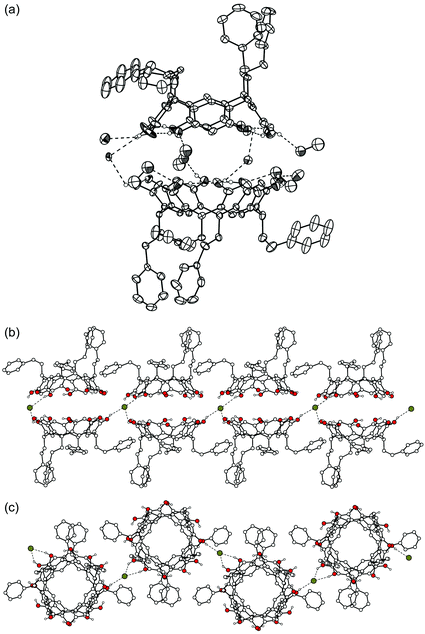
Instead of dimeric capsule structures, crystals of open complexes of 1 with both the chloride and the bromide salts of 42+ are easily obtained by slow evaporation from alcohol solutions. Here, we report four structures of such complexes: complex II (1b·242+·4Br−·n-PrOH·2H2O), complex III (1b·42+·2Br−·EtOH), complex IV (1c·42+·2Cl−·2MeOH) and complex V (1d·1.542+·3Br−·EtOH·H2O). The asymmetric unit of complex II comprises one resorcinarene (1b) and two 42+ cations with their bromide counterions, together with two water molecules and one n-propanol molecule [Fig. 2(a)]. One of the two cations is complexed within the cavity of 1b, practically in a horizontal position, forcing 1b to adopt a somewhat flattened crown conformation (distances of the centroids of the opposite aromatic rings are 6.6 and 7.2 Å). The other cation is located outside the resorcinarene cavity, interacting mostly with the counterions [Fig. 2(c)]. The shortest distances between the cation carbons and the centres of the closest aromatic rings of 1b are 3.4 Å for CH2, 3.6 Å for CH3 and 4.4 Å for N+, indicating C–H⋯π and N+⋯π interactions. The 42+ dication is not located as deeply in the cavity as 2+ in complex I, since the closest distance between the cation carbon and the plane formed by the methine bridges in 1b is 3.6 Å for complex I and 4.4 Å for complex II. In the crystal packing hydrophobic and hydrophilic layers are formed [Fig. 2(c)], similarly to what is observed in the crystal structures of various other resorcinarene complexes.6,15
 | ||
| Fig. 2 X-Ray crystal structure of complex II. Non-hydrogen bonding hydrogen atoms are omitted for clarity. (a) A thermal ellipsoid (50% probability level) illustration of the asymmetric unit. One of the two bromide anions and one n-propanol solvent are H-bonded to hydroxyl groups. Hydrogen bonds are shown by dashed lines. (b) Molecules of 1b are hydrogen-bonded to adjacent host molecules via H-bonds with n-propanol molecules or bromide anions. Cations are omitted for clarity. (c) Side view of the packing with guest ions, non-hydrogen bonding hydrogen atoms and solvent molecules omitted for clarity. | ||
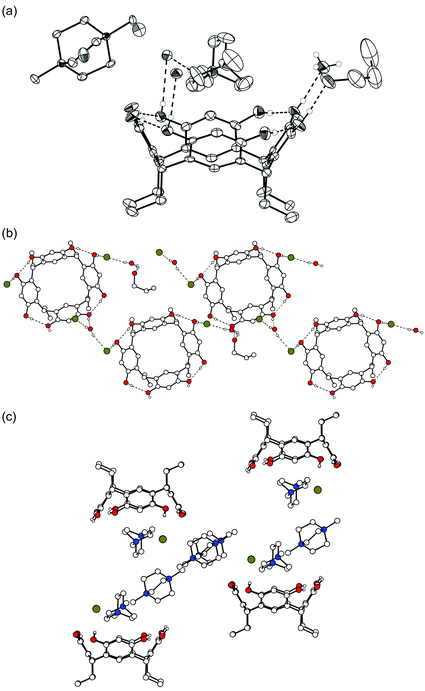
Crystallization of 1b from aqueous ethanol with 42+ dibromide results in crystals of an open 1∶1 complex III [1b·42+·2Br−·EtOH, Fig. 3(a)]. Again, the co-crystallized EtOH solvent molecule and the bromide anions are hydrogen-bonded to 1b, while the 42+ cation is located inside the slightly distorted 1b with distances between centres of facing aromatic rings being 6.5 and 7.2 Å. In this case, the cation is in an upright position, slightly tipped away from the symmetry axis of 1b to allow better interaction between the methyl group of the cation and an aromatic ring of 1b. The shortest distances between CH3, CH2 and N+ and the closest aromatic ring are 3.5, 3.3 and 3.9 Å, respectively. The upright position allows the cation to dive more deeply into the cavity with a distance of 2.8 Å between the closest guest's methyl carbon and the methine plane in 1b, 0.8 Å shorter than the corresponding value for 2+ in complex I. Each 1b molecule is hydrogen-bonded to the adjacent 1bvia EtOH molecules and/or Br− anions. The structure forms a wave-like H-bonded chain and hydrophilic and hydrophobic areas are layered [Fig. 3(b)].
 | ||
| Fig. 3 X-Ray crystal structure of complex III. Non-hydrogen bonding hydrogens are omitted for clarity. Hydrogen bonds are shown by dashed lines. (a) A thermal ellipsoid (50% probability level) plot of the asymmetric unit. One of the two bromide anions and one EtOH are H-bonded to 1b. (b) Resorcinarenes are connected via hydrogen bonds from phenolic OH groups to EtOH molecules, bromide anions and finally adjacent resorcinarenes, forming infinite chains. | ||
Complex IV (1c·42+·2Cl−·2MeOH) resulted from crystallizing 1c with 42+ dichloride in aqueous methanol (Fig. 4). It closely resembles complex III with respect to the cation binding and cation positions relative to the host. Also, the interactions of the chloride anions and MeOH solvent molecules with the upper rim OH groups of 1c are similar. Adjacent resorcinarenes are also H-bonded to each other similarly, that is, they are mediated via hydrogen bonds to Cl− and to methanol molecules.
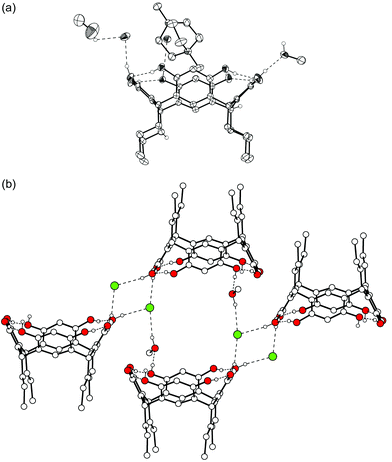 | ||
| Fig. 4 X-Ray crystal structure of complex IV with hydrogen bonds shown by dashed lines. (a) Thermal ellipsoids with 50% probability. (b) Packing and H-bond network in the complex. The Br− anions and one methanol molecule mediate the H-bonds between the hosts. | ||
It is interesting to compare the only dimeric capsule encapsulating 42+ (i.e., a dimer of 1b6f ) with complex IV because its components differ only by the counteranion, which is a bromide instead of a chloride, and the host, which is 1b instead of 1c. The hydrogen-bonding scheme of the closed 1b capsule is symmetrical and involves two water molecules in every other solvent bridge and one methanol molecule in every other bridge (Fig. 5). The cation position is horizontal and it is disordered over two positions with equal population parameters so that the methyl groups point into the corners of the hydrogen-bonding seam. This arrangement provides a good and tight fit for the dication inside the extended capsule's cavity (Fig. 5). Since the solvent system in both crystallization experiments was the same, it seems that the anion plays a pivotal role. In the capsular structure, the Br− anions are located in-between the ethyl chains of the host, fairly far away from their counterions (7.9 Å), while in complex IV, the Cl− ions are hydrogen-bonded to 1c and located directly next to the cations [4.4 Å, Fig. 4(a)]. Since Cl− is a harder anion than Br−, and thus a better hydrogen-bond acceptor, it is hydrogen-bonded to the OH groups of the host. In contrast, the softer Br− prefers the more lipophilic environment between the ethyl side chains. Also, the energy required for the charge separation of cation and anion is larger for Cl−, favouring anion binding in the direct proximity of the cation.
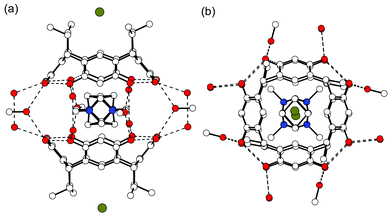 | ||
| Fig. 5 X-Ray crystal structure of the dimeric 1b capsule encapsulating disordered 42+. (a) Side view of the capsule showing the hydrogen-bonding pattern with dashed lines. The bromide anion is located in between the ethyl chains of 1b. (b) Top view of the capsule. Hydrogens are omitted for clarity.6f | ||
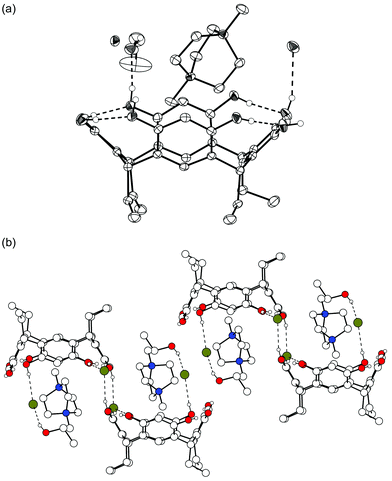
Finally, as the last complex of N,N′-dimethylated DABCO, complex V (1d·1.542+·3Br−·EtOH·H2O) was obtained by slow crystallization from aqueous ethanol. V is similar to complex II in the sense that there are two cations in different environments and only one of them is complexed inside the resorcinarene bowl, and this in a similar horizontal position as in complex II (Fig. 6). The other cation is part of a hydrophilic salt layer stabilized by electrostatic interactions. As typically observed with long alkyl chain resorcinarenes,16 the hexyl chains of one layer are staggered with chains from adjacent layers. Thus, hydrophilic and hydrophobic layers are formed [Fig. 6(b)]. Two of the three bromide anions in the asymmetric unit are disordered over two positions, which makes a detailed study of the hydrogen bonding basically impossible. The 1d molecules are directly hydrogen-bonded to two other 1d molecules and are additionally indirectly bridged by hydrogen bonds between a bromide anion and two OH groups of the adjacent resorcinarenes [Fig. 6(c)]. Three intramolecular hydrogen bonds stabilize the clearly flattened crown conformation of 1d (distances between centroids of opposite aromatic groups are 6.4 and 7.4 Å). This distortion of the 1d bowl and the fact that only one of the three DABCO bridges is complexed in the bowl (instead of two of them as in complex II) allows the dication 42+ to come a little closer to the aromatic groups of the host (closest distances for CH2, CH3 and N+ are 3.3, 3.4 and 4.3 Å, respectively) and to the plane of the methine bridges of 1d (3.5, 3.8 and 4.2 Å for CH2, CH3, and N+, respectively).
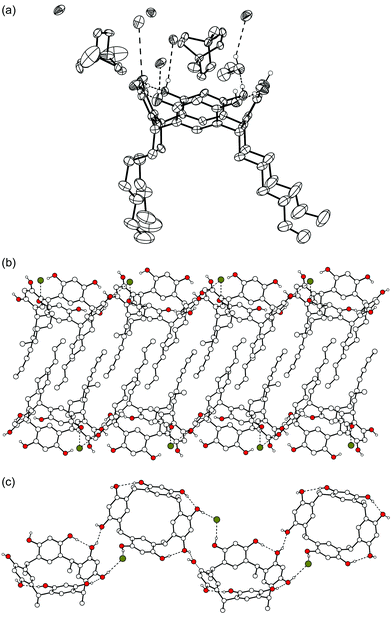 | ||
| Fig. 6 X-Ray crystal structure of complex V, where only OH hydrogens are shown. (a) Thermal ellipsoid plot of the complex. (b) The alkyl chains of 1d stagger together forming hydrophilic and hydrophobic layers. (c) Hydrogen bonds connect adjacent 1d molecules. Cations and solvent molecules are omitted for clarity. | ||
Salts of poor hydrogen bond acceptors such as I−, PF6− and BF4− were used in crystallization experiments, but, so far, no suitable crystals were obtained for analysis. The co-crystallization of 1f with 42+ halide salts from ethanol resulted only in crystals of the known ethanol solvate of 1f.16 This effect was also observed with methanol.
Two solid state structures, complex VI (21c·1.552+·3Br−·0.5H2O·7MeOH) and complex VII (1f·0.552+·Br−·2.5EtOH), provide examples of the two competing binding modes. In complex VI, which was crystallized from methanol, two independent resorcinarenes and two dications with different environments are found. One of the cations clearly interacts with a 1c molecule and is complexed with the DABCO moiety in a horizontal position, similarly to complexes II and V. The two benzyl groups point away from the resorcinarene [Fig. 7(a)]. The resorcinarene again adopts a flattened crown conformation (distances between the opposite aromatic groups are 6.3 and 7.5 Å), held together via intramolecular H-bonds, while the remaining OH groups are hydrogen-bonded to methanol and disordered water molecules or to the bromide anions. The other cation is located in close proximity to the benzyl group of the first cation, the distance of the centroids of the aromatic groups being ca. 4.3 Å. This indicates offset face-to-face π⋯π interactions between them. The second disordered dication also complexes to resorcinarenes with both of its benzyl groups [Fig. 7(b) and 7(c)]. Due to the severe disorder of the cation, the DABCO moiety could not be unambiguously assigned for the second cation.
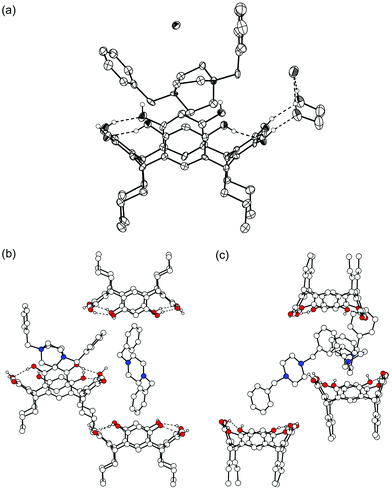 | ||
| Fig. 7 X-Ray crystal structure of complex VI. (a) Thermal ellipsoid plot drawn with 50% probability level of one of the cations and resorcinarenes in the structure. (b) View of the second, disordered dication and its interactions with 1c molecules. (c) Side view of the arrangement. | ||
In complex VII, the asymmetric unit comprises one 1f, half a 52+ and 2.5 EtOH molecules, and grows into a dimeric assembly of 1f mediated by cation complexation to both resorcinarenes via its phenyl groups (Fig. 8). All of the EtOH molecules are disordered, making the study of hydrogen bonding difficult. Also, the DABCO moiety is severely disordered and it could not be unambiguously assigned from the electron density map. The phenyl groups interact with the aromatic groups of 1f mostly face-to-face, the distance between the centroids of the interacting aromatic groups being 3.9 Å. Again the complex packs to form hydrophobic and hydrophilic layers throughout the crystal.
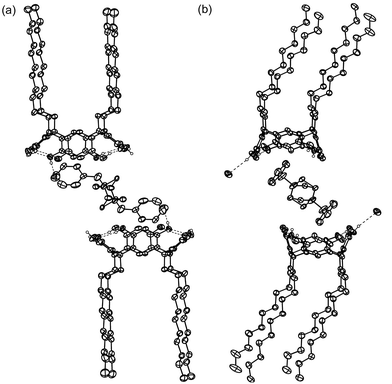 | ||
| Fig. 8 Thermal ellipsoid plot of complex VII with 50% probability level. Solvents and non-hydrogen bonding hydrogens are omitted for clarity. (a) The 2∶1 complex of 1d and 52+. (b) View of the bending of the alkyl chains and the offset π⋯π interactions of 52+ benzyl groups with the interior of 1d aromatic rings. | ||
NMR titration studies
When resorcinarene 1b was mixed with one of the guests 2+, 42+ or 52+, significant complexation-induced upfield shifts of the guest signals were observed, as expected from the shielding effects of the aromatic rings of the bowl-shaped host cavity. This clearly points at fast guest exchange compared to the NMR time scale at room temperature and even down to 233 K. Job plot experiments18 were performed in order to study the stoichiometry of the complexes (Fig. 9) and they indicate a clear 1∶1 complexation model in methanol-d4, irrespective of which of the guest signals was used for the evaluation of the experiment. The fact that 2∶1 dimer formation is not observed, even in the case of tetramethyl ammonium 2+, may be due to the efficiency with which the solvent competes with the hydrogen-bonding system and is included in the resorcinarene cavity, and that it also solvates quite well the ions present. Unfortunately, less polar solvents could not be used in the titration experiments due to the poor solubility of the corresponding salts in aprotic solvents.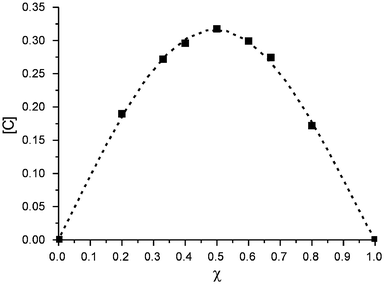 | ||
| Fig. 9 Job plot of the complexation of 42+ by 1b, as an example of a 1∶1 complex stoichiometry. | ||
Subsequently, 1H NMR titrations of 1b with the bromide salts of 2+–52+ were performed in the same solvent (e.g.,42+in Fig. 10). The binding constants for 2+, 42+ and 52+ at 233 K were determined to be 460 ± 40, 570 ± 50 and 600 ± 45 M−1 by the least-squares global fitting of the titration curve with the Specfit program.19 A model including only 1∶1 complexes and free host and guest species was used, giving excellent fits, and thus confirming the Job plot analysis. The binding constants for singly and doubly charged guests are within a narrow range, with the binding of the dication being slightly stronger. Due to the small change in chemical shifts observed for the guest signals and overlapping solvent signals, the binding constant for 32+ could not be determined precisely. The binding constants for similar quaternary alkyl ammonium cations determined by Atwood and Szumna10b at 298 K [KS = 93 M−1 for Me4N+ (2+), KS = 81 M−1 for Et4N+ and KS = 25 M−1 for Pr4N+] are in close agreement with our results.
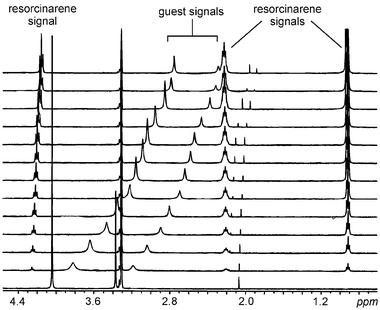 | ||
| Fig. 10 1H NMR titration of 42+ with a solution of 1b in methanol-d4 at 233 K. At each step, 0.3 equiv. of a 60 mM resorcinarene solution were added to a 4 mM solution of 42+(Br−)2. | ||
In order to determine the enthalpic and entropic contributions to the binding of the cations inside the resorcinarene cavity, temperature-dependent measurements of the complexation constants of 2+ and 42+ with 1b were performed in the temperature range from 233 to 313 K (Table 1). Van't Hoff plots of lnK versus 1/T (Fig. 11) exhibit linear relationships for both 2+ and 42+ cations, so that the separation of the enthalpic and entropic contributions to the binding is possible. The binding is largely enthalpic in nature with rather small binding entropies (ΔS). Both entropy values are slightly positive, although one might have expected negative values, since binding in a host-guest complex combines two molecules into one. However, solvation needs to be taken into account and methanol may well be present as a guest in the resorcinarene cavity20 before addition of the ammonium cations. Consequently, expulsion of methanol from the cavity upon cation binding and removal of at least part of the solvation shell around the cation are plausible reasons for the slightly positive entropy.
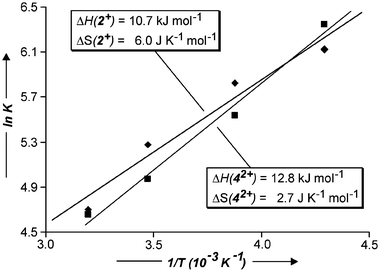 | ||
| Fig. 11 Van't Hoff plot allowing to evaluate enthalpic and entropic contributions to the binding of ammonium ions 2+ and 42+ with 1b. | ||
| K S/M−1 at T/K | ΔH/kJ mol−1 | ΔS/J mol−1 K−1 | ||||
|---|---|---|---|---|---|---|
| 233 | 258 | 288 | 313 | |||
| 2 + | 460 ± 40 | 340 ± 35 | 195 ± 35 | 110 ± 25 | −10.7 | 6.0 |
| 4 2+ | 570 ± 50 | 255 ± 30 | 145 ± 25 | 105 ± 30 | −12.8 | 2.7 |
In the NMR titrations of the bromide salt of 52+ with 1b, complexation-induced chemical shifts were observed for both the central dicationic DABCO moiety as well as for the aromatic protons of the benzyl groups (Fig. 12). In view of the Job plots, this suggests that a mixture of two different 1∶1 complexes is formed, which is in line with the solid state structures of complexes VI and VII, in which either the DABCO moiety is oriented into the resorcinarene cavity or one benzyl group is complexed in the cavity by π⋯π interactions, respectively. The binding constant can therefore only be regarded as the weighted average of those of the two complexes.
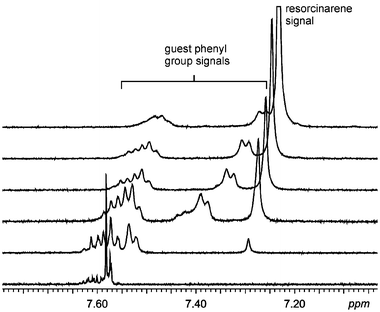 | ||
| Fig. 12 The aromatic region of the 1H NMR spectra of the titration of 52+ with a methanol-d4 solution of 1b at 233 K. The shift of the signals corresponding to the benzyl groups of the dication indicates binding interactions between the aromatics and the resorcinarene cavity. | ||
Finally, no complexation of any of the cations was observed in acetonitrile-d3, likely due to the formation of acetonitrile complexes with the resorcinarenes. Acetonitrile inclusion is common in calix[4]arene type cavities17b,20 and competes with the complexation of the cations.
Mass spectrometric studies
Previously, we reported mass spectral studies of complexes of 1a and 1b with singly charged alkyl ammonium cations Me4N+ (2+), Me2Et2N+, Et4N+ and Pr4N+, showing that dimeric molecular capsules of resorcinarene exist also in the gas phase, with the largest encapsulated guest being Et4N+.6e Similar experiments were conducted with doubly charged guests 32+–52+. Since these guest cations are somewhat larger than tetramethyl ammonium, it is still an open question whether they would provide capsules detectable by mass spectrometry. In our previous studies in the crystalline state, two of the smallest of these guests, 32+ and 42+, were shown to form solvent-extended capsules similar to those containing singly charged guest ions in their cavity.6e In the gas phase, however, the capsules of singly charged cations are not solvent-mediated. In fact, we suspect the resorcinarenes in the Me4N+-containing capsules to be directly hydrogen-bonded to each other via the upper rim hydroxyl groups.For electrospray ionization, solutions of the tetrafluoroborate salts of guests 32+–52+ (50 µM) and the corresponding resorcinarenes were used. Doubly protonated DABCO 32+ turned out not to be the guest of choice for ESI-MS experiments, although it would have made comparison to the crystal structures and NMR data more straightforward. During the electrospray process, it loses one of the two protons and the major signals in the mass spectrum are due to complex formation with its singly protonated analogue. Only minor signals were observed for doubly charged complexes.
Consequently, we switched to the dimethylated DABCO 42+ as the guest cation (Fig. 13). In the spectrum of this guest with resorcinarene 1b, the base peak corresponds indeed to the desired 1∶2 complex of guest ion and resorcinarene 42+·1b2 (m/z 672). This ion is doubly charged as evidenced by the spacings of Δm = 0.5 amu between two isotope peaks.
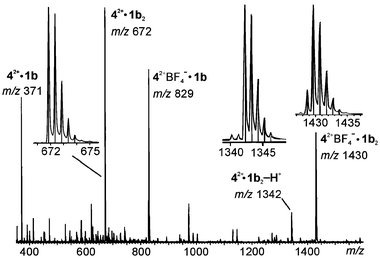 | ||
| Fig. 13 Positive ion ESI mass spectrum of a mixture of 42+BF4− and 1b in acetonitrile. The insets show the experimental isotope patterns (curves) and those calculated on the basis of the natural abundance (line spectra), which agree well with each other. Note that the peak spacing is Δm = 0.5 amu for the signal at m/z 672, indicating a doubly charged ion. | ||
Besides two monomeric ions, that is, 42+·1b (m/z 371) and 42+BF4−·1b (m/z 829), two more singly charged dimeric complexes are formed. In the first, 42+·1b2-H+ (m/z 1342), one of the positive charges of the guest is compensated through deprotonation of one of the resorcinarene's OH groups. Since its elemental composition differs from that of 42+·1b2 only by one hydrogen, the isotope pattern is basically the same, but the spacings between isotope peaks are now Δm = 1 amu, indicating the singly charged nature. The second singly charged dimer bears an additional BF4− anion and appears at m/z 1430. Since boron has two naturally occurring isotopes, 10B and 11B, the latter being the more abundant, the first signal in the isotope pattern is smaller than the second one. The ratio of these two indicates the presence of one boron atom and thus confirms that one anion is incorporated in the complex. Finally, a manifold of salt cluster signals, which we will not discuss in detail here, are observed at lower intensities. Resorcinarenes 1a and 1c behave quite similarly: the signals are just shifted according to the differences in mass for these derivatives.
Heterodimers again form quickly upon mixing solutions of 1a and 1b with 42+(BF4−)2 as the guest salt. Fig. 14 depicts the region of the spectra in which the doubly charged dimer complexes 42+·1a2 (m/z 616) and 42+·1b2 (m/z 672) appear. Traces (a) and (b) in Fig. 14 correspond to the two solutions before mixing, and trace (c) shows the result after mixing. The two homodimers and the heterodimer form in almost exactly the expected statistical 1∶2∶1 ratio. The exchange of monomers is complete in less than a minute. It should be noted that, although not shown, the other dimeric complexes also give rise to heterodimers.
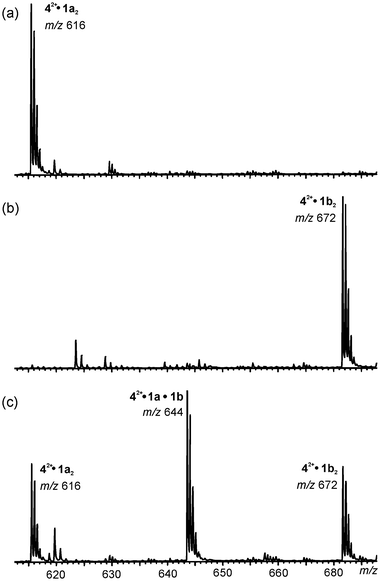 | ||
| Fig. 14 Partial ESI mass spectra of: (a) 42+BF4− with 1a, (b) 42+BF4− with 1b and (c) 42+BF4− with a 1∶1 mixture of 1a and 1b in acetonitrile. Heterodimer formation is clearly observed in the statistical 1∶2∶1 ratio. | ||
Also, the size selectivity was tested with bisbenzylated DABCO 52+, which is definitively too large to be trapped inside a dimeric capsule. Indeed, in the spectrum obtained from a competition experiment with 42+ and 52+, all signals correspond to complexes with 42+ as a subunit. In addition, monomeric complexes with 52+ are also observed, but no signal could be detected for any dimeric complex with this cation.
We hoped that it would be possible to use in-source fragmentation of the dimeric capsules to gather some information on their structure, the same way as this was possible for the singly charged capsules discussed above. Unfortunately, these results were somewhat ambiguous. Upon increasing the sample cone voltage, the dimeric 42+·1b2 ions vanished almost completely, indicating that these complexes do not represent very stable aggregates. However, HBF4 is lost from 42+BF4−·1b2 to generate 42+·1b2-H+, which gives an intense signal even at the highest possible sample cone voltage, while most of the other complexes were destroyed. Potentially, this suggests a much higher hydrogen bonding strength between a neutral and a singly deprotonated resorcinarene due to the charged nature of the O–H⋯O− hydrogen bond. Here, the negative ion spectrum (Fig. 15) is more informative. In general, two different series of ions are observed: (i) resorcinarenes attach a single BF4− anion and yield 1bnBF4− complexes (n = 1–4) at m/z 687, 1287, 1887 and 2487, respectively; (ii) a second series is observed in which one doubly charged guest cation is bound together with two additional BF4− counterions. This gives rise to 42+·1bn(BF4−)3 complexes (n = 1–3) at m/z 1003, 1603 and 2203, respectively. From all these ions, loss of HBF4 is possible, leading to ions containing a singly deprotonated resorcinarene (asterisks in Fig. 15). First of all, the complexes bearing three (and four) resorcinarene monomers are more prominent in this spectrum as compared to the positive ion spectrum, where they appeared with intensities hardly above the noise. They indicate that some unspecific aggregation occurs. However, at higher sample cone voltages they vanish completely and can thus be destroyed easily. Most interestingly, the 42+·1b2(BF4−)3 ion at m/z 1603, upon increase of the sample cone voltage, shows three consecutive losses of HBF4 rather than expulsion of the guest cation (inset in Fig. 15). The intensity of the signal at m/z 1603 decreases significantly in favour of that at m/z 1516 when collisions are induced by voltage changes. While no signal for additional losses of HBF4 are observed at a sample cone voltage of −40 V, they appear at a voltage of −70 V and further increase somewhat, even when the voltage is increased to −150 V. The fact that the guest cation is still present in these complexes strongly indicates that it must be bound rather strongly and one reasonable explanation would be the formation of a capsule-like structure.
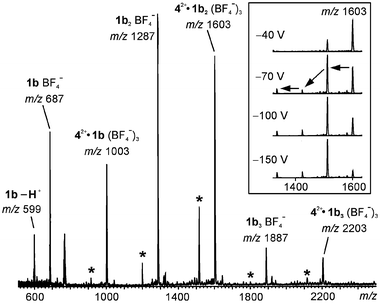 | ||
| Fig. 15 Negative ion ESI mass spectrum of the acetonitrile solution mixture of 42+BF4− and 1b. Asterisks indicate HBF4 loss related signals. The inset shows a series of spectra recorded at different sample cone voltages. At higher voltage, fragmentation is more pronounced and several consecutive HBF4 losses are observed. | ||
Consequently, the results for complexes containing doubly charged ammonium ions remain somewhat less convincing than those obtained for monocationic guests. Still, they point to the formation of capsules as far as size selectivity and fragmentation pattern for negatively charged species are concerned. The question remains though, on how a dimeric capsule, which has a preference for binding Me4N+ over Et4N+, 6e could easily accommodate an even larger guest such as 42+. We suggest that the dication allows tighter binding due to its dicationic nature. Since each side of the dication may bind one resorcinarene with one charge, cation-π interactions will be stronger than those operative for the monocations. This would allow a higher packing coefficient, that is, the size of the guest relative to the empty cavity size, and thus, the formation of capsules would be possible despite the larger size of the guest cation. An impressive increase in packing coefficient from a standard 0.55 to 0.78, due to interactions of a cationic guest with a calixarene dimer has been reported previously.13b,21 Similar effects would help to bind a larger, doubly charged guest inside a resorcinarene dimer. Another explanation would be that the seam of hydrogen bonds is not completely intact, when 42+ is encapsulated in 1b2. An overall capsular structure would still be energetically not too unfavourable, because it may optimize the number of hydrogen bonds in combination with cation-π interactions with the guest cation. Such a structure, which is only close to a perfect capsule, would then also help to rationalize why the results obtained for the doubly charged guests speak somewhat less clearly in favour of a capsule.
AM1 semi-empirical calculations
Since the mass spectrometric results are somewhat ambiguous and different explanations may be invoked to rationalize them, we performed AM1 semi-empirical calculations of the resorcinarenes, their capsules, and their host–guest complexes with 2+ and 42+ in the gas phase in order to get an idea of the structures of the ions involved. These calculations should also clarify whether a fully closed capsule of two resorcinarenes is possible, even in the absence of solvent molecules and counterions, when any of the ammonium cations included in this study is inside the cavity. The binding of the cations inside a capsule involves cation-π interactions, which rules out most force fields for molecular modelling, because they usually do not include the necessary parameters to describe these interactions reasonably. Indeed, some preliminary tests with several of them immediately showed that they did not give results consistent with the experimental data.Fig. 16 shows the optimized structures of the empty monomer and dimer of 1a and their complexes with tetramethyl ammonium 2+. The resorcinarenes are held in a crown conformation through intramolecular hydrogen bonding. The empty dimer shows a capsular structure in which the aromatic rings of the two resorcinarenes adopt a staggered conformation. The eclipsed arrangement, which has been found in the crystal structures of several solvent-mediated capsules,6e suffers from steric repulsion of the aromatic hydrogen atoms located between the resorcinarene OH groups on each aromatic ring. Eight O–H⋯O hydrogen bonds connect the two halves of the dimer. The calculated binding energy for the empty dimeric capsule amounts to ca. 88 kJ mol−1. This value (as most others included here) seems to be quite high, but if one considers that the calculations represent gas-phase structures, which do not suffer from competing solvent molecules, the energies appear to be reasonable. In methanol solution, this binding energy is likely reduced to almost zero and thus, binding of two resorcinarenes is not observed in the NMR experiments. However, empty dimers can be clearly detected in the gas phase as protonated species, when sprayed from methanol. The binding energy of the cation to the resorcinarene monomer is ca. 49 kJ mol−1. The cation is almost exactly centred inside the cavity with one methyl group pointing into the resorcinarene bowl.
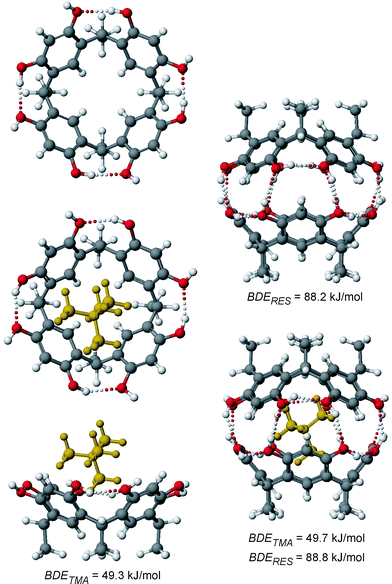 | ||
| Fig. 16 AM1-optimized geometries of the resorcinarene monomer 1a in its crown conformation (top left), its empty dimer (top right), the monomeric 2+·1a complex (bottom left, shown as top and side views), and the dimer-guest complex 2+·1a2 (bottom right). Hydrogen bonds are shown as dotted lines. The bond dissociation energies represent the calculated binding energies of the tetramethyl ammonium cation (BDETMA) and of the two resorcinarene monomers (BDERES). | ||
The most favourable cation-dimer complex 2+·1a corresponds to a closed capsule, whose geometry and hydrogen-bonding pattern do not change significantly with respect to the empty capsule. For the small tetramethyl ammonium cation, formation of a capsule without solvent molecules mediating the hydrogen-bonding seam is thus possible, which is in good agreement with the mass spectral data published previously.6e It is quite interesting to see that none of the binding energies discussed above changes much when the inclusion complex is compared to the monomer-guest complex or the empty capsule. The examination of the binding of dication 42+ requires first to consider different orientations of the guest inside the host. Three different monomer-guest complexes 42+·1a are shown in Fig. 17. It turns out that an arrangement corresponding to an orientation of 2+ in which one methyl group points into the resorcinarene bowl does not correspond to the most energetically favourable structure. Instead, the insertion of one CH2CH2 bridge of the dication is optimal, because (i) it maximizes electrostatic interactions of the cation with the partially negative oxygen atoms by minimizing the distance and (ii) the four hydrogen atoms from the bridge inside the cavity can favourably interact with the four aromatic rings of the resorcinarene. They are almost perfectly arranged on top of the four rings and thus, correspond much better to the fourfold symmetry of the host than a methyl group. Consequently, CH-π interactions can contribute significantly to the stability of the complex. This arrangement is found in many of the crystal structures of resorcinarenes with 42+ (Figs. 2, 5 and 6). The dication in its upright arrangement is also found, although the calculations predict it to be energetically less favourable. This may be due to the counterions being close to the dication in these structures (Figs. 3 and 4). The calculations are thus in excellent agreement with our experimental results.
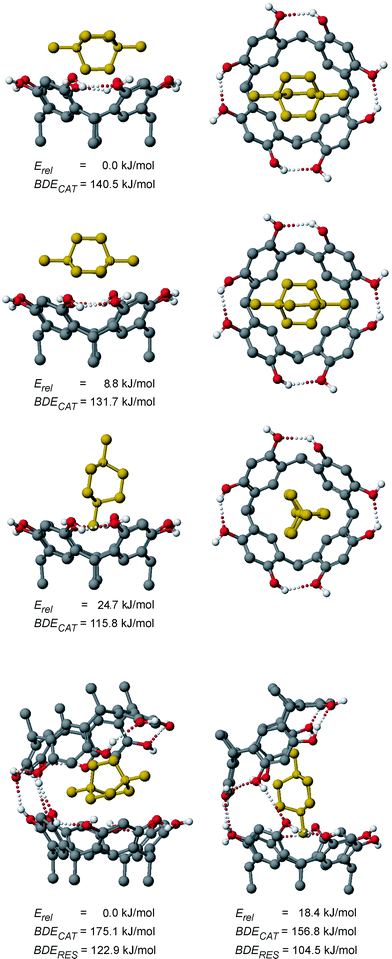 | ||
| Fig. 17 From top to bottom: three AM1-optimized geometries of different conformations of 42+·1a shown as side (left) and top (right) views, which differ from each other only with respect to the orientation of the encapsulated dication, and AM1-optimized structures (left and right, bottom) of two conformations of the dimer-guest complex 42+·1a2. Erel provides the calculated relative stability of the structures, BDECAT refers to the calculated binding energies of the dication, and BDERES refers to the dication binding energies relative to the empty dimer shown in Fig. 16. Hydrogen bonds shorter than 3.5 Å are shown as dotted lines. It should be noted that relative energies can only be compared within a series of species with the same elemental formula. | ||
When 42+ was inserted into the closed capsule of the resorcinarene dimer shown in Fig. 16, the calculation optimized to two conformers for 42+·1a2, which differ by the orientation of the dication (Fig. 17, bottom). Obviously, the dication is too large to fit inside a fully closed capsule without solvent participation and part of the hydrogen bonding seam is thus broken. In both structures, three hydrogen bonds connecting the two halves remain. Such structures are in line with the results obtained by mass spectrometry.
The binding energy of one resorcinarene monomer to the monomeric host–guest complex is higher than that calculated for the empty dimer shown in Fig. 16, although the seam of hydrogen bonds is not intact in 42+·1a2. This suggests that a significant part of the binding energy comes from the interaction with the dicationic guest rather than the other resorcinarene monomer. Similarly, a more than three times higher binding energy is calculated for the monomer-guest complex 42+·1a compared to the monocationic analogue 2+·1a. Apparently, the horizontal orientation of the dication provides quite a significant optimization of the binding interactions, compared to the situation where a methyl group is diving into the cavity.
Conclusions
The present paper collects structural information on host–guest complexes of alkyl ammonium ions and resorcinarenes: in the solid state, where packing and space filling is an important factor; in solution, where interactions with solvents come into play; in the gas phase, where no environment is present at all; and complements them with semi-empirical calculations.When small quaternary cations are complexed by resorcinarenes, solvent-mediated dimeric capsules are formed quite often in the solid state, while such capsules are stable in the gas phase even without solvent connecting the hydrogen-bonding seams.6e The use of a polar competitive solvent such as methanol or acetonitrile destroys these capsules in solution in favour of either 1∶1 complexes or resorcinarenes occupied solely by solvent molecules.
The use of the larger, doubly charged N,N’-bisalkylated DABCO derivatives prevents, in most cases, the formation of a fully closed capsule. In the crystal, 1∶1 complexes are co-crystallized, while capsules are scarcely found,6f and in solution, only 1∶1 complexes are formed. In the mass spectra, however, signals appear for 2∶1 complexes, which very likely do not have a capsular structure, but rather incorporate the cation in a “half-open capsule”. Part of the seam of hydrogen bonds remains intact and the interactions with the dicationic guests are maximized in such structures. In terms of energies, the situation in the gas phase differs radically from that in methanol solution. In the gas phase, no competition of the hydrogen bonds between two resorcinarenes with those formed with methanol occurs.
The counterions play a significant role in each of the solid state structures as hydrogen bond acceptors and only complexes of alkyl ammonium halide salts have successfully been crystallized, while salts with poor H-bond acceptors, such as BF4 and PF6, have resulted as precipitates or crystals of the starting compounds. In solution, the counterions stabilize the cations and thus diminish the binding energies significantly, while methanol, as a good hydrogen-bond donor and acceptor, interferes with the binding of the two resorcinarenes. In the gas phase, the counterions are absent and thus the intrinsic binding energies fully support the formation of 2∶1 complexes of resorcinarenes, even with larger guests, which destroy part of the seam of hydrogen bonds.
In view of earlier reports,6,10 which showed additional effects coming from the counterions or the length of the resorcinarene side chain, we are now able to conclude that resorcinarenes, although quite rigid, offer a rich structural chemistry full of subtleties, which depend on the details of their particular features as well as on the properties of their environment.
The differences in behaviour in all three states makes it difficult to draw conclusions from, for example, an X-ray crystal structure and extrapolate to the properties in solution. On the other hand, the amount of information on noncovalent forces gathered from these systems is particularly high because of these differences, which provide more insight into the role of the environment than many other systems. Understanding these subtle effects certainly contributes to obtaining significant insight into noncovalent interactions, which are important in many other fields of chemistry.
Experimental
Crystal structure determination
The solid state structures were crystallized by slow evaporation of the solvent from 2∶1 mixtures of the corresponding resorcinarenes and alkylammonium salts in aqueous MeOH, EtOH or n-propanol. In all cases the crystals formed within one week. A selection of crystal data and collection parameters are compiled in Table 2.†The X-ray crystallographic data for all complexes were recorded with a Nonius Kappa CCD diffractometer. Graphite monochromated MoKα radiation (λMoKα = 0.710 73 Å) and a temperature of 173.0 ± 0.1 K were used in all cases. The CCD data were processed with Denzo-SMN v. 0.95.373 22a and all structures were solved by direct methods (SHELXS-97 22b) and refined on F2 by full-matrix least-squares techniques (SHELXL-97 22c). Whenever possible, the hydrogen atoms of the resorcinarene groups were located from the electron density map and the rest of the hydrogen atoms were calculated in their idealized positions with isotropic temperature factors (1.2 or 1.5 times the carbon temperature factor) and refined as riding atoms. The MeOH molecule and the cation in complex I, one EtOH molecule in V, one of the cations in VI and one EtOH molecule in VII were disordered and treated isotropically, while the disordered Br− and H2O in complex I, the ethyl group of 1b in II, the hexyl group of 1d, the Br−and H2O in V, the Br− in VI and the EtOH molecule and cation in VII could all be treated anisotropically, despite the disorder.
| I | II | III | IV | V | VI | VII | |
|---|---|---|---|---|---|---|---|
|
1e·0.52+·0.5Br−·2.5MeOH·0.5H2O |
1b·242+·4Br−·n-PrOH·2H2O |
1b·42+·2Br−·EtOH |
1c·42+·2Cl−·2MeOH |
1d·1.542+·3Br−·EtOH·H2O |
21c·1.552+·3Br−·0.5H2O·7MeOH |
1f·0.552+·Br−·2.5EtOH |
|
| a With I > 2σ( I0). | |||||||
| Formula | C64.5H73O11N0.5Br0.5 | C55H88O11N4Br4 | C46H64O9N2Br2 | C50H74O10N2Cl2 | C66H107O10N3Br3 | C117H164O23.5N3Br3 | C87H140O10.5NBr |
| M r | 2142.4 | 1300.9 | 948.8 | 934.0 | 1342.3 | 2228.2 | 1447.9 |
| a/Å | 29.4551(5) | 12.4513(2) | 12.0544(1) | 11.3877(2) | 53.2560(8) | 15.8558(3) | 9.8246(1) |
| b/Å | 15.4086(2) | 13.6703(2) | 12.8536(2) | 12.5699(3) | 12.6490(3) | 17.8230(4) | 15.7155(2) |
| c/Å | 25.4790(5) | 19.7836(4) | 15.3163(2) | 17.5983(4) | 20.5761(7) | 20.2560(4) | 26.8439(4) |
| α/° | 90 | 71.716(5) | 85.953(1) | 99.359(1) | 90 | 85.693(1) | 91.387(1) |
| β/° | 99.503(3) | 79.631(5) | 84.491(1) | 91.183(1) | 97.955(1) | 88.540(1) | 91.438(1) |
| γ/° | 90 | 72.394(1) | 69.520(1) | 98.780(1) | 90 | 86.632(1) | 92.686(1) |
| U/Å3 | 11405.3(3) | 3034.2(1) | 2211.1(1) | 2453.7(1) | 13727.4(6) | 5697.0(2) | 4137.5(1) |
| Z | 8 | 2 | 2 | 2 | 4 | 2 | 2 |
| Space system | Monoclinic | Triclinic | Triclinic | Triclinic | Monoclinic | Triclinic | Triclinic |
| Space group | C2/c (no. 15) | P-1 (no. 2) | P-1 (no. 2) | P-1 (no. 2) | C2/c (no. 15) | P-1 (no. 2) | P-1 (no. 2) |
| ρ calcd/Mg m−3 | 1.248 | 1.424 | 1.425 | 1.264 | 1.299 | 1.299 | 1.162 |
| μ/mm−1 | 0.432 | 2.71 | 1.890 | 0.191 | 1.818 | 1.133 | 0.553 |
| Reflections measured | 17408 | 24116 | 22107 | 13446 | 32427 | 32249 | 21336 |
| Unique reflections | 10066 | 10107 | 8301 | 8605 | 11413 | 17331 | 14265 |
| Reflections used in refinementa | 5632 | 6452 | 7209 | 5747 | 3703 | 8271 | 9062 |
| R int | 0.058 | 0.046 | 0.029 | 0.035 | 0.137 | 0.094 | 0.032 |
| R | 0.108 | 0.049 | 0.030 | 0.061 | 0.068 | 0.074 | 0.063 |
| wR | 0.276 | 0.077 | 0.065 | 0.128 | 0.134 | 0.139 | 0.118 |
1H NMR titration
NMR titrations were performed in methanol-d4 by titrating 1 ml of a 4 mM solution of the bromide salt of the corresponding alkyl ammonium salt with a 60 mM solution of ethyl resorcin[4]arene. The 1H NMR spectra of each titration step was measured on a 500 MHz Bruker Avance DRX spectrometer equipped with temperature regulation. Binding constants were obtained by fitting the titration curves with the Specfit program package.19ESI-TOF mass spectroscopy
The mass spectrometric experiments were performed with a Micromass LCT ESI-TOF instrument equipped with a Z geometry electrospray ion source. For both positive and negative ion spectra, the samples were introduced into the source as acetonitrile solution mixtures of 1a (50 µM) and the salts of doubly charged cations 42+ and 52+ (50 µM) at flow rates of 15 µl min−1. Tetrafluoroborate salts of these guests gave the best results, probably due to increased solubility in acetonitrile, but bromides could also be used. Acetone was an alternative spray solvent and gave almost identical results. The addition of methanol is not advantageous, but a minor amount might help to dissolve the salts without hampering the measurements.Constant spray and highest intensities were achieved with a capillary voltage of 3700 V at a source temperature of 80 °C and a dissolvation temperature of 120 °C. Other selected source parameters were as follows: sample cone voltage 20–30 V; extraction cone voltage 3–6 V; flow of cone gas 10 l h−1; flow of dissolvation gas 150 l h−1. Other parameters did not influence much the ion intensities. The sample cone voltage was optimum at a somewhat higher value of 50 V for negative ion spectra. For the examination of heterodimer formation, two of the sample solutions were mixed in a 1∶1 ratio and then subjected to the same experiments. Multiple scans (50–200) were recorded and averaged for each spectrum in order to improve the signal-to-noise ratio. Since the instrument does not permit MS/MS experiments, the fragmentation behaviour of the samples was examined by in-source fragmentation, induced by collisions with the gas molecules present in the ion source. For this purpose, the ions were accelerated to different kinetic energies by varying the sample cone voltage. At high voltages, the ions undergo collisions at higher velocities and usually pronounced fragmentation is observed.
AM1 calculation
Several alternative conformations of the molecules and complexes shown in Fig. 16 and Fig. 17 were optimized at the AM1 level of theory23 as implemented in the MOPAC version delivered with the CaChe 5.0 program package.24Acknowledgements
The Graduate School of Bio-organic and Medicinal Chemistry (H.M.) and Academy of Finland (M.N.) are gratefully acknowledged. C.A.S. wishes to thank the Deutsche Forschungsgemeinschaft for a Heisenberg fellowship, the Fonds der Chemischen Industrie for a Dozentenstipendium, and the Deutscher Akademischer Austauschdienst for travel support. We also wish to thank Mr Reijo Kauppinen and Prof. Erkki Kolehmainen for assistance with the NMR experiments and Dr Minna Luostarinen for providing the resorcinarenes.References
- (a) M. M. Conn and J. Rebek, Jr., Chem. Rev., 1997, 97, 1647 CrossRef CAS; (b) C. A. Schalley, Adv. Mater., 1999, 11, 1535 CrossRef CAS; (c) J. Rebek, Jr., Acc. Chem. Res., 1999, 32, 278 CrossRef; (d) C. A. Schalley and J. Rebek, Jr., in Chemical Encapsulation in Self-Assembling Capsules, Stimulating Concepts in Chemistry, eds. J. F. Stoddart, F. Vögtle and M. Shibasaki, Wiley–VCH, Weinheim, 2000, p. 199 Search PubMed.
- T. Heinz, D. M. Rudkevich and J. Rebek, Jr., Nature (London), 1998, 394, 764 CrossRef CAS.
- (a) J. M. Rivera, T. Martín and J. Rebek, Jr., Science, 1998, 279, 1021 CrossRef CAS; (b) R. K. Castellano, C. Nuckolls and J. Rebek, Jr., J. Am. Chem. Soc., 1999, 121, 11156 CrossRef CAS.
- (a) J. Kang and J. Rebek, Jr., Nature (London), 1997, 385, 50 CrossRef CAS; (b) J. Kang, J. Santamaría, G. Hilmersson and J. Rebek, Jr., J. Am. Chem. Soc., 1998, 120, 7389 CrossRef CAS.
- K. Murayama and K. Aoki, Chem. Commun., 1997, 119 RSC.
- (a) K. Murayama and K. Aoki, Chem. Commun., 1998, 607 RSC; (b) K. N. Rose, L. J. Barbour, G. W. Orr and J. Atwood, Chem. Commun., 1998, 407 RSC; (c) A. Shivanyuk, K. Rissanen and E. Kolehmainen, Chem. Commun., 2000, 1107 RSC; (d) A. Shivanyuk and J. Rebek, Jr., Chem. Commun., 2001, 2374 RSC; (e) H. Mansikkamäki, M. Nissinen and C. A. Schalley, New. J. Chem., 2003, 27, 88 RSC; (f) H. Mansikkamäki, M. Nissinen and K. Rissanen, Chem. Commun., 2002, 1902 RSC.
- (a) G. W. Orr, L. J. Barbour and J. L. Atwood, Science, 1999, 285, 1049 CrossRef; (b) H. Mansikkamäki, M. Nissinen and K. Rissanen, Angew. Chem., Int. Ed., 2004, 43, 1243 CrossRef.
- L. R. MacGillvray and J. L. Atwood, Nature (London), 1997, 389, 469 CrossRef CAS.
- (a) A. Shivanyuk and J. Rebek, Jr., Chem. Commun., 2001, 2424 RSC; (b) A. Shivanyuk and J. Rebek, Jr., Proc. Natl. Acad. Sci. USA, 2001, 98, 7662 CrossRef CAS; (c) L. Avram and Y. Cohen, Org. Lett., 2002, 4, 4365 CrossRef CAS; (d) L. Avram and Y. Cohen, J. Am. Chem. Soc., 2002, 124, 15148 CrossRef CAS; (e) A. Shivanyuk and J. Rebek, Jr., J. Am. Chem. Soc., 2002, 4, 3433; (f) A. Schivanyuk and J. Rebek, Jr., J. Am. Chem. Soc., 2003, 125, 3432 CrossRef CAS.
- For earlier NMR studies on interactions of resorcinarenes with alkyl ammonium cations, see: (a) H. J. Schneider, D. Güttes and U. Schneider, Angew. Chem., Int. Ed. Engl., 1986, 25, 647 CrossRef; (b) J. L. Atwood and A. Szumna, J. Supramol. Chem., 2002, 2, 479 Search PubMed.
- J. C. Ma and D. A. Dougherty, Chem. Rev., 1997, 97, 1303 CrossRef CAS.
- (a) M. Przybylski and M. O. Glocker, Angew. Chem., Int. Ed. Engl., 1996, 35, 806 CrossRef CAS; (b) J. S. Brodbelt, Int. J. Mass Spectrom., 2000, 200, 57 CrossRef CAS; (c) C. A. Schalley, Int. J. Mass Spectrom., 2000, 194, 11 CrossRef CAS; (d) C. B. Lebrilla, Acc. Chem. Res., 2001, 34, 653 CrossRef CAS; (e) C. A. Schalley, Mass Spectrom. Rev., 2001, 20, 253 CrossRef CAS.
- (a) C. A. Schalley, J. M. Rivera, T. Martín, J. Santamaría, G. Siuzdak and J. Rebek, Jr., Eur. J. Org. Chem., 1999, 1325 CrossRef CAS; (b) C. A. Schalley, R. K. Castellano, M. S. Brody, D. M. Rudkevich, G. Siuzdak and J. Rebek, Jr., J. Am. Chem. Soc., 1999, 121, 4568 CrossRef CAS; (c) M. S. Brody, D. M. Rudkevich, C. A. Schalley and J. Rebek, Jr., Angew. Chem., Int. Ed., 1999, 38, 1640 CrossRef CAS; (d) C. A. Schalley, T. Martín, U. Obst and J. Rebek, Jr., J. Am. Chem. Soc., 1999, 121, 2133 CrossRef CAS; (e) A. Lützen, A. R. Renslo, C. A. Schalley, B. M. O'Leary and J. Rebek, Jr., J. Am. Chem. Soc., 1999, 121, 7455 CrossRef; (f) B. M. O'Leary, T. Szabo, N. Svenstrup, C. A. Schalley, A. Lützen and J. Rebek, Jr., J. Am. Chem. Soc., 2001, 123, 11519 CrossRef.
- (a) J. A. Bryant, M. T. Blanda, M. Vincenti and D. J. Cram, J. Am. Chem. Soc., 1991, 113, 2167 CrossRef CAS; (b) L. M. Nuwaysir, J. A. Castoro, C. L.-C. Yang and C. L. Wilkins, J. Am. Chem. Soc., 1992, 114, 5748 CrossRef CAS; (c) F. Inokuchi, Y. Miyahara, T. Inazu and S. Shinkai, Angew. Chem., 1994, 34, 1364; (d) P. S. H. Wong, X. Yu and D. V. Dearden, Inorg. Chim. Acta, 1996, 246, 259 CrossRef CAS; (e) E. Ventola, K. Rissanen and P. Vainiotalo, Chem. Commun., 2002, 1110 RSC; (f) M. Mäkinen, P. Vainiotalo and K. Rissanen, J. Am. Soc. Mass Spectrom., 2002, 7, 851 CrossRef CAS; (g) M. Mäkinen, M. Nissinen, K. Rissanen and P. Vainiotalo, J. Am. Chem. Soc., 2003, 14, 143 CAS; (h) M. C. Letzel, B. Decker, A. B. Rozhenko, W. W. Schoeller and J. Mattay, J. Am. Chem. Soc., 2004, 126, 9669 CrossRef CAS.
- (a) L. R. MacGillivray and J. Atwood, J. Am. Chem. Soc., 1997, 119, 6931 CrossRef CAS; (b) L. R. MacGillivray, G. S. Papaefstathiou, J. L. Reid and J. A. Ripmeester, Cryst. Growth Des., 2001, 1, 373 CrossRef CAS; (c) B.-Q. Ma and P. Coppens, Chem. Commun., 2003, 504 Search PubMed; (d) I. Gregoriev and E. Bosch, Cryst. Growth Des., 2004, 4, 235 CrossRef.
- D. E. Hibbs, M. B. Hursthouse, K. M. A. Malik, H. Adams, C. J. M. Stirling and F. Davis, Acta Crystallogr., Sect. C, 1996, 54, 987 CrossRef.
- (a) W. Iwanek, R. Frölich, M. Urbaniak, C. Näther and J. Mattay, Tetrahedron, 1998, 54, 14031 CrossRef CAS; (b) M. Nissinen and K. Rissanen, Supramol. Chem., 2003, 15, 581 CrossRef CAS.
- (a) K. A. Connors, Binding Constants, Wiley, New York, 1987 Search PubMed; (b) K. Hirose, J. Inclusion Phenom. Macrocycl. Chem., 2001, 39, 193 CrossRef CAS and references cited therein.
- Specfit, version 3.0.31, Spectrum Software Associates, Chapel Hill, NC, USA, 2002 Search PubMed. See also: H. Gampp, M. Maeder, C. J. Meyer and A. D. Zuberbühler, Talanta, 1986, 33, 943 Search PubMed and references cited therein.
- J. C. Shivanyuk, S. Friese, S. Döring and J. Rebek, Jr., J. Org. Chem., 2003, 68, 6489 CrossRef.
- This has been confirmed by a crystal structure, see: I. Thondorf, F. Broda, K. Rissanen, M. Vysotsky and V. Böhmer, J. Chem. Soc., Perkin Trans. 2, 2002, 1796 Search PubMed.
- References to programs used for crystallographic work: (a) Z. Otwinowski and W. Minor, Processing of X-ray Diffraction Data Collected in Oscillation Mode in Methods in Enzymology, Macromolecular Chrystallography, Part A, eds. C. W. Carter, Jr. and R. M. Sweet, Academic Press, New York, 1997, vol. 276, p. 307 Search PubMed; (b) G. M. Sheldrick, SHELXS-97, Program for solution of crystal structures, University of Göttingen, Germany, 1997 Search PubMed; (c) G. M. Sheldrick, SHELXL-97, Program for refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
- M. J. S. Dewar, E. G. Zoebisch, E. F. Healy and J. J. P. Stewart, J. Am. Chem. Soc., 1985, 107, 3902 CrossRef.
- CACHE 5.0 for Windows, Fujitsu Ltd., Krakow, Poland, 2001 Search PubMed.
Footnote |
| † CCDC reference numbers 254958–254964. See http://www.rsc.org/suppdata/nj/b4/b415401k/ for crystallographic data in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2005 |