The syntheses and structures of some main group complexes of the sterically hindered N,N′-bis(2,6-diisopropylphenyl)-4-toluamidinate ligand
René T.
Boeré
a,
Marcus L.
Cole†
b and
Peter C.
Junk
*b
aDepartment of Chemistry and Biochemistry, University of Lethbridge, Lethbridge, Alberta, Canada T1K 3M4
bSchool of Chemistry, Monash University, Victoria, 3800, Australia. E-mail: peter.junk@sci.monash.edu.au
First published on 14th December 2004
Abstract
The stoichiometric reaction of the bulky benzamidine N,N′-bis(2,6-diisopropylphenyl)-4-toluamidine (HDippAm) with the metal alkyls BunLi (1∶1 in THF), Bu2Mg (2∶1 in THF) and Me3Al (1∶1 in Et2O) is presented. This provides the mononuclear dihapto benzamidinate compounds [Li(DippAm)(THF)2] (1), [Mg(DippAm)2] (2) and [Al(DippAm)Me2] (3), respectively. Compound 3 was also obtained by salt elimination using dimethylaluminium chloride and 1. All three compounds exhibit sterically strained geometries that are maintained in solution at increased temperatures. Compound 3 displays exceptional thermal and aerobic stability, while 2 constitutes a rare example of non-porphyrin supported square planar magnesium.
Introduction
Since we reported the first N,N′-bis(2,6-diisopropylphenyl)amidines in 1998 (Fig. 1A),1 the area of metal amidinate chemistry has undergone a significant expansion in scope and application.2–5 In part, this can be attributed to a report from Jordan in 1997 describing the treatment of dimethylaluminium amidinates ([R1NC(R2)NR3]−) with methide abstraction reagents,6e.g. tris(pentafluorophenyl)borane, to afford cationic ethene polymerisation catalysts. Since, interest in amidinate supported alkylaluminium species has understandably flourished.2,7–14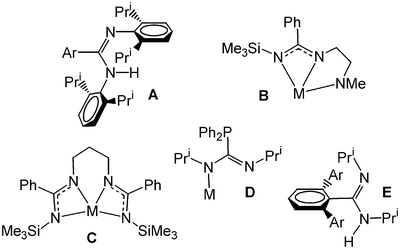 | ||
| Fig. 1 Recently developed amidinate ligand types (Ar = alkyl substituted or unsubstituted aryl, M = metal). | ||
Despite the implications of this landmark discovery, attempts to soundly identify the catalytic species have not been conclusive.2,15 However, dinuclear trisalkyl species have been put forward as candidates.2 If indeed these are the active species, the substituents about the amidinate will have a considerable influence on the performance of this system not just electronically but also sterically. This is borne out by [Al(Benzamidinate)R2] (R = alkyl) species with large aryl substituents at the NCN backbone carbon. These generate catalytically inactive species upon alkide abstraction.12 Similar studies for dialkylaluminiums bearing amidinates with encumbered substituents at nitrogen have not been reported.8
As a side benefit of the above, the metalloamidinate chemistries of groups 1–5,3,4,16–20 especially that of group 3,21–26 have received significant interest. This has been bolstered by the size-charge characteristics of amidinates, which can be considered comparable to cyclopentadienides.27 Here, a major aim has been the steric and electronic tuning of the amidine/ate. To attain this the traditional synthetic paths to such species,28,29 these being: (i) protonolysis of a neutral amidine using a metal alkyl30 or organoamide31 of appropriate basicity; (ii) insertion of a carbodiimide into a metal alkyl32 or pnictide33 (the latter to render ‘guanidinate’ type species); and (iii) salt elimination between a group 1 amidinate (typically generated by path (i) or (ii)) and a metal halide,34 have been tailored to introduce supplementary donors, e.g. pendant amides35 or amidinates25 (Fig. 1, B and C respectively), heteroatoms36 (D) and sterically bulky substituents9,10,12,17,21 (E) to the amidinate frame. Owing to our interest in ligands of type A (Fig. 1) developments toward the last of these interest us most.
So far, the introduction of bulky substituents has been executed by two means. The first of these, the method that we chose in our original contribution,1 is to generate neutral amidines with bulky substituents at nitrogen using classical amidine synthetic protocols.37 However, from the prodigious work of Power et al.,38 preparative routes to terphenyl halides have been popularised giving ready access to lithiated meta-terphenyls.39 These include the lithiated 2,6-Tripp2C6H3, 2,6-Mes2C6H3, 2,4,6-Ph3C6H2 and 2,6-(4-ButC6H4)C6H3 species (Tripp = 2,4,6-triisopropylphenyl, Mes = 2,4,6-trimethylphenyl), which have been applied to amidinate synthesis path (ii) (the insertion of a carbodiimide into a metal alkyl or pnicitide), to generate amidinates with terphenyl groups at carbon (see E, Fig. 1).9,10,12,17,21 According to the group of Arnold, this generates ‘bowl-like’ metal coordination environments with steric encumbrance coplanar and orthogonal to the diazaallyl donor.21 A space-fill depiction of one of these coordinated ligands (that of [Al(PriNC(2,6-MesC6H3)NPri)(CH3)2]),12 viewed both above and in the plane of the MNCN metallacycle, can be seen in Fig. 2. Recent reports of lithium,17,21 magnesium17 and aluminium9–10,12 complexes bearing this ligand-type provide a firm basis for comparison with the bulk of our N,N′-bis(2,6-diisopropylphenyl)amidinates, represented here by N,N′-bis(2,6-diisopropylphenyl)-4-toluamidinate (DippAm).1 Herein we describe the synthesis of [Li(DippAm)(THF)2] (1), [Mg(DippAm)2] (2) and [Al(DippAm)(CH3)2] (3), generated by stoichiometric treatment of the appropriate metal alkyl with HDippAm, and relate the observed solution/solid-state behaviour of these species to relevant compounds from the groups of Arnold and Jordan.
![Space-fill illustration of [Al(PriNC(2,6-MesC6H3)NPri)(CH3)2] above (left) and in (right) the metallacyclic plane. Isopropyl and dimethylaluminium carbons coloured teal and yellow respectively (POV-RAY illustration, 100% van der Waals radii).](/image/article/2005/NJ/b409086a/b409086a-f2.gif) | ||
| Fig. 2 Space-fill illustration of [Al(PriNC(2,6-MesC6H3)NPri)(CH3)2] above (left) and in (right) the metallacyclic plane. Isopropyl and dimethylaluminium carbons coloured teal and yellow respectively (POV-RAY illustration, 100% van der Waals radii). | ||
Results and discussion
The treatment of HDippAm with nbutyllithium (1∶1) or dibutylmagnesium (2∶1) in THF at low temperature results in clean alkane elimination to give compounds devoid of the FTIR stretches (Nujol) and 1H NMR resonances (C6D6) attributable to the N–H moiety of the parent ligand (see Scheme 1). These ‘trademark’ stretches and signals occur at 3428 cm−1, 3366 cm−1 (N–H stretch)1 and 5.98 ppm, respectively and are associated with a shift in the NCN backbone C–N stretching frequencies from five strong absorbances at 1650, 1611, 1584, 1567 and 1510 cm−1 to two distinct stretches at 1610 and 1573 cm−1 for lithium compound 1, and 1609 and 1573 cm−1 for magnesium compound 2. In addition, the FTIR spectrum of 2, unlike that of 1, is free of stretches associated with THF, e.g. a broad C–O stretch at ca. 1050 cm−1, indicating the absence of included solvent. 1H and 13C NMR Spectra confirm this and, from singular NCN resonances located at 154.3 and 158.9 ppm (1 and 2 respectively, HDippAm; 141.5 ppm), infer the presence of a singular benzamidinate isomer in solution. Furthermore, for both 1 and 2, the number of aromatic singlets in the 13C NMR spectra (8 for both) suggests symmetrical binding about the NCN donor. This arrangement invokes isopropyl methyl substituents with two separate chemical shifts (1; 1.20 and 1.30 ppm, 2; 1.16 and 1.34 ppm). The existence of one broadened septet methyne resonance for both 1 and 2 (1; virtual septet 3.84 ppm, 2; virtual septet 3.79 ppm) suggests these methyl environments emanate from restricted rotation about the aryl–Pri bond rather than two distinct isopropyl environments. Also, a ratio of 2 THF donors∶1 DippAm ligand can be deduced from the 1H NMR spectrum of compound 1. This gives 1 and 2 the empirical formulae [Li(DippAm)(THF)2] and [Mg(DippAm)2].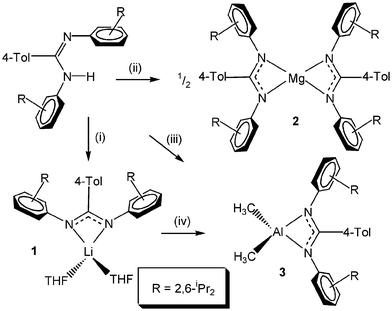 | ||
| Scheme 1 Reagents and conditions: (i) 1.0 nBuLi, −1.0 nBuH, THF, −30 °C to ambient temperature, overnight; (ii) 0.5 Bu2Mg, −1.0 nBuH, THF, −30 °C to ambient temperature, 2 h; (iii) 1.0 Me3Al, −1.0 CH4, Et2O, −50 °C to ambient temperature, overnight; (iv) 1.0 Me2AlCl, −1.0 LiCl, Et2O/hexane, 0 °C to ambient temperature, ca. 5 h. | ||
While HDippAm exists as a dynamic equilibrium of at least two isomers at ambient temperature in deutero benzene solution,40 the NMR spectra of 1 and 2 indicate the absence of fluxional processes in solution as is consistent with deprotonated ligands of this type. Presuming the steric bulk of the 2,6-diisopropyl N-substituents is too great to permit a Z-syn41 DippAm ligand, the symmetrical spectra of 1 and 2 must result from E-anti isomerism,41 wherein placement of the 2,6-diisopropylphenyl rings orthogonal to the metal–NCN metallacycle demands projection of one isopropyl methyl toward and the other away from the NCN backbone. To gain unequivocal proof of this and to elucidate the nuclearity of 1 and 2, both compounds were recrystallised to provide samples suitable for single crystal X-ray structure determination. The outcome of these can be seen in Figs. 3 and 4a
(POV-RAY illustrations, 40% thermal ellipsoids, see captions for selected bond lengths and angles), while unit cell and refinement parameters are listed in Table 1. As illustrated, 1 and 2 do indeed exhibit symmetrical DippAm ligands, in which both chelate a single metal centre. For compound 1, two THF donors complete four-coordination of the lithium centre, while delocalisation of the anionic charge across the bulky benzamidinate is evidenced by the NCN backbone carbon–nitrogen lengths of 1.327(4) and 1.330(3)
Å
(identical within experimental error) and a NCN angle of 116.7(3)°
(HDippAm; 1.317(3), 1.344(3)
Å and 119.4(2)° resp.).1 These parameters are similar to those of the monomeric four-coordinate TMEDA complex; [Li(PriNC{2,6-(4-ButC6H4)2C6H3}NPri)(TMEDA)]
(NCN parameters; 1.329(5), 1.327(5)
Å and 116.3(4)°),17 which displays a dihaptic benzamidinate. This ‘bowl-like’ complex possesses Li-amidinate nitrogen bond lengths of 1.995(9) and 1.998(9)
Å,17 suggesting greater ligand–metal proximity than the DippAm ligand of 1
(Li–N; 2.032(6) and 2.057(6)
Å). Presumably this arises from the bite of the TMEDA donor for the former (86.5(4)°), which eases approach of the benzamidine relative to the THF donors of 1
(O–Li–O bite 96.8(3)°). By contrast, the lithium–benzamidinate nitrogen bond length of the three coordinate lithium species [Li(PriNC(2,6-Tripp2C6H3)NPri)(TMEDA)] is expectedly shorter due to decreased lithium coordination (1.978(8)
Å). Here the significant bulk of the benzamidinate forces a singular (monohapto) lithium–amide nitrogen contact, and retention of discrete C–N and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond character across the backbone (1.309(5) and 1.361(5)
Å, respectively).17
N bond character across the backbone (1.309(5) and 1.361(5)
Å, respectively).17
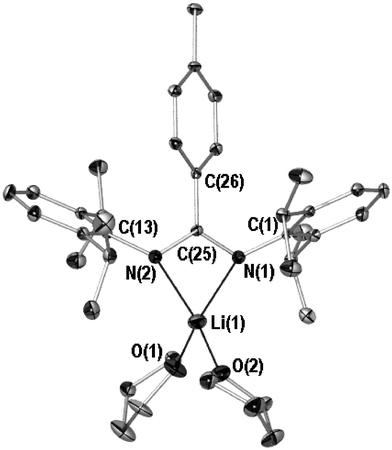 | ||
| Fig. 3 X-ray crystal structure of 1 (POV-RAY illustration, 40% thermal ellipsoids). All hydrogen atoms omitted for clarity. Selected bond (Å) lengths and angles (°): Li(1)–N(1) 2.032(6), Li(1)–N(2) 2.057(6), Li(1)–O(1) 1.955(6), Li(1)–O(2) 1.966(6), N(1)–C(25) 1.327(4), N(2)–C(25) 1.330(3), C(25)–C(26) 1.508(4), N(1)–Li(1)–N(2) 67.2(2), N(1)–Li(1)–O(1) 126.3(3), N(1)–Li(1)–O(2) 119.9(3), O(1)–Li(1)–O(2) 96.8(3), N(1)–C(25)–N(2) 116.7(3), tolyl plane∶metallacyclic plane 49.6(1). | ||
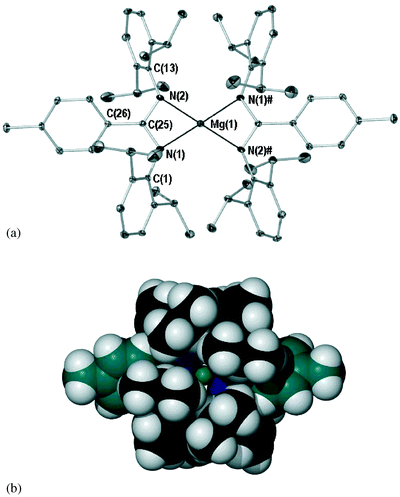 | ||
| Fig. 4 (a) X-ray crystal structure of 2 (POV-Ray illustration, 40% thermal ellipsoids). All hydrogen atoms and lesser occupancy disordered atoms (C(24B)) omitted for clarity. Selected bond (Å) lengths and angles (°): Mg(1)–N(1) 2.047(2), Mg(1)–N(2) 2.069(2), N(1)–C(25) 1.347(3), N(2)–C(25) 1.343(3), C(25)–C(26) 1.497(4), N(1)–Mg(1)–N(2) 65.5(1), N(1)–Mg(1)–N(1)# 171.2(1), N(1)–Mg(1)–N(2)# 114.9(1), N(2)–Mg(1)–N(2)# 174.6(1), N(1)–C(25)–N(2) 111.8(2), tolyl plane∶metallacyclic plane 34.1(1). Symmetry transformation used to generate # atoms: 1 − x, y, 1/2 − z. (b) Space-fill illustration of 2 (same orientation as 4a). Tolyl carbons coloured teal (POV-RAY illustration, 100% van der Waals radii). | ||
| [Li(DippAm)(THF)2] (1) | [Mg(DippAm)2] (2) | [Al(DippAm)(CH3)2] (3) | |
|---|---|---|---|
| Formula | C40H57N2O2Li | C32H42N2Mg0.5 | C34H47N2Al |
| Formula Weight | 604.82 | 465.82 | 510.72 |
| Temperature (K) | 123(2) | 123(2) | 123(2) |
| Space Group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
C2/c | P31 |
| a/Å | 10.922(3) | 23.4090(11) | 15.2283(4) |
| b/Å | 11.627(2) | 10.6758(4) | 15.2283(4) |
| c/Å | 15.562(4) | 25.0402(12) | 12.0910(2) |
| α/° | 80.772(9) | 90 | 90 |
| β/° | 77.567(13) | 114.337(3) | 90 |
| γ/° | 77.758(11) | 90 | 120 |
| Volume/Å3 | 1872.3(8) | 5701.7(4) | 2428.26(10) |
| Z | 2 | 8 | 3 |
| D c/g cm−3 | 1.073 | 1.085 | 1.048 |
| μ/mm−1 | 0.064 | 0.072 | 0.085 |
| Reflections collected | 22064 | 30267 | 17025 |
| Unique reflections | 8589 | 6614 | 7271 |
| Parameters varied | 415 | 332 | 365 |
| R(int) | 0.1829 | 0.1902 | 0.0674 |
| R 1 | 0.0719 | 0.0666 | 0.0477 |
| wR 2 | 0.1563 | 0.1387 | 0.0946 |
Magnesium compound 2 (see Fig. 4) crystallises with one half molecule in the asymmetric unit, wherein the magnesium lies on a two-fold rotation axis perpendicular to the metallacyclic plane. This generates a planar “Mg(NCN)2” core. The bond lengths and angles within the metallacycles, in particular the NCN parameters, suggest the metal centre is more congested (NCN C–N lengths and NCN angle; 1.347(3), 1.343(3) Å and 111.8(2)°, the former two are identical within experimental error) than its lithium analogue. Furthermore, the square planar geometry adopted is highly unorthodox42 (N(1)–Mg(1)–N(2) 65.5(1)°, N(1)–Mg(1)–N(2)# 114.9(1)°, sum of angles about Mg = 360.9°). This geometry has only been observed before in rigidly held porphyrin coordinated43 magnesium metal centres and two unusual compounds from Lappert and Sachdev; the 1-azallyl species [Mg(Me3SiNC(But)C(H)SiMe3)2]44 and the hydrazide compound [Mg{PhNN(SiMe3)2}2].45 The former of these exhibits considerable closing of the planes created by the NCC 1-azallyl donor and the MgN2C2 metallacycle (ca. 46°),44 while the latter displays amide nitrogen to magnesium contacts (mean value; 1.92 Å) that radically contrast with the mean Mg–N amine contacts (2.42 Å).45 These ‘secondary’ lengths are outside of the combined covalent radii of magnesium and nitrogen (ca. 2.1 Å)46 and do not suggest explicit four-coordination of the metal. Accordingly, neither ‘non-porphyrin’43–45 example demonstrates the ‘symmetrical’ planarity of 2, wherein the MgNCN metallocycles exhibit a maximum rms deviation from planarity of below 0.001 Å (N(1) and N(2) exhibit deviations of 0.0001(11) Å). Further, there are no obvious Mg⋯H–C agostic contacts that could rationalise the adopted geometry, the closest Mg⋯C and Mg⋯H distances being those of C(7) and H(7) (3.888(3) and 3.07 Å, respectively).47 Aside from magnesium bis(amidinate) species bearing apical THF donors,18,48 the known Mg(Amidinate)2 species49–53 possess ligands in which the NCN units are disposed near orthogonal to one another to minimise steric interaction. In the absence of any close intermolecular interactions (including π–π arene stacking), we propose that the DippAm ligand prohibits a tetrahedral geometry by unfavourable interaction of the 2,6-diisopropyl substituents.42 This coerces metallacycle to metallacycle coplanarity. A space-fill diagram of 2 is displayed in Fig. 4b (same orientation as Fig. 4a). From this it is apparent that the tolyl ring (in teal) sits coplanar to the diazaallyl fragment, thereby inducing significant buttressing with the 2,6-diisopropylphenyl rings. This impedes aryl–Pri bond rotation and NCN backbone lengths (see above) that are extended relative to those of the homoleptic tetrahedral magnesium bis(amidinate) species [Mg(PriN(C(2,6-Mes2C6H3)NPri)2] (both C–N NCN lengths 1.31(2) Å),17 [Mg(ButNC(Ph)NBut)2] (mean C–N NCN length; 1.33 Å)49 and [Mg(MesNC(But)NMes)2] (mean C–N NCN length; 1.33 Å).52 Likewise, the Mg–N lengths of 2 (2.047(2) and 2.069(2) Å) are longer than those of these compounds (mean values 2.04, 2.04 and 2.04 Å respectively),17,49,52 which in turn are considerably shorter than those of five/six coordinate magnesium bis(amidinates), e.g. [Mg{(4-MeC6H4)NC(H)N(4-MeC6H4)}2(THF)2] (mean Mg–N; 2.153 Å).18
Variable temperature 1H NMR experiments in the temperature range 0–70 °C were conducted for 1 and 2 to assess the observed impeded rotation. For 1, the two isopropyl-methyl doublets coalesce to one broadened singlet between 60 and 65 °C. A similar measurement for 2 could not be made in the temperature range used (b.p. of C6D6 79.1 °C), with distinct isopropyl-methyl doublets plainly evident at 70 °C. This suggests considerable hindrance about the aryl–Pri bond. As suggested above, it is apparent from this and the solid-state data that the driving force for decreased bond mobility in both 1 and 2 is the non-orthogonal placement of the 4-tolyl group relative to the NCN backbone (see Fig. 4b).54 A similar interaction is sterically forbidden for the benzamidinates of Arnold (Fig. 1E), i.e. placement of the backbone phenyl perpendicular to the diazaallylic plane is favoured.
Like 1 and 2, the reaction of trimethylaluminium with one equivalent of HDippAm at low temperature results in clean alkane elimination to form the dimethylaluminium compound [Al(DippAm)(CH3)2] (3). Compound 3 was also prepared via salt elimination using dimethylaluminium chloride and 1 under similar conditions (see Scheme 1). The FTIR and 1H, 13C NMR spectra of solvent donor free 3 display DippAm stretches and resonances consistent with those for 1 and 2 (C–N stretches at 1612, 1576 cm−1, Pri methyl doublets at 1.09 and 1.39 ppm, virtual methyne septet at 3.75 ppm, discrete resonances maintained at 70 °C without coalescence) while the NCN backbone resonance is shifted upfield by ca. 20 ppm to 173.5 ppm. This placement compares well to the NCN resonances of [Al(PriNC(2,6-Tripp2C6H3)NPri)(CH3)2]12 and [Al(AdNC(CH3)NAd)(CH3)2]8 (Ad = 1-adamantyl), which appear at 169.5 and 172.6 ppm respectively. Likewise, the location of the “Al(CH3)2” 1H, 13C and 27Al NMR resonances at 0.04, −9.5 and 69.5 ppm (Al2Me627Al NMR resonance in toluene; 157 ppm)55 compare well to those of the above species (Tripp benzamidinate; −0.37, −4.34,12 adamantyl acetamidinate; −0.82, −9.6 ppm,827Al NMR not reported) and a closely related dimethylaluminium N,N′-bis(2,6-diisopropylphenyl)pivalamidinate (−0.76 and −6.8 ppm respectively, 27Al NMR not reported).8
Like compounds 1 and 2, the ready availability of crystalline samples of 3 permitted a single crystal X-ray structure determination. As can be seen in Fig. 5 (POV-RAY illustration, 40% thermal ellipsoids, see Table 1 for unit cell and refinement parameters), 3 crystallises as a monomeric [Al(DippAm)(CH3)2] unit with a dihaptic DippAm. Akin to 1 and 2, the NCN C–N bond lengths suggest significant delocalisation of the anionic charge across the backbone (1.338(3) and 1.338(3) Å, HDippAm; 1.317(3) and 1.344(3) Å).1 Meanwhile, the NCN angle of 110.3(2)° and Al–N bond lengths of 1.942(2) and 1.942(2) Å are reasonably typical for bulky amidinates coordinated to dimethylaluminium (analogous angles and lengths for [Al(PriNC(2,6-Mes2C6H3)NPri)(CH3)2]; 108.2(3)°, 1.953(4) and 1.951(4) Å)12 and thus open and extended (resp.) relative to less bulky species like [Al{(c-C6H11)NC(But)N(c-C6H11)}(CH3)2] (analogous angles and lengths; 107.8°, 1.927(2) and 1.912(1) Å).7
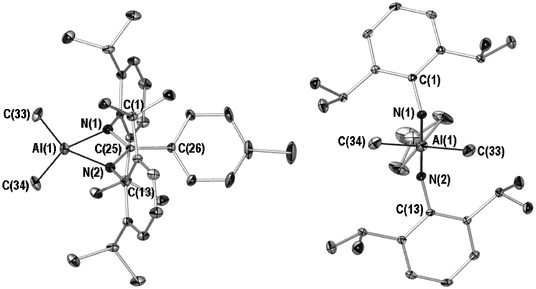 | ||
| Fig. 5 X-ray crystal structure of 3. Depiction at an angle to (left) and in the plane of the metallocycle (right) (POV-RAY illustration, 40% thermal ellipsoids). All hydrogen atoms and lesser occupancy disordered atoms (C(29A) and C(32A)) omitted for clarity. Selected bond (Å) lengths and angles (°): Al(1)–N(1) 1.942(2), Al(1)–N(2) 1.941(2), Al(1)–C(33) 1.951(3), Al(1)–C(34) 1.952(3), N(1)–C(25) 1.338(3), N(2)–C(25) 1.338(3), C(25)–C(26) 1.476(3), N(1)–Al(1)–N(2) 68.9(1), N(1)–Al(1)–C(33) 117.2(2), N(1)–Al(1)–C(34) 112.8(1), C(33)–Al(1)–C(34) 118.4(1), N(1)–C(25)–N(2) 110.3(2), tolyl plane:∶metallacyclic plane 39.9(1). | ||
Aside from these structural and spectroscopic characteristics, the most striking aspect of compound 3 is its exceptional aerobic and thermal stability. Solid samples show no sign of decomposition up to 360 °C (limit of apparatus used) under a dinitrogen atmosphere56 and, similarly, no decomposition (as evidenced by 1H NMR) upon exposure to air for periods in excess of 2 h. This stability is also evident in deutero benzene solution where, in the absence of moisture (sample stored over 3 Å molecular sieves), there is no decomposition after exposure to air overnight. It appears this robustness is borne out of considerable steric bulk about the AlNCN metallacycle (see Fig. 6, aspects chosen same as Fig. 2). The significant bending of the 2,6-diisopropylphenyl ipso-carbons out of the AlNCN metallacyclic plane (see Fig. 5) testifies to this bulk (ipso-carbons sit 0.341(3) and 0.343(3) Å out of the AlNCN plane).
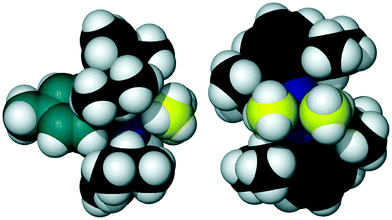 | ||
| Fig. 6 Space-fill illustration of compound 3 above (left) and in (right) the metallacyclic plane (as per Fig. 2). Tolyl and dimethylaluminium carbons coloured teal and yellow respectively (POV-RAY illustration, 100% van der Waals radii). | ||
Conclusion
We have demonstrated that the ligand DippAm is capable of exerting considerable steric influence when complexed to lithium, magnesium and aluminium. In the special instance of magnesium; this results in a rare ‘square planar’ magnesium compound. For dimethylaluminium, the steric protection afforded by the DippAm ligand manifests as exceptional thermal and aerobic stability. As demonstrated by Figs. 2 and 6, the protection afforded by ligands of type A may rival that of the ‘Arnold-type’ ligands; E (Fig. 1).56 All three compounds suggest the 4-tolyl backbone group, placed such that it sits near coplanar to the metallacycles generated, increases the hindrance experienced at the metal.54 This incites heavily impeded rotation about the aryl–isopropyl bonds, and is maintained at increased temperatures.Our use of the ligand frame A (see Fig. 1) in the stabilisation and preparation of NCP, PCP and NCAs anionic donor sets will form the basis of forthcoming publications.
Experimental
General
N,N′-Bis(2,6-diisopropylphenyl)-4-toluamidine (HDippAm) was prepared by a literature procedure.1,37nButyllithium (1.6 M in hexane), dibutylmagnesium (1.0 M in heptane), trimethylaluminium (2.0 M in hexanes) and dimethylaluminium chloride (1.0 M in hexanes) were purchased from Aldrich and used as received. Tetrahydrofuran (THF), diethyl ether and hexane were dried over sodium, freshly distilled from sodium benzophenone ketyl and freeze-thaw degassed prior to use. All manipulations were performed using conventional Schlenk techniques under an atmosphere of high purity dinitrogen in flame-dried glassware. Infrared spectra were recorded as Nujol mulls using sodium chloride plates on a Nicolet Nexus FTIR spectrophotometer. 1H NMR spectra were recorded at 300.1 MHz, 13C NMR spectra at 75.5 MHz and 27Al NMR at 78.2 MHz using a Bruker DPX 300 spectrometer. Chemical shifts were referenced to the 13C or residual 1H resonances of the d6-benzene solvent employed (13C and 1H NMR respectively), or an external [Al(H2O)6]3+ standard (1.1 M [Al(H2O)6][NO3]3 in H2O/D2O) (27Al NMR). Melting points were determined in sealed glass capillaries under dinitrogen and are uncorrected. Microanalyses (C, H and N) were undertaken at the University of Otago, P.O. Box 56, Dunedin, New Zealand.[Li(η2-N,N′-DippAm)(THF)2] (1)
nButyllithium (0.70 cm3, 1.12 mmol) was added dropwise to a stirred colourless solution of HDippAm (0.50 g, 1.10 mmol) in THF (30 cm3) at −30 °C. The resulting solution, which developed a yellow hue upon addition, was gradually warmed to ambient temperature and stirred overnight. Removal of all volatiles under reduced pressure rendered a light yellow powder that was washed with cold (0 °C) hexane (2 × 3 cm3). Addition of fresh THF (ca. 10 cm3) followed by filtration and placement at −10 °C rendered 1 as colourless rectangular blocks (0.57 g, 86%), m.p. 161 °C. 1H NMR (d6-benzene, 303 K): δ 1.20 (br d, 12H, CH(CH3)(CH3), 3JHH = 6.0 Hz), 1.30 (br d, 12H, CH(CH3)(CH3), 3JHH = 5.9 Hz), 1.36 (m, 8H, CH2 THF), 1.86 (br s, 3H, 4-CH3), 3.48 (m, 8H, OCH2 THF), 3.84 (br septet, 4H, CH(CH3)(CH3), 3JHH = 6.1 Hz), 6.66–7.38 (br m, 8H, Ar–H), 7.66 (br d, 2H, Ar–H, JHH = 7.6 Hz). 13C NMR (d6-benzene, 303 K): δ 21.4 (CH(CH3)(CH3)), 24.9 (CH(CH3)(CH3)), 25.4 (4-CH3), 23.9 (CH2 THF), 28.7 (CH(CH3)(CH3)), 68.4 (OCH2 THF), 121.3, 123.4, (Ar–CH), 128.0 (Ar–C), 129.7, 131.0 (Ar–CH), 138.7, 141.5, 143.6 (Ar–C), 154.3 (NCN). FTIR (Nujol)/cm−1: 1610 (sh w), 1573 (sh m), 1464 (br s), 1356 (sh m), 1316 (s), 1254 (s), 1234 (s), 1184 (sh m), 1174 (sh m), 1142 (sh w), 1098 (sh m), 1072 (m), 1048 (sh s), 941 (m), 895 (m), 835 (sh w), 818 (sh m), 808 (w), 796 (w), 760 (sh s), 731 (sh w), 686 (w), 652 (w), 632 (w). Anal. Calc. For Li1C40H57N2O2: C, 79.43; H, 9.50; N, 4.63. Found: C, 80.21; H, 9.76; N, 4.90.[Mg(η2-N,N′-DippAm)2] (2)
Dibutylmagnesium (0.50 cm3, 0.50 mmol) was added dropwise to a stirred colourless solution of HDippAm (0.50 g, 1.10 mmol) in THF (30 cm3) at −30 °C. The resulting colourless solution was gradually warmed to ambient temperature over a period of ca. 2 hours, concentrated in vacuo (to ca. 3 cm3), filtered and left for several days. During this period small light yellow blocks of 2 deposited. These were collected by filtration and dried under a flow of dinitrogen after washing with cold hexane (0 °C, 1 × 2 cm3) (0.16 g, 34%), m.p. 278 °C (decomposition). 1H NMR (d6-benzene, 303 K): δ 1.16 (d, 12H, CH(CH3)(CH3), 3JHH = 7.0 Hz), 1.34 (d, 12H, CH(CH3)(CH3), 3JHH = 6.9 Hz), 1.78 (s, 3H, 4-CH3), 3.79 (virtual septet, 4H, CH(CH3)(CH3), 3JHH = 7.0 Hz), 6.78 (d, 2H, Ar–H, JHH = 7.8 Hz), 6.94–7.32 (m, 8H, Ar–H)HH. 13C NMR (d6-benzene, 303 K): δ 19.8 (CH(CH3)(CH3)), 22.6 (CH(CH3)(CH3)), 25.6 (4-CH3), 31.3 (CH(CH3)(CH3)), 122.2, 123.4, 125.9 (Ar–CH), 126.6 (Ar–C), 130.7 (Ar–CH), 136.2, 139.9, 143.1 (Ar–C), 158.9 (NCN). FTIR (Nujol)/cm−1: 1609 (sh m), 1573 (sh m), 1458 (br s), 1383 (s), 1361 (s), 1319 (m), 1278 (m), 1232 (s), 1194 (sh m), 1159 (m), 1109 (s), 1059 (m), 1045 (m), 1022 (w), 965 (sh m), 952 (sh m), 932 (sh w), 840 (m), 823 (sh m), 795 (m), 775 (m), 761 (m), 738 (w), 698 (w), 688 (w), 676 (w), 657 (w), 636 (w). Anal. Calc. for Mg1C64H82N4: C, 82.51; H, 8.87; N, 6.01. Found: C, 82.41; H, 9.38; N, 6.14.[Al(η2-N,N′-DippAm)(CH3)2] (3)
Method (i): A solution of HDippAm (0.30 g, 0.66 mmol) in diethyl ether (20 cm3) was added dropwise to a cooled (−50 °C) stirred solution of trimethylaluminium (0.40 cm3, 0.80 mmol), also in diethyl ether (40 cm3) over a period of 30 minutes. The resulting colourless solution was stirred overnight and permitted to warm to ambient temperature. Filtration and removal of all volatiles in vacuo gave a colourless powder. This was washed with cold hexane (3 × 1.50 cm3, −10 °C) to remove traces of trimethylaluminium, and the remaining colourless solid recrystallised from fresh diethyl ether (ca.10 cm3) at 0 °C. This gave the title compound as irregular colourless plates around the periphery of the solution (0.14 g, 42%), m.p. >360 °C.Method (ii): A diethyl ether solution of 1 (0.30 g, 0.50 mmol, 10 cm3) was added to a cooled (0 °C) stirred solution of dimethylaluminium chloride (0.50 cm3, 0.50 mmol) in hexane (10 cm3). After stirring for several hours, volatiles were removed in vacuo to render a light yellow solid. This was extracted into diethyl ether (ca. 20 cm3) and separated from the remnant lithium chloride by filtration. Concentration (ca. 10 cm3), followed by placement at −10 °C gave 3 as colourless irregular plates (0.21 g, 82%), m.p. >360 °C. 1HNMR (d6-benzene, 303 K): δ 0.04 (s, 6H, Al(CH3)2), 1.09 (d, 12H, CH(CH3)(CH3), 3JHH = 7.0 Hz), 1.39 (d, 12H, CH(CH3)(CH3), 3JHH = 6.8 Hz), 1.77 (s, 3H, 4-CH3), 3.75 (virtual septet, 4H, CH(CH3)(CH3), 3JHH = 6.8 Hz), 6.53 (d, 2H, Ar–H, JHH = 8.1 Hz), 7.13–7.27 (m, 8H, Ar–H). 13C NMR (d6-benzene, 303 K): δ 9.5 (Al(CH3)2), 21.3 (CH(CH3)(CH3)), 23.3 (4-CH3), 26.2, 29.0 (CH(CH3)(CH3)), 124.4, 126.4 (Ar–CH), 126.9 (Ar–C), 129.0, 130.9 (Ar–CH), 139.1, 141.5, 144.3 (Ar–C), 173.5 (NCN). 27Al NMR (C6D6, 303 K): δ 69.5 (br s, width at half peak height 4300 Hz). FTIR (Nujol)/cm−1: 1612 (sh m), 1576 (sh m), 1517 (sh w), 1446 (s), 1394 (s), 1363 (sh s), 1342 (m), 1321 (sh s), 1284 (sh m), 1252 (sh m), 1216 (sh w), 1188 (sh m), 1161 (w), 1125 (w), 1098 (m), 1056 (sh m), 1045 (sh m), 979 (sh m), 951 (w), 935 (w), 855 (sh w), 824 (m), 800 (m), 767 (s), 736 (s), 722 (s), 702 (s), 676 (s), 644 (m), 628 (m), 611 (m), 591 (sh m). Anal. Calc. for Al1C34H47N2: C, 79.96; H, 9.28; N, 5.48. Found: C, 79.99; H, 9.23; N, 5.65.
X-ray crystallography
Crystalline samples of compounds 1–3 were mounted on glass fibres in viscous hydrocarbon oil at −150 °C (123 K). Crystal data were obtained using an Enraf-Nonius Kappa CCD. X-ray data were processed using the DENZO-SMN program.57 Structural solution and refinement was carried out using the SHELX suite of programs58,59 with the graphical interface X-Seed.60For compound 2, both methyl groups of one isopropyl were found to be disordered over two sites of partial occupancy (C(23) and C(24)). Modelling of this disorder was attempted proving successful for C(23) (modelled as 72:28 occupancy; C(23A):C(23B)) and unsuccessful for C(24) (failed to give satisfactory thermal parameters). C(24) left prolate.
For compound 3, the para-tolyl group was found to exhibit significant libration orthogonal to the arene plane using C(25) as a fulcrum. Disorder was modelled successfully for the para-carbon (C(29)) and tolyl methyl (C(32)) carbons (partial occupancies of 47∶53% for C(29A)/C(32A)∶C(29B)/C(32B)), while the lesser disorder exhibited by the ipso-, ortho- and meta-carbons (C(26), C(27)/C(31) and C(28)/C(30) respectively) could not be modelled satisfactorily. Owing to the unusual disorder of 3, and as its molecular units lie on a 31 screw axis with near two-fold rotational symmetry about the aluminium–tolyl methyl vector perpendicular to this axis, doubling of the c-axis (the axis which the above disorder lies along) and refinement in the space group P3(1)12 (same systematic absences as P3(1))61 was attempted. This failed to provide satisfactory refinement parameters.
Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre. CCDC reference numbers 242105–242107. See http://www.rsc.org/suppdata/nj/b4/b409086a/ for crystallographic data in .cif or other electronic format.
Acknowledgements
The authors would like to thank the Australian Research Council for continued financial support. RTB acknowledges the ARC for a Linkage-International award and Prof. Alan M. Bond for a fruitful research visit to Monash University. Dr Craig M. Forsyth and Ms Kristina Konstas are sincerely thanked for their assistance during the collection of 27Al NMR data, and Mr Jason D. Masuda is thanked for performing some preliminary experiments.References
- R. T. Boeré, V. Klassen and G. Wolmershäuser, J. Chem. Soc., Dalton Trans., 1998, 4147 RSC.
- S. Dagorne, I. A. Guzei, M. P. Coles and R. F. Jordan, J. Am. Chem. Soc., 2000, 122, 274 CrossRef CAS.
- E. A. C. Brussee, A. Meetsma, B. Hessen and J. H. Teuben, Chem. Commun., 2000, 497 RSC.
- J. Arnold and J. R. Hagadorn, Angew. Chem. Int. Ed., 1998, 37, 1729 CrossRef CAS.
- H. Nagashima, H. Kondo, T. Hayashida, Y. Yamaguchi, M. Gondo, S. Masudam K. Miyazaki, K. Matsubara and K. Kirchner, Coord. Chem. Rev., 2003, 245, 177 CrossRef CAS.
- M. P. Coles and R. F. Jordan, J. Am. Chem. Soc., 1997, 119, 8125 CrossRef CAS.
- M. P. Coles, D. C. Swenson, R. F. Jordan and V. G. Young, Organometallics, 1997, 16, 5183 CrossRef CAS.
- M. P. Coles, D. C. Swenson, R. F. Jordan and V. G. Young, Organometallics, 1998, 17, 4042 CrossRef CAS.
- D. Abeysekera, K. N. Robertson, T. S. Cameron and J. A. C. Clyburne, Organometallics, 2001, 20, 5532 CrossRef CAS.
- H. A. Jenkins, D. Abeysekera, D. A. Dickie and J. A. C. Clyburne, J. Chem. Soc., Dalton Trans., 2002, 3919 RSC.
- J. Grundy, M. P. Coles and P. B. Hitchcock, J. Organomet. Chem., 2002, 662, 178 CrossRef CAS.
- J. A. R. Schmidt and J. Arnold, Organometallics, 2002, 21, 2306 CrossRef CAS.
- C.-T. Chen, C.-A. Huang, Y.-R. Tzeng and B.-H. Huang, Dalton Trans., 2003, 2585 RSC.
- B. Clare, N. Sarker, R. Shoemaker and J. R. Hagadorn, Inorg. Chem., 2004, 43, 1159 CrossRef CAS.
- G. Talarico, V. Busico and P. H. M. Budzelaar, Organometallics, 2001, 20, 4721 CrossRef CAS.
- M. L. Cole, D. J. Evans, P. C. Junk and M. K. Smith, Chem. Eur. J., 2003, 9, 415 CrossRef CAS.
- J. A. R. Schmidt and J. Arnold, J. Chem. Soc., Dalton Trans., 2002, 2890 RSC.
- M. L. Cole, D. J. Evans, P. C. Junk and L. M. Louis, New J. Chem., 2002, 1015 RSC.
- A. Lisovskii, M. Botoshansky and M. S. Eisen, J. Chem. Soc., Dalton Trans., 2001, 1692 RSC.
- E. A. C. Brussee, A. Meetsma, B. Hessen and J. H. Teuben, Organometallics, 1998, 17, 4090 CrossRef CAS.
- J. A. R. Schmidt and J. Arnold, Chem. Commun., 1999, 2149 RSC.
- J. Zhang, R. Y. Ruan, Z. H. Shao, R. F. Cai, L. H. Weng and X. G. Zhou, Organometallics, 2002, 21, 1420 CrossRef CAS.
- D. Doyle, Y. K. Gun’ko, P. B. Hitchcock and M. F. Lappert, J. Chem. Soc., Dalton Trans., 2000, 4093 RSC.
- S. Bambirra, M. J. R. Brandsma, E. A. C. Brusee, A. Meetsma, B. Hessen and J. H. Teuben, Organometallics, 2000, 19, 3197 CrossRef CAS.
- S. Bambirra, A. Meetsma, B. Hessen and J. H. Teuben, Organometallics, 2001, 20, 782 CrossRef CAS.
- S. Bambirra, D. van Leusen, A. Meetsma, B. Hessen and J. H. Teuben, Chem. Commun., 2003, 522 RSC.
- R. Kempe, Angew. Chem. Int. Ed., 2000, 39, 468 CrossRef CAS.
- J. Barker and M. Kilner, Coord. Chem. Rev., 1994, 133, 219 CrossRef CAS.
- F. T. Edelmann, Coord. Chem. Rev., 1994, 137, 403 CrossRef.
- For example see: F. A. Cotton, S. C. Haefner, J. H. Matonic, X. P. Wang and C. A. Murillo, Polyhedron, 1997, 16, 541 Search PubMed.
- For example see: J. Baldamus, C. Berghof, M. L. Cole, D. J. Evans, E. Hey-Hawkins and P. C. Junk, J. Chem. Soc., Dalton Trans., 2002, 2802 Search PubMed.
- For example see: T. Chivers, A. Downward and M. Parvez, Inorg. Chem., 1999, 38, 3447 Search PubMed.
- For example see: M. L. Cole and P. C. Junk, J. Chem. Soc., Dalton Trans., 2003, 2109 Search PubMed.
- For example see: S. Dagorne, R. F. Jordan and V. G. Young, Organometallics, 1999, 18, 4619 Search PubMed.
- For example see: M. J. R. Brandsma, E. A. C. Brussee, A. Meetsma, B. Hessen and J. H. Teuben, Eur. J. Inorg. Chem., 1998, 1867 Search PubMed.
- For example see: M. P. Coles and P. B. Hitchcock, Chem. Commun., 2003, 2794 Search PubMed.
- The chemistry of amidines and imidates, ed. S. Patai and Z. Rappoport, 1991, Wiley, Chichester Search PubMed.
- For example see: L. H. Pu, A. D. Phillips, A. F. Richards, M. Stender, R. S. Simons, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 2003, 125, 11626 Search PubMed.
- C. J. F. Du, H. Hart and K. K. D. Ng, J. Org. Chem., 1986, 51, 3162 CrossRef CAS.
- From single crystal X-ray structure determination and isomer trapping experiments using molybdenum hexacarbonyl to create ‘rigid’ [Mo(Arene)(CO3)] ‘piano-stool’ species, HDippAm is most stable as the asymmetric Z-anti and E-syn tautomers1.
- Using the nomenclature of C. L. Perrin, Ch. 3., p. 152, reference 38.
- Preliminary semi-empirical calculations (PM3) performed using the crystal geometry of 2 suggest the optimum gas phase structure distorts to yield an inter-NCN angle of 41.3°, however this is still a significant departure from the true tetrahedral value of 90°.
- Examples of rigid planar geometry in porphyrin coordinated magnesium centres: (a) M. P. Byrn, C. J. Curtis, I. Goldberg, Y. Hsiou, S. I. Khan, P. A. Sawin, S. K. Tendick and C. E. Strouse, J. Am. Chem. Soc., 1991, 113, 6549 CrossRef CAS; (b) M. P. Byrn, C. J. Curtis, Y. Hsiou, S. I. Khan, P. A. Sawin, S. k. Tendick, A. Terzis and C. E. Strouse, J. Am. Chem. Soc., 1993, 115, 9480 CrossRef CAS; (c) N. S. Mani, L. S. Beall, T. Miller, O. P. Anderson, H. Hope, S. R. Parkin, D. J. Williams, A. G. M. Barrett and B. M. Hoffman, J. Chem. Soc., Chem. Commun., 1994, 2095 RSC; (d) J. Mizuguchi, J. Phys. Chem. A, 2001, 105, 1121 CrossRef CAS; (e) J. Janczak and R. Kubiak, Polyhedron, 2001, 20, 2901 CrossRef CAS.
- C. F. Caro, P. B. Hitchcock and M. F. Lappert, Chem. Commun., 1999, 1433 RSC.
- H. Sachdev, Eur. J. Inorg. Chem., 2001, 2681.
- E. Fluck and K. G. Heumann, Periodic Table of the Elements, VCH, Weinheim, Germany, 1991 Search PubMed.
- An example where metal to hydrogen agostic interactions have been elegantly identified as the driving force for such geometries can be found in: F. G. N. Cloke, P. B. Hitchcock, M. F. Lappert, G. A. Lawless and B. Royo, J. Chem. Soc., Chem. Commun., 1991, 724 Search PubMed.
- B. Srinvas, C.-C. Chang, C.-H. Chen, M. Y. Chiang, I.-T. Chen, Y. Wang and G.-H. Lee, J. Chem. Soc., Dalton Trans., 1997, 957 RSC.
- A. R. Saddique, M. J. Hegg and C. H. Winter, Inorg. Chem., 2001, 40, 6349 CrossRef.
- M. Westerhausen and H.-D. Hausen, Z. Anorg. Allg. Chem., 1992, 27, 615.
- D. Walther, P. Gebhardt, R. Fischer, U. Kreher and H. Gorls, Inorg. Chim. Acta, 1998, 281, 181 CrossRef CAS.
- A. Xia, H. M. El-Kaderi, M. J. Heeg and C. H. Winter, J. Organomet. Chem., 2003, 682, 224 CrossRef CAS.
- K. Kincaid, C. P. Gerlach, G. R. Giesbrecht, J. R. Hagadorn, G. D. Whitener, A. Shafir and J. Arnold, Organometallics, 1999, 18, 5360 CrossRef CAS.
- An N,N′-di(aryl)formamidinate ([RNC(H)NR]−) analogue of 1 has been prepared. 1H NMR spectra of this compound indicate free rotation about the aryl–isopropyl bonds and a single methyl environment, M. L. Cole, P. C. Junk, unpublished data.
- R. Benn, A. Rufinska, H. Lehmkuhl, E. Jannsen and C. Krueger, Angew. Chem., Int. Ed. Engl., 1983, 22, 779 CrossRef.
- Unfortunately, references 8, 9 and 13 do not report the thermal or aerobic stability of the compounds included. Accordingly, no complete comparison of stability can be made.
- Z. Otwinowski, W. Minor, in “Processing of X-ray Diffraction Data Collected in Oscillation Mode”, Methods in Enzymology, 276: Macromolecular Crystallography, Part A, eds. C. W. Cater and R. M. Sweet, 1997, 307, Academic Press, New York Search PubMed.
- G. M. Sheldrick, SHELXL-97, Program for refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXS-97, Program for solution of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189 Search PubMed.
- International Tables for X-Ray Crystallography, eds. J. A. Ibers and W. C. Hamilton, Kynoch Press, Birmingham, UK, 1974, Vol. 4 Search PubMed.
Footnote |
| † Present address: Department of Chemistry, University of Adelaide, South Australia 5005, Australia. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2005 |