Novel carbon dioxide and carbonyl carbonate complexes of molybdenum. The X-ray structures of trans-[Mo(CO2)2{HN(CH2CH2PMe2)2}(PMe3)] and [Mo3(μ2-CO3)(μ2-O)2(O)2(CO)2(H2O)(PMe3)6]·H2O
Leopoldo
Contreras
a,
Margarita
Paneque
a,
Murielle
Sellin
a,
Ernesto
Carmona
*a,
Pedro J.
Pérez
b,
Enrique
Gutiérrez-Puebla
c,
Angeles
Monge
c and
Caridad
Ruiz
c
aDepartamento de Química Inorgánica, Instituto de Investigaciones Químicas, Universidad de Sevilla–Consejo Superior de Investigaciones Científicas, Avda. Américo Vespucio s/n, Isla de la Cartuja, E-41092, Sevilla, Spain. E-mail: guzman@us.es
bDepartamento de Química y Ciencia de los Materiales, Universidad de Huelva, Campus de El Carmen s/n, E-21007, Huelva, Spain. E-mail: perez@dqcm.uhu.es
cInstituto de Ciencia de Materiales de Madrid, Consejo Superior de Investigaciones Científicas, Campus de Cantoblanco, E-28049, Madrid, Spain
First published on 13th December 2004
Abstract
A new bis(carbon dioxide) adduct of molybdenum containing the tridentate, bis(phosphine) ligand, HN(CH2CH2PMe2)2 (NP2), in a mer conformation has been synthesized and structurally characterized. Its carbonyl carbonate isomer [Mo(CO3)(CO)(NP2)(PMe3)] has also been prepared. In addition, the reactivity of the complex [Mo(CO3)(CO)(PMe3)4] toward CO has been studied, a transformation that has led to the formation of the dicarbonyl complex [Mo(CO3)(CO)2(PMe3)3]. This complex liberates CO2 upon heating under a carbon monoxide atmosphere by means of a CO–CO3 conproportionation reaction. The new trinuclear complex [Mo3(μ2-CO3)(μ2-O)2(O)2(CO)2(H2O)(PMe3)6]·H2O, resulting from the interaction of [Mo(CO3)(CO)(PMe3)4] and water, has been isolated and structurally characterized.
Introduction
The serious environmental problems associated with global warming have stimulated further academic and industrial research into carbon dioxide, as this substance is a major component of the greenhouse gases.1 In addition, there is growing interest in the use of CO2 as a C1 source for chemicals and fuels, as well as in a variety of technological applications.2 A key step in the chemical, photochemical and electrochemical transformations of CO2 is the formation of a M–CO2 complex, since coordination to a metal centre activates this rather unreactive molecule toward a number of chemical reactions. It is noteworthy that despite the importance of the M–CO2 functionality, the number of structurally characterized mononuclear complexes is still limited.Of the three coordination modes that may be envisaged for a M–CO2 unit, namely the side-on κ2-C,O (Scheme 1, structure A) and the end-on κ1-C or κ1-O (B and C, respectively), only A and B have been authenticated by X-ray crystallography. κ1-C coordination has been found in [RhCl(CO2)(diars)]3a and Ru(CO2)(CO)(bipy)2]·3H2O,3b whereas side-on binding (A) has been demonstrated in complexes of both the early and the late transition elements.4,5 All these compounds contain a single M–CO2 unit, with the only exception of some Mo complexes prepared by our group some years ago, which possess a trans-Mo(CO2)2 functionality.5
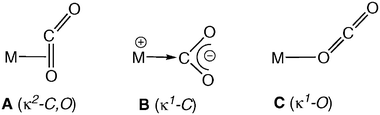 | ||
| Scheme 1 Coordination modes of carbon dioxide. | ||
One of the reactions a coordinated molecule of CO2 may undergo is reduction to CO by means of oxygen atom transfer to another substrate. The latter can be an oxophilic metal6 or a phosphine ligand7 (or other readily oxidized ligand). But it is also possible for CO2 to act as its own oxygen sink, giving rise to CO and CO32− [eqn. (1)] in a reaction called reductive disproportionation of CO2:7,8
| 2 CO2 + 2 e− → CO32− + CO | (1) |
Following previous work on the generation of Mo–CO2 adducts5 and Mo carbonyl carbonate complexes,8 we would like to report new findings in this area that include the structural characterization by X-ray methods of the bis(CO2) adduct, trans-[Mo(CO2)2(NP2)(PMe3)] (1), which contains the tridentate ligand HN(CH2CH2PMe2)2 (NP2). An X-ray study on the trinuclear, mixed-valence Mo(II)–Mo(VI) complex of composition [Mo3(μ2-CO3)(μ2-O)2(O)2(CO)2(H2O)(PMe3)6]·H2O (4) is also reported.
Results and discussion
Synthesis and molecular structure of trans-[Mo(CO2)2(NP2)(PMe3)] (1)
We have shown earlier that the Mo(0) compound trans-[Mo(CO2)2(PMe3)4] exhibits a characteristic substitution chemistry that allows stepwise replacement of the labile PMe3 groups, without alteration of the trans-Mo(CO2)2 linkage.5 During formation of compounds of composition trans-[Mo(CO2)2(PMe3)3(CNR)], trans-[Mo(CO2)2(PMe3)2(Me2PCH2CH2PMe2)], and others,5b the two CO2 ligands remain staggered with respect to one another and eclipse the trans-L–Mo–L′ vectors of the plane perpendicular to the trans-Mo(CO2)2 bond axis. This conformation, found to be the most favourable by ab initio calculations with the model compound trans-[Mo(CO2)2(PH3)4], maximizes the Mo(CO2)2 bonding interaction.10As previous substitution chemistry involved either mono- or bidentate ligands,5 we wondered if the use of a tridentate ligand could force the two molecules of CO2 into a cis geometry, and perhaps induce their subsequent coupling to give the corresponding head-to-tail or head-to-head dimers.11 Edwards’ HN(CH2CH2PMe2)2 (NP2) was synthesized12 and used for this purpose. Nonetheless, and despite the flexibility of this and related ligands that allows them to coordinate to the metal either in facial or meridional forms,13 the addition of NP2 to solutions of trans-[Mo(CO2)2(PMe3)4] gives the trans-Mo(CO2)2 compound 1 [eqn. (2)] as the only detectable product:
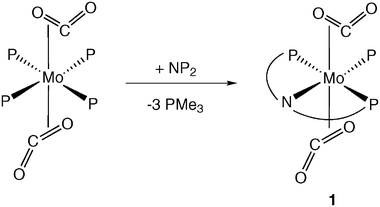 | (2) |
The IR spectrum of 1 shows bands at 1660, 1155 and 1100 cm−1, which can be assigned to vibrations arising from the coordinated molecules of CO2 by comparison with the spectrum of 1* (ca. 30% 13CO2-enriched). These absorptions have strikingly similar energies to those of the starting material and of other trans-Mo(CO2)2 adducts.5 In the 1H NMR spectrum, inequivalency of the P–Me groups of the tridentate ligand is evidenced by the observation of four independent doublets in the range δ 0.9–1.4, whereas the molecule of PMe3 gives a doublet centred at δ 1.33 (JP-H = 8.7 Hz). As already indicated, resonances attributable to an AMX spin system are discerned in the 31P{1H} NMR spectrum. Those due to the 31P termini of the NP2 ligand (A and X) show the effect of a strong J(PA-PX) coupling of 178 Hz, indicative of a mutual trans geometry. The single PMe3 group (PM), cis with respect to PA and PX, appears as a triplet due to accidentally degenerate PA-PM and PA-PX couplings of 12 Hz. The room temperature 13C{1H} NMR spectrum of 1* consists of two broad, partially unresolved signals centred at about δ 213.5 and 217, suggestive of fluxionality. Accordingly, upon cooling to −80 °C, two well-resolved signals, each consisting of a doublet of doublets of doublets, are observed (see Experimental). The fluxionality of related trans-Mo(CO2)2 adducts has been studied in detail and demonstrated to consist of a synchronous motion of the two CO2 ligands in which both molecules rotate in the same direction.5b
The spectroscopic data discussed in the preceding paragraph supports the geometry proposed for complex 1 in eqn. (2). Unequivocal confirmation has been provided by single-crystal X-ray studies, whose results are presented in Fig. 1 and Tables 1 and 2. Compound 1 has a distorted octahedral geometry, with the CO2 ligands side-on bonded to molybdenum through one of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds. In agreement with theory,10 and with structures of analogous derivatives,5a the two CO2 ligands are mutually staggered, each one eclipsing a corresponding trans-L–Mo–L′ vector in the plane perpendicular to the CO2–Mo–CO2 axis. Bonding parameters within the Mo–CO2 units are similar to those found for trans-Mo(CO2)2(PMe3)3(CN–i-Pr) and reveal, likewise, strong Mo–CO2 bonding interactions. The Mo–C bond lengths in 1 (2.08 Å ave) are identical within experimental error to those in the above CN–i-Pr derivative and approach normal Mo–CO distances {1.970(4) and 2.03(1) Å, average values for the two types of CO groups in cis-[Mo(CO)4(PMe3)2]14}. The same argument applies to the Mo–O bonds in 1 (2.12 Å ave). The two C–O bonds of each molecule of CO2 appear to be of identical length within experimental error (Table 2) but are longer than in free CO2 (1.16 Å).
O bonds. In agreement with theory,10 and with structures of analogous derivatives,5a the two CO2 ligands are mutually staggered, each one eclipsing a corresponding trans-L–Mo–L′ vector in the plane perpendicular to the CO2–Mo–CO2 axis. Bonding parameters within the Mo–CO2 units are similar to those found for trans-Mo(CO2)2(PMe3)3(CN–i-Pr) and reveal, likewise, strong Mo–CO2 bonding interactions. The Mo–C bond lengths in 1 (2.08 Å ave) are identical within experimental error to those in the above CN–i-Pr derivative and approach normal Mo–CO distances {1.970(4) and 2.03(1) Å, average values for the two types of CO groups in cis-[Mo(CO)4(PMe3)2]14}. The same argument applies to the Mo–O bonds in 1 (2.12 Å ave). The two C–O bonds of each molecule of CO2 appear to be of identical length within experimental error (Table 2) but are longer than in free CO2 (1.16 Å).
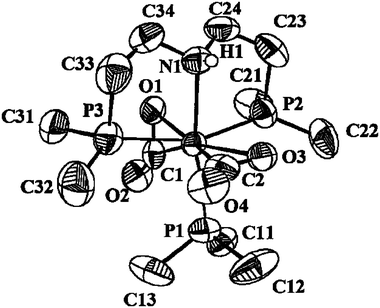 | ||
| Fig. 1 ORTEP view of the molecule in compound 1. | ||
| 1 | 4 | |
|---|---|---|
| Empirical formula | C13H30MoNO4P3 (C3H8 )0.5 | C21H56MoO10P6·H2O |
| Formula weight | 474.25 | 960.3 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | C2/c | P21/n |
| a/Å | 19.947(5) | 9.769(1) |
| b/Å | 11.670(2) | 16.333(1) |
| c/Å | 20.004(7) | 24.682(1) |
| β/° | 101.89(2) | 98.99(1) |
| U/Å3 | 4557 (2) | 3889.9(5) |
| Z | 8 | 4 |
| T/K | 295 | 294 |
| μ/mm−1 | 0.801 | 1.244 |
| Reflexions measured | 5616 | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 422 422 |
| Unique reflexions | 5459 | 11177 |
| R 1 (all data) | 0.236 | 0.0760 |
| wR 2 (all data) | 0.238 | 0.1038 |
| R 1 [I > 2σ(I)] | 0.064 | 0.0327 |
| wR 2 [I > 2σ(I)] | 0.173 | 0.0889 |
| a Esd are given in parentheses | |||
|---|---|---|---|
| Mo–Pl | 2.434(3) | C24–N1 | 1.45(2) |
| Mo–P2 | 2.481(3) | C34–N1 | 1.46(2) |
| Mo–P3 | 2.443(3) | C1–O1 | 1.284(11) |
| Mo–N1 | 2.270(7) | Cl–O2 | 1.199(10) |
| Mo–C1 | 2.088(9) | C2–O3 | 1.245(11) |
| Mo–O1 | 2.111(6) | C2–O4 | 1.234(11) |
| Mo–C2 | 2.085(9) | Hl–N1 | 0.91(11) |
| Mo–O3 | 2.138(6) | ||
| C1–Mo–C2 | 148.6(4) | C1–Mo–P2 | 92.7(3) |
| C1–Mo–O1 | 35.6(3) | C2–Mo–P2 | 113.0(3) |
| C2–Mo–O1 | 157.1(4) | O1–Mo–P2 | 84.1(2) |
| C1–Mo–O3 | 156.5(3) | O3–Mo–P2 | 78.9(2) |
| C2–Mo–O3 | 34.3(3) | N1–Mo–P2 | 79.5(2) |
| O1–Mo–O3 | 158.9(3) | P1–Mo–P2 | 97.8(1) |
| C1–Mo–N1 | 116.2(4) | P3–Mo–P2 | 156.0(1) |
| C2–Mo–N1 | 87.2(3) | C24–N1–C34 | 110.0(8) |
| O1–Mo–N1 | 80.7(3) | C24–N1–Mo | 113.7(6) |
| O3–Mo–N1 | 84.0(4) | C34–N1–Mo | 115.0(6) |
| C1–Mo–P1 | 76.2(3) | C24–N1–H1 | 105.8 |
| C2–Mo–P1 | 82.3(2) | C34–N1–H1 | 105.8 |
| O1–Mo–P1 | 111.6(2) | Mo–N1–H1 | 105.8 |
| O3–Mo–P1 | 83.2(2) | O2–C1–O1 | 131.2(9) |
| N1–Mo–P1 | 167.2(2) | O2–C1–Mo | 155.6(8) |
| C1–Mo–P3 | 84.9(3) | O1–C1–Mo | 73.2(5) |
| C2–Mo–P3 | 78.6(3) | C1–O1–Mo | 71.2(5) |
| O1–Mo–P3 | 80.2(2) | O4–C2–O3 | 131.6(10) |
| O3–Mo–P3 | 111.5(2) | O4–C2–Mo | 153.2(9) |
| N1–Mo–P3 | 80.1(2) | O3–C2–Mo | 75.2(5) |
| P1–Mo–P3 | 104.8(1) | C2–O3–Mo | 70.5(5) |
The rigidity of the Mo(NP2) chelate causes significant deviations of the corresponding bond angles from the ideal 90° and 180° values expected for an octahedral geometry. For instance, the two N–Mo–P angles approach 80°, whereas that between the two, supposedly trans, Mo–P bonds amounts to only 155.9(1)°. Considering this, the mer arrangement of the NP2 ligand, although not unprecedented,13 may seem unexpected. It appears that the flexibility of the N–CH2–CH2–P arms allow the tetrahedral nitrogen donor to be accommodated in a mer structure [angles centred on N: C24–N–Mo, 113.7(6)°; C34–N–Mo, 115.0(6)°; C24–N–C34, 110.0(8)°], in this way keeping the trans-Mo(CO2)2 moiety unaltered. The Mo–N and Mo–P separations have normal values (Table 2). An additional interesting feature of the solid state structure of 1 is the intermolecular hydrogen bond interaction that involves the aminic HNP2 hydrogen and an exo (i.e., non-coordinated) oxygen atom of a neighbouring molecule (Fig. 2). The observed H1–Oexo distance of 2.2 Å (ave) is shorter that the sum of the van der Waals’ radii15 [1.40 (O) + 1.2 (H) = 2.60 Å], clearly in support of this proposal. Protic solvents like methanol (vide supra) may also participate in hydrogen bonding. An extended 3D network of hydrogen bonds has been shown to exist in the solid state structure of the η1-CO2 adduct [Ru(CO2)(CO)(bipy)2]·3H2O.3b
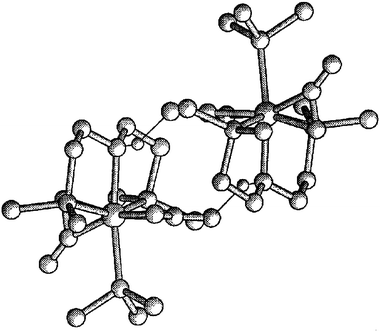 | ||
| Fig. 2 Molecular view of two molecules of 1 showing the hydrogen bonds. | ||
Carbonyl carbonate complexes of Mo. Molecular structure of [Mo3(μ2-CO3)(μ2-O)2(O)2(CO)2(H2O)(PMe3)6]·H2O (4).
In coordinating solvents, cis-[Mo(N2)2(PMe3)4] induces the reductive disproportionation of CO2 with formation of the carbonyl carbonates [Mo(CO3)(CO)(PMe3)4] and [Mo(CO3)(CO)(PMe3)3]2. Both complexes interconvert readily by loss or addition of PMe3, and react with chelating diphosphine ligands (P-P) to afford the substitution products [Mo(CO3)(CO)(P-P)(PMe3)2] and [Mo(CO3)(CO)(P-P)2].8bIn a like manner, addition of NP2 to solutions of either of the above carbonato compounds allows the isolation of a yellow microcrystalline species [eqn. (3)] of composition [Mo(CO3)(CO)(NP2)(PMe3)] (2):
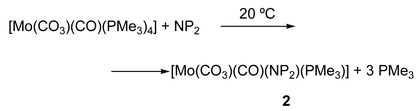 | (3) |
At variance with this result, only one PMe3 ligand is substituted by CO when [Mo(CO3)(CO)(PMe3)4] is reacted at room temperature, with 4 atm of carbon monoxide [eqn. (4)]:
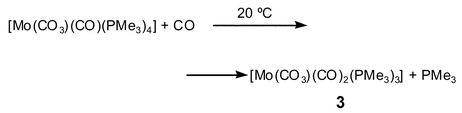 | (4) |
O) of the carbonate shifts slightly to lower energy (1580 cm−1), while the two CO groups are at the origin of absorptions at 1920 and 1820 cm−1.
Somewhat unexpectedly, reduction to Mo(0) occurs upon mild heating (60 °C) of solutions of carbonato 3. The reaction becomes cleaner when performed in the presence of CO; a mixture of the known carbonyl derivatives [Mo(CO)n(PMe3)6−n] (n = 2, 3)17 is obtained under these conditions. Analysis of the volatiles reveal the presence of CO2 further identified by its known, characteristic reaction9a,b with the complex 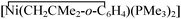 , in the presence of small amounts of water, to give the carbonate [Ni2(CH2CMe2Ph)2(μ-CO3)(PMe3)3]. These observations indicate that, upon heating, the CO32− and CO ligands of 3 undergo an oxidative conproportionation to CO2 [eqn. (5)], with concomitant reduction of the metal from Mo(II) to Mo(0):
, in the presence of small amounts of water, to give the carbonate [Ni2(CH2CMe2Ph)2(μ-CO3)(PMe3)3]. These observations indicate that, upon heating, the CO32− and CO ligands of 3 undergo an oxidative conproportionation to CO2 [eqn. (5)], with concomitant reduction of the metal from Mo(II) to Mo(0):
| CO32− + CO − 2 e− → 2 CO2 | (5) |
Besides [Mo(CO3)(CO)(PMe3)4] and [Mo(CO3)(CO)(PMe3)3]2, the reaction of cis-[Mo(N2)2(PMe3)4] and CO2 provides, under certain conditions, small amounts of another carbonato derivative, namely the known8b tetranuclear [Mo4(CO3)(μ-O)2(μ-OH)4(CO)2(PMe3)6]. As depicted schematically in D and E, this compound contains a unique bridging carbonate group. Heating a THF solution of [Mo(CO3)(CO)(PMe3)3]2 plus added water, at 50 °C, gives the same Mo4 species, albeit once more in low yields (ca. 10%).
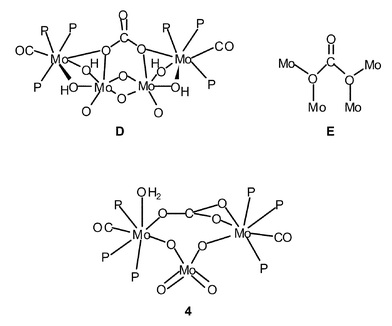
In an attempt to find a reliable, higher-yield synthetic procedure, [Mo(CO3)(CO)(PMe3)4] and [Mo(CO3)(CO)(PMe3)3]2 have been reacted with H2O under different experimental conditions, including in the presence of air. Acetone, methanol and tetrahydrofuran, as well as some of their mixtures, have been tested with controlled addition of H2O at room temperature and at 50–60 °C. A sufficiently better route to the Mo4 complex has not been found but nevertheless a somewhat related Mo3 complex of composition [Mo3(μ2-CO3)(μ2-O)2(O)2(CO)2(H2O)(PMe3)6]·H2O (4) has been isolated. Due to its insolubility in common organic solvents, reliable NMR data have not been recorded. Its IR spectrum shows a band at ca. 3500 cm−1 due to a coordinated molecule of water, along with carbonyl absorptions at 1770 and 1755 cm−1 and others attributable to the carbonate (1515 and 1280 cm−1) and PMe3 (945 cm−1) ligands. The structure of the molecule of 4 has been unambiguously determined by a single-crystal X-ray study, whose results are presented in Fig. 3 and Tables 1 and 3.
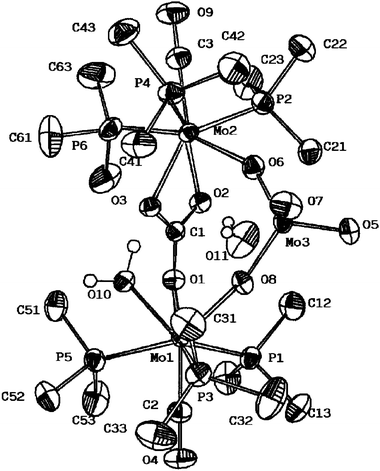 | ||
| Fig. 3 ORTEP view of the molecule of compound 4. | ||
| a Esd are given in parentheses | |||
|---|---|---|---|
| Mo1–Pl | 2.437(1) | Mo2–O6 | 2.189(2) |
| Mo1–P3 | 2.438(1) | Mo2–C1 | 2.598(3) |
| Mo1–P5 | 2.452(1) | Mo2–C3 | 1.918(4) |
| Mo1–O1 | 2.232(2) | Mo3–O5 | 1.749(3) |
| Mo1–O8 | 2.167(2) | Mo3–O6 | 1.773(2) |
| Mo1–O10 | 2.271(2) | Mo3–O7 | 1.739(3) |
| Mo1–C2 | 1.887(4) | Mo3–O8 | 1.783(2) |
| Mo2–P2 | 2.412(1) | O1–C1 | 1.265(4) |
| Mo2–P4 | 2.424(1) | O2–C1 | 1.284(4) |
| Mo2–P6 | 2.442(1) | O3–C1 | 1.304(4) |
| Mo2–O2 | 2.202(2) | O4–C2 | 1.188(4) |
| Mo2–O3 | 2.204(2) | O9–C3 | 1.181(4) |
| O10–Mo1–C2 | 130.1(1) | P6–Mo2–C3 | 76.7(1) |
| O8–Mo1–C2 | 131.5(1) | P6–Mo2–O6 | 158.75(7) |
| O8–Mo1–O10 | 81.98(9) | P6–Mo2–O3 | 79.45(6) |
| O1–Mo1–C2 | 127.3(1) | P6–Mo2–O2 | 82.53(6) |
| O1–Mo1–O10 | 86.35(8) | P4–Mo2–C3 | 75.2(1) |
| O1–Mo1–O8 | 82.81(8) | P4–Mo2–O6 | 76.50(7) |
| P5–Mo1–C2 | 73.6(1) | P4–Mo2–O3 | 90.10(6) |
| P5–Mo1–O10 | 79.43(7) | P4–Mo2–O2 | 145.35(6) |
| P5–Mo1–O8 | 154.94(7) | P4–Mo2–P6 | 109.66(4) |
| P5–Mo1–O1 | 79.4(6) | P2–Mo2–C3 | 73.0(1) |
| P3–Mo1–C2 | 74.5(1) | P2–Mo2–O6 | 79.27(6) |
| P3–Mo1–O10 | 79.99(6) | P2–Mo2–O3 | 140.30(6) |
| P3–Mo1–O8 | 78.04(6) | P2–Mo2–O2 | 84.13(6) |
| P3–Mo1–O1 | 157.81(6) | P2–Mo2–P6 | 113.30(4) |
| P3–Mo1–P5 | 114.70(3) | P2–Mo2–P4 | 117.31(3) |
| P1–Mo1–C2 | 74.3(1) | O7–Mo3–O8 | 109.5(1) |
| P1–Mo1–O10 | 155.59(7) | O6–Mo3–O8 | 110.9(1) |
| P1–Mo1–O8 | 79.98(6) | O6–Mo3–O7 | 108.0(1) |
| P1–Mo1–Ol | 75.22(6) | O5–Mo3–O8 | 111.1(1) |
| P1–Mo1–P5 | 111.92(3) | O5–Mo3–O7 | 108.9(1) |
| P1–Mo1–P3 | 111.89(3) | O5–Mo3–O6 | 108.3(1) |
| O6–Mo2–C3 | 124.4(1) | O2–C1–O3 | 115.3(3) |
| O3–Mo2–C3 | 145.5(1) | O1–C1–O3 | 122.1(3) |
| O3–Mo2–O6 | 80.24(8) | O1–C1–O2 | 122.7(3) |
| O2–Mo2–C3 | 139.4(1) | Mo1–C2–O4 | 179.2(3) |
| O2–Mo2–O6 | 81.84(9) | Mo2–C3–O9 | 179.1(3) |
| O2–Mo2–O3 | 59.49(8) | ||
As can be seen, complex 4 is a mixed-valence Mo(II)–Mo(VI) trinuclear species that consists of two seven-coordinate Mo(II) atoms bridged by a κ2-O,O′-MoO42− unit. The structural relation of the moiety comprised by the two Mo(II) atoms with [Mo(CO3)(CO)(PMe3)3]2 and the structurally characterized PMe2Ph analogue is evident.7a One of the μ-CO32− ligands of [Mo(CO3)(CO)(PMe3)3]2 is lost, being formally replaced by the molybdate group. As the remaining CO32− acts as a κ2-O,O’ toward Mo(2) and as κ1-O” toward Mo(1), the coordination of the latter is completed by a molecule of H2O. Note that the “Mo(CO)(PMe3)3” terminal fragments are maintained both in 4 and in the Mo4 complex referred to above. In complex 4, the CO32− group that bridges these fragments is planar: the three O–C–O angles have similar values that add together to practically 360°. The three C–O bonds do not differ appreciably in length and the same can be said for the three Mo–OCO2 separations.
Despite its bidentate nature, the bridging MoO42− group has a nearly tetrahedral geometry, with O6–Mo3–O7 [108.0(1)°] and O5–Mo3–O8 [111.1(1)°] showing the largest deviations with respect to the 109.5° ideal value. The two Mo–Ot bonds (Mo3–O5 and Mo3–O7) are identical within experimental error and average 1.74 Å. The bridging Mo–O bonds are also of equal length (1.77 Å, average value) and only slightly longer than the Mo–Ot. For comparative purposes, the Mo–O distance in K2MoO4 is 1.76(1) Å.18 Trimetallic units are bonded among themselves, via intermolecular hydrogen bonds (Table 4) through the coordinated water molecules in the [010] direction and through the uncoordinated ones in the [100] direction. This arrangement gives rise to infinite layers perpendicular to the [001] direction of composition {[Mo3(CO)2(PMe3)6(H2O)(μ2-CO3)(O)2(μ2-O2)]H2O}∞. The only two oxygen atoms that are not involved in the connectivity inside these layers, those of the carbonyl groups O(4) and O(9), are pointing outside, toward the interlayer space (Fig. 4).
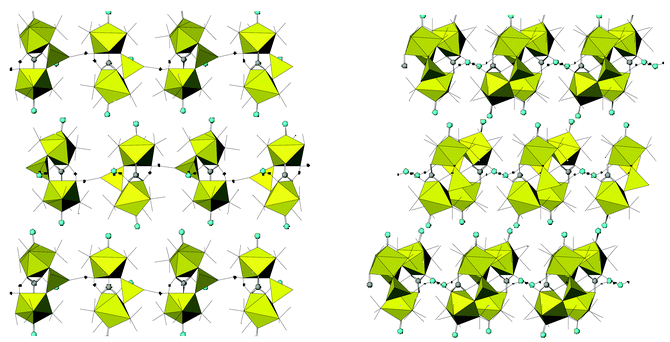 | ||
| Fig. 4 View of the packing showing the connections among the Mo trimetallic units of 4 through the coordinated and uncoordinated water molecules in the b (left) and a (right) directions, respectively. Blue circles are terminal oxygen atoms (O4 and O9) and uncoordinated water oxygen (O11), grey circles represent the carbon atoms of the carbonate (C1). | ||
| O–H⋯Oa | d(O–H) | d(H⋯O) | d(O⋯O) | ∠(O–H⋯O) |
|---|---|---|---|---|
| a Esd are given in parentheses. Symmetry code: (′) 3/2 − x, y − 1/2, 1/2 − z. (′′) x − 1, y, z. | ||||
| O10–H101⋯O5′ | 0.77(6) | 1.94(6) | 2.683(4) | 164(5) |
| O10–H102⋯O3 | 0.96(6) | 1.74(6) | 2.599(4) | 148(5) |
| O11–H111⋯O7′′ | 0.92(6) | 1.99(7) | 2.902(6) | 172(5) |
| O11–H112⋯O2 | 0.94(6) | 1.98(6) | 2.890(5) | 160(5) |
The identification of the molybdate group in the structure of 4 allows the design of a rational preparative route, consisting in the reaction of stoichiometric amounts of [Mo(CO3)(CO) (PMe3)3]2, dissolved in THF, and of Na2MoO4, dissolved in water (yields are about 50%). Even though 4 may be an intermediate along the path leading to the Mo4 compound, we have been unable to find consistent proof for this hypothesis. An alternative possibility is that they form independently. Regardless of this, their insolubility in common solvents permits their isolation and ulterior characterization.
Conclusion
In summary, the electronically favourable trans-Mo(CO2)2 conformation, with staggered CO2 ligands, characteristic of the complex trans-[Mo(CO2)2(PMe3)4] is retained during the substitution of three molecules of PMe3 by the tridentate HN(CH2CH2PMe2)2 ligand (NP2), to give compound 1. The isomeric carbonyl carbonate complex, [Mo(CO3)(CO)(NP2)(PMe3)], has also been prepared, along with an unusual trinuclear complex 4, shown by X-ray studies to consist of two seven-coordinate Mo(II) atoms bridged by a bidentate MoO42− unit. The trimetallic units of 4 are bonded among themselves by means of intermolecular hydrogen bonds that involve both the coordinated and the uncoordinated water molecules of 4, forming the infinite layers schematically represented in Fig. 4.During the revision of this work, a linear, O-coordinated carbon dioxide complex of uranium has been reported.19
Experimental
All preparations and manipulations were performed under an oxygen-free nitrogen atmosphere using conventional Schlenk techniques. Solvents were rigorously dried and degassed before use. The complexes trans-[Mo(CO2)2(PMe3)4],5 [Mo(CO3)(CO)(PMe3)4],8 [Mo(CO3)(CO)(PMe3)3]2,8 and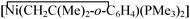 9 and the phosphine HN(CH2CH2PMe2)212 were prepared according to literature methods. NMR spectra were acquired with Varian XL-200 and Bruker DRX 500 MHz spectrometers. Microanalyses were carried out by the Microanalytical Service of the University of Seville. Despite all our attempts, satisfactory analytical data have been obtained only for 1 and 4.
9 and the phosphine HN(CH2CH2PMe2)212 were prepared according to literature methods. NMR spectra were acquired with Varian XL-200 and Bruker DRX 500 MHz spectrometers. Microanalyses were carried out by the Microanalytical Service of the University of Seville. Despite all our attempts, satisfactory analytical data have been obtained only for 1 and 4.
Syntheses
O); 950, ν(P–C). NMR (see F for numbering scheme): 1H (CD3OD, 500 MHz) δ 3.96 [br t, N–H, J(HH6) = 11.6, J(HH5) = 11.6, J(HH7) = 3.5, J(HH8) = 3.5, J(HPX) = 37.2 Hz ], 3.19 [m, H8, J(H8H6) = 11.6, J(H8-H1) = 4.8, J(H8-H7) = 2.1, J(H8-H4) = 1.2 Hz], 3.14 [m, H7, J(H7-H5) = 11.6, J(H7-H2) = 4.8, J(H7-H3) = 2.1, J(H7-PA) = 35.8 Hz], 2.58 [m, H6, J(H6-H1) = 14.7, J(H6-H4) = 3.2, J(H6-PX) = 7.8 Hz ], 2.50 [m, H5, J(H5-H1) = 14.8, J(H5-H3) = 3.2, J(H5-PA) = 37.2 Hz], 2.13 [br t, H4, J(H4-H1) = 14.4, J(H4-PX) = 13.2 Hz], 1.99 [br t, H3, J(H3-H2) = 12.9, J(H-PA) = 12.9 Hz], 1.86 [m, H2, J(H2-PA) = 4.1 Hz], 1.78 [d, 3H, PA–Me, J(H-P) = 9.1 Hz], 1.66 [m, H1, J(H1-PX) = 4.3 Hz], 1.39 [d, 3H, PX–Me, J(H-P) = 7.8 Hz], ], 1.33 [d, 9H, P–Me3, J(H-P) = 8.7 Hz], ], 1.30 [d, 3H, PX–Me, J(H-P) = 8.3 Hz], 0.94 [d, 3H, PA–Me, J(H-P) = 8.1 Hz]; 13C{1H} (75 MHz, CH3OH–CD3COCD3, −80 °C, sample 30% enriched in 13CO2) δ 218.6 [ddd, 1 CO2, J(C-P) = 49.1, J(C-P) = 15.5, J(C-P) = 7.8 Hz], 214.1 [ddd, 1 CO2, J(C-P) = 43.7, J(C-P) = 18.5, J(C-P) = 10.5 Hz]. At 25 °C, these resonances appear as two multiplets with the same chemical shifts. 13C{1H} (125 MHz, C6D6, 25 °C, non-enriched sample) δ 210.5 [m, 1 CO2], 208.0 [m, 1 CO2], 49.3 [s, CH2], 48.0 [s, CH2], 34.1 [d, P–CH2, J(C-P) = 35 Hz ], 31.8 [d, P–CH2, J(C-P) = 28 Hz ], 20.0 [d, P(CH3)3, J(C–P) = 40 Hz ], 12.2 [d, PA–Me, J(C-P) = 39 Hz], 9.5 [d, PX–Me, J(C-P) = 28.5 Hz], 9.0 [d, PX–Me, J(C-P) = 28.1 Hz], 8.7 [d, PA–Me, J(C-P) = 29.1 Hz]; 31P{1H} (C6D6, 121 MHz), AMX spin system, δA 28.3 [PA, J(PA-PM) = 12, J(PA-PX) = 178 Hz], δM 22.9 [PMe3, J(PM-PX) = 12 Hz], δX 15.0 [PX].
Heating a methanolic solution of complex 3 at 60 °C under CO (1 atm) for 3 h results in a change of colour from the initial orange to dark yellow. The reaction gases are trapped inside of a liquid nitrogen-cooled ampoule, a white solid (CO2) being obtained. The former solution was investigated by NMR, a mixture of carbonyls of composition [Mo(CO)6(PMe3)6−n] being detected. The trapped gas was transferred into a flask containing a wet solution of the nickelacycle compound and the mixture stirred for 2 h, before volatile removal and NMR investigation of the crude sample.
An alternative procedure has been developed. A solution of Na2MoO4 in water (5 ml) was transferred into a flask containing [Mo(CO3)(CO)(PMe3)4] (0.2 g, 0.4 mmol) suspended in acetone (10 ml). Upon addition, the starting material dissolved at the same time that a new orange-coloured solid came out of the solution. The mixture was stirred overnight, the solid filtered off, washed with THF and Et2O, and dried under vacuum. The resulting orange solid displayed an IR spectrum identical to that described above. Yield: 50%.
Crystal structure determinations for compounds 1 and 4†
A summary of the fundamental crystal data is given in Table 1. In both cases, a crystal of prismatic shape was coated with resin epoxy and mounted in a Kappa diffractometer. Data were collected using graphite monochromated Mo-Kα (λ = 0.71069 Å) radiation. The cell dimensions were refined by least-squares fitting of the θ values of 25 reflections. The intensities were corrected for Lorentz and polarization effects. Scattering factors for neutral atoms and anomalous dispersion corrections for Mo and P were taken from the International Tables for X-ray Crystallography.20 The structure was solved by Patterson and Fourier methods. An empirical absorption correction was applied at the end of the isotropic refinement. A final refinement was undertaken with anisotropic thermal parameters for the non-hydrogen atoms. In the case of complex 1, the H1 atom was located in a difference synthesis and its coordinates were refined. Final difference synthesis showed three peaks that were assigned to 3-carbon chains originating from the petroleum ether used as the solvent. Most of the calculations were carried out with the SHELXTL system.21Acknowledgements
We thank the Junta de Andalucía for financial support (Acciones Coordinadas).References
- T. G. Spiro and W. M. Stigliani, in Chemistry of the Environment, Prentice Hall, Upper Saddle River, New Jersey, 1996, ch. 2 Search PubMed.
- (a) S. L. Wells and J. DeSimone, Angew. Chem., Int. Ed., 2001, 40, 518 CrossRef CAS; (b) X. Yin and J. R. Moss, Coord. Chem. Rev., 1999, 181, 27 CrossRef CAS; (c) D. H. Gibson, Chem. Rev., 1996, 96, 2063 CrossRef CAS; (d) K. Tanaka, Adv. Inorg. Chem., 1995, 43, 409 CAS; (e) P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1995, 95, 259 CrossRef CAS; (f) W. Leitner, Angew. Chem., Int. Ed. Engl., 1995, 34, 2207 CrossRef CAS.
- (a) J. C. Calabrese, T. Herskovictz and J. B. Kinney, J. Am. Chem. Soc., 1983, 105, 5914 CrossRef CAS; (b) H. Tanaka, H. Nagano, S. Peng and K. Tanaka, Organometallics, 1992, 11, 1450 CrossRef CAS.
- For some examples of κ2-C,O–M, see: (a) M. Aresta, C. F. Nobile, V. G. Albano, E. Forni and M. Manassero, J. Chem. Soc., Chem. Commun., 1975, 36 RSC; (b) M. Aresta and C. F. Nobile, J. Chem. Soc., Dalton Trans., 1977, 708 RSC; (c) G. S. Bristow, P. B. Hitchcock and M. F. Lappert, J. Chem. Soc., Chem. Commun., 1981, 1145 RSC; (d) S. Gambarotta, C. Floriani, A. Chiesi-Villa and C. Gaustini, J. Am. Chem. Soc., 1985, 107, 2985 CrossRef CAS.
- (a) R. Alvarez, E. Carmona, J. M. Marín, M. L. Poveda, E. Gutiérrez-Puebla and A. Monge, J. Am. Chem. Soc., 1986, 108, 2286 CrossRef CAS; (b) E. Carmona, A. K. Hughes, M. A. Muñoz, D. O'Hare, P. J. Pérez and M. L. Poveda, J. Am. Chem. Soc., 1991, 113, 9210 CrossRef CAS.
- (a) G. Fachinetti, C. Floriani, A. Chiesi-Villa and C. Gaustini, J. Am. Chem. Soc., 1979, 101, 1767 CrossRef CAS; (b) J. C. Bryan, S. J. Geib, A. L. Rheingold and J. M. Mayer, J. Am. Chem. Soc., 1987, 109, 2826 CrossRef CAS.
- (a) J. Chatt, M. Kubotta, G. J. Leigh, T. C. March, R. Mason and D. J. Yarrow, J. Chem. Soc., Chem. Commun., 1974, 1033 RSC; (b) S. Inoue and N. Yamazaki, Organic and Bioinorganic Chemistry of Carbon Dioxide, Halsted Press, Tokyo, Japan, 1982 Search PubMed; (c) A. D. Palmer and R. Van Edik, Chem. Rev., 1983, 83, 651 CrossRef CAS; (d) L. K. Fong, J. R. Fox and N. J. Cooper, Organometallics, 1987, 6, 223 CrossRef CAS.
- (a) R. Alvarez, E. Carmona, A. Galindo, E. Gutiérrez, J. M. Marín, A. Monge, M. L. Poveda, C. Ruíz and J. M. Savariault, Organometallics, 1989, 8, 2430 CrossRef CAS; (b) R. Alvarez, J. L. Atwood, E. Carmona, P. J. Pérez, M. L. Poveda and R. D. Rogers, Inorg. Chem., 1991, 30, 1493 CrossRef CAS.
- (a) E. Carmona, P. Palma, M. Paneque, M. L. Poveda, E. Gutiérrez-Puebla and A. Monge, J. Am. Chem. Soc., 1986, 108, 6424 CrossRef CAS; (b) E. Carmona, E. Gutiérrez-Puebla, J. M. Marín, A. Monge, M. Paneque, M. L. Poveda and C. Ruíz, J. Am. Chem. Soc., 1989, 111, 2883 CrossRef CAS; (c) E. Carmona, J. M. Marín, P. Palma, M. Paneque and M. L. Poveda, Inorg. Chem., 1989, 28, 1895 CrossRef CAS; (d) M. K. Reinking, J. Ni, P. E. Fanwick and C. P. Kubiak, J. Am. Chem. Soc., 1989, 111, 6454 CrossRef CAS.
- E. Sánchez-Marcos, R. Caballol, G. Trinquier and J.-C. Barthelat, J. Chem. Soc., Dalton Trans., 1987, 2373 RSC.
- T. Herskovitz and L. J. Guggenberger, J. Am. Chem. Soc., 1976, 98, 1615 CrossRef CAS.
- A. A. Danopoulos and P. G. Edwards, Polyhedron, 1989, 8, 1767 CrossRef CAS.
- (a) A. R. Willis, P. G. Edwards, H. Harman and M. B. Hursthouse, Polyhedron, 1989, 9, 1457 CrossRef CAS; (b) A.-R. H. Al-Soudani, A. S. Batsanov, P. G. Edwards and J. A. K. Howard, J. Chem. Soc., Dalton Trans., 1994, 987 RSC; (c) P. G. Edwards, P. W. Read, M. B. Hursthouse and K. M. Abdul Malik, J. Chem. Soc., Dalton Trans., 1994, 971 RSC; (d) M. D. Fryzuk, T. S. Haddad and S. J. Rettig, Organometallics, 1991, 10, 2026 CrossRef CAS; (e) M. D. Fryzuk and C. D. Montmongery, Coord. Chem. Rev., 1989, 95, 1 CrossRef CAS; (f) C. Bianchini, D. Masi, A. Romerosa, F. Zanobini and M. Peruzzini, Organometallics, 1999, 18, 2376 CrossRef CAS; (g) C. Bianchini, M. Peruzzini, F. Zanobini, C. Lopez, I. de los Ríos and A. Romerosa, Chem. Commun., 1999, 443 RSC; (h) F. G. N. Cloke, P. B. Hitchcock and J. B. Love, J. Chem. Soc., Dalton Trans., 1995, 25 RSC; (i) R. R. Schrock, A. L. Casado, J. T. Goodman, L.-Ch. Liang, P. J. Bonitatebus, Jr. and W. M. Davis, Organometallics, 2000, 19, 5325 CrossRef CAS.
- F. A. Cotton, D. J. Darensbourg, S. Klein and B. W. S. Kolthammer, Inorg. Chem., 1982, 21, 2661 CrossRef CAS.
- L. Pauling, The Nature of the Chemical Bond, Cornell University Press, New York, 3rd edn., 1960 Search PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, John Wiley and Sons, New York, 5th edn., 1997 Search PubMed.
- R. Mathieu, M. Lenzi and R. Poilblanc, Inorg. Chem., 1970, 9, 2030 CrossRef CAS.
- B. M. Gatehouse and P. Leverett, J. Chem. Soc. A, 1969, 848 Search PubMed.
- I. Castro-Rodríguez, H. Nakai, L. N. Zakharov, A. L. Rheingold and K. Meyer, Science, 2004, 305, 1757 CrossRef CAS.
- International Tables for X-Ray Crystallography, Kynoch Press, Birmingham, UK, 1974, vol. IV, p. p. 72 Search PubMed.
- Software for the SMART System, V5.04, and SHELXTL, V5.1, Bruker–Siemens Analytica X-ray Instrument, Inc., Madison, WI, 1998 Search PubMed.
Footnote |
| † CCDC reference numbers 231960 (1) and 231961 (4). See http://www.rsc.org/suppdata/nj/b4/b409385b/ for crystallographic data in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2005 |