Preparation of α,β-unsubstituted meso-arylbidipyrrins via metal-templated, oxidative coupling of dipyrrins†
Hubert S.
Gill
,
Isaac
Finger
,
Ivana
Božidarević
,
Florence
Szydlo
and
Michael J.
Scott
*
Department of Chemistry, University of Florida, P.O. Box 117200, Gainesville, FL 32611, USA. E-mail: mjscott@chem.ufl.edu; Fax: +1 352 392 3255; Tel: +1 352 846 1165
First published on 9th December 2004
Abstract
A three-step, one-pot procedure provides meso-aryl-α,β-unsubstituted bisdipyrrinato nickel complexes; oxidative coupling of these ligands followed by demetallation affords an unprecedented class of meso-aryl-α,β-unsubstituted bidipyrrins.
Oxidative biaryl coupling reactions have been employed in the preparation of numerous classes of compounds, including polycyclic aromatic hydrocarbons,1a fused porphyrin arrays,1b and natural products.1c Direct carbon-carbon bond-forming reactions are among the most challenging transformations in synthetic chemistry, often requiring harsh reaction conditions and unpleasant metal reagents or expensive catalysts. Furthermore, due to the lack of regiospecificity, intermolecular dehydrogenation reactions of aromatic compounds frequently result in polymers or complex product mixtures, unless directing or protecting groups are utilized. Inspired by the successful oxidative biaryl coupling methodology recently employed for the synthesis of large, flat porphyrins,2 we were interested in applying these concepts to a divergent area of synthetic chemistry. In this system, a late transition metal was incorporated into the macrocycle to lower the oxidation potential of the ligand, and the pre-organization of the carbons to be coupled facilitated the cyclo-dehydrogenation under very mild oxidative conditions. With these issues in mind, we examined the reactivity of metal complexes with meso-aryl-α,β-unsubstituted bidipyrrins (Fig. 1) since coupling of two of the α-carbons would produce a bidipyrrin, a difficult molecule to prepare by known methodology.
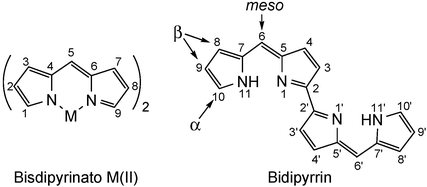 | ||
| Fig. 1 General bidipyrrin and bisdipyrrinato compounds and their respective numbering schemes. | ||
Bidipyrrins represent the class of bile pigments with two dipyrromethene units linked directly by α-carbons. With an eye towards utilizing these tetrapyrroles as synthons for corrole forming reactions, Johnson and Price first described their preparation in 1960,3 but little work was pursued with this ligand prior to the report of a dimeric zinc complex (M2L2) by Dolphin and co-workers in 1998.4 In the following years, Bröring and co-workers isolated a similar metal-free octaalkylbidipyrrin using modified procedures,5 and they expanded the scope of their methodology to include the preparation of the previously unprecedented meso-arylbidipyrrins. Their approach, employing diacylbipyrroles, represents the only synthetic scheme for the preparation of meso-arylbidipyrrins to date. Unfortunately, the reaction requires harsh conditions, involving a reflux in neat POCl3, and the methodology is not compatible with either reactive meso-aryl groups or with the preparation of α- or β-unsubstituted meso-arylbidipyrrins.
For reasons enumerated by Bröring, meso-arylbidipyrrins offer advantages over their meso-unsubstituted analogs.5 Aryl moieties incorporated at the meso-positions of bidipyrrins provide points for synthetic elaboration, allow for the fine-tuning of steric and electronic properties. The range of aryl substituents that may be incorporated at the bidipyrrin meso-positions by the methodology of Bröring is limited to para- and meta-substituted aryl moieties, as ortho-substituted aryl groups hinder the acylation reaction.6 In addition, the absence of α-alkyl substituents on bidipyrrins is a desirable feature for many applications, including monomers for electropolymerization, and the ability to further derivatize at these free α positions may allow for the preparation of macrocycles.
In order to couple dipyrrinato groups, two properties are important for the metal template: a propensity towards square-planar coordination and the formation of charge neutral complex with both the bisdipyrrinato and bidipyrrinato ligands. Nickel offers several advantages over metals such as palladium or platinum for this purpose. The stronger Lewis acidity of Ni(II) weakens the C–H bonds of coordinated ligands to a greater degree than Pd(II) or Pt(II), and the small ionic radius of Ni(II) brings the α-hydrogens of bisdipyrrinato Ni(II) complexes into close contact (2.481 and 2.532 Å), as illustrated in the solid-state structure of Ni-2b depicted in Fig. 2. Additionally, the removal of the metal template should be facile, since Ni(II) complexes are typically easier to demetallate than analogous chelates of Pd(II) or Pt(II).
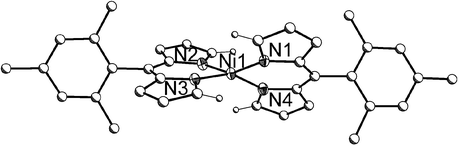 | ||
| Fig. 2 Solid-state structure of Ni-2b (40% ellipsoids). Carbon atoms are drawn with arbitrary radii and hydrogen atoms other than those on the α-carbons have been omitted for clarity. | ||
α,β-Unsubstituted-5-aryldipyrromethanes are needed for the synthesis presented in Fig. 3. These readily available molecules offer several desirable attributes for precursors, including their ease of preparation, numerous choices for aryl functional groups, and the well-established coordination chemistry of their 2H+, 2e− oxidized dipyrromethene derivatives.7,8 Preparations of α,β-unsubstituted-5-arylbisdipyrrinato M(II) complexes have been recently described by the groups of both Dolphin and Lindsey.8b,e For the preparations of Ni-2a and Ni-2b, a room temperature, three-step, one-pot procedure similar to that utilized by Lindsey and co-workers was employed (Fig. 3). Isolation of the resulting bisdipyrrinato Ni(II) complexes is straightforward and the complexes Ni-2a and Ni-2b are obtained in good yields as crystalline solids, despite the multi-step nature of the synthetic methodology.
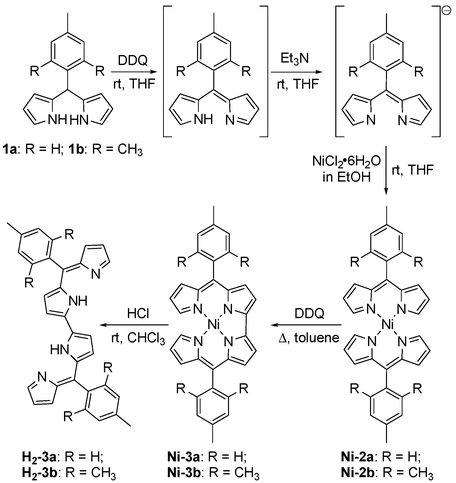 | ||
| Fig. 3 Illustration of the three-step, one-pot reaction sequence employed for the preparation of bisdipyrrinato Ni(II) complexes followed by oxidation and demetallation to afford dibipyrrins. | ||
The cyclic voltammograms measured for the bisdipyrrinato nickel complexes revealed irreversible oxidative processes at 1.10 V for Ni-2a and 1.08 V for Ni-2b (vs. Ag/AgCl). Various reaction conditions were attempted to affect this oxidative change chemically, but the nickel complexes proved to be incompatible with the Lewis acids often employed for dehydrogenation reactions. Due to this difficulty, alternative procedures were pursued; treatment of Ni-2a or Ni-2b with a slight excess of DDQ in refluxing toluene afforded the desired bidipyrrinato nickel complexes (Fig. 3). Filtration through a plug of silica, followed by recrystallization, provides Ni-3a and Ni-3b as purple microcrystalline solids. Treatment of CHCl3 solutions of Ni-3a and Ni-3b with HCl results in an immediate color change from wine-red to bright-green. Aqueous workup causes a change in color to indigo upon deprotonation and solvent removal provides the metal-free ligands, H2-3a and H2-3b, in nearly quantitative yields.
Typically, tetrahedral Ni(II) complexes have an S = 1 ground state while square-planar Ni(II) complexes are diamagnetic, and often Ni(II) complexes with an intermediate geometry will exhibit temperature-dependent spin equilibrium.9 Other than Ni-2a, Ni-2b, and the related phenyl and p-nitrophenyl-α-unsubstituted analogs described by Dolphin and co-workers,8b all reported bisdipyrrinato nickel complexes bear methyl or phenyl groups at their α-carbons. The steric clash between these groups induces a severe distortion towards tetrahedral geometry and the complexes are paramagnetic.10 Based upon these precedents and the realization that even hydrogen atoms on the α-carbons sterically preclude a square-planar environment, Dolphin and co-workers were surprised to observe sharp signals in the 1H and 13C NMR spectra of the phenyl and p-nitrophenyl complexes.8b The dihedral angle between the planes containing N(1)–Ni(1)–N(4) and N(2)–Ni(1)–N(3) indicates the degree of tetrahedral distortion for square-planar complexes, and the structural characterization of the phenyl derivative revealed a dihedral angle of 38.5°, which is considerably smaller than the tetrahedral angle of 76.3° calculated from the X-ray structure of [bis(1,3,7,9-tetramethyldipyrrinato)]Ni(II).10c In view of the properties of the phenyl and p-nitrophenyl congeners, Ni-2a and Ni-2b were expected to be diamagnetic, low-spin complexes and the 1H NMR spectrum of Ni-2a (see electronic supplementary information, ESI) did indeed exhibit sharp signals. In contrast, the resonances in the spectrum of Ni-2b were broadened and many were shifted downfield relative to their analogous positions in the spectrum of Ni-2a (see ESI). The α-carbon protons experienced the most significant shift, from a value of 9.23 ppm in Ni-2a to 15.62 ppm in Ni-2b. When Ni(II) complexes adopt geometries in an intermediate range between square planar and tetrahedral, they often exhibit a temperature-dependent magnetic moment; in the structure of Ni-2b the angle between N(1)–Ni(1)–N(4) and N(2)–Ni(1)–N(3) planes has opened up in comparison to the phenyl derivative to a value of 49.6°.8b This 11.1° increase is apparently sufficient to induce thermal population of a paramagnetic state for the d8 Ni(II) ion and preliminary magnetic data is consistent with this observation.11
The coupling of the α-carbons upon going from Ni-2b to Ni-3b appears to relieve some of the steric strain in the complex; the Ni(II) cation assumes a coordination geometry closer to square planar (Fig. 4), with a smaller angle between the planes containing N(1)–Ni(1)–N(2) and N(3)–Ni(1)–N(4) for Ni-3b (11.8°) in comparison to Ni-2b (38.3°). As apparent from the sharp signals in the 1H NMR spectrum of Ni-3b, the paramagnetism observed for Ni-2b is lost and the resonance for the hydrogens on the uncoupled α-carbons experiences a dramatic change upon conversion to Ni-3b, changing from a broad singlet at 15.62 ppm to a doublet of doublets at 6.02 ppm (see ESI). Although conversion to the bidipyrrin causes a slight increase in the separation of the uncoupled α-carbons from 3.025 Å to 3.093 Å, their associated hydrogens are brought in closer proximity to each other (2.284 Å for Ni-3b) relative to the α-protons in the uncoupled complex (2.507 Å for Ni-2b).
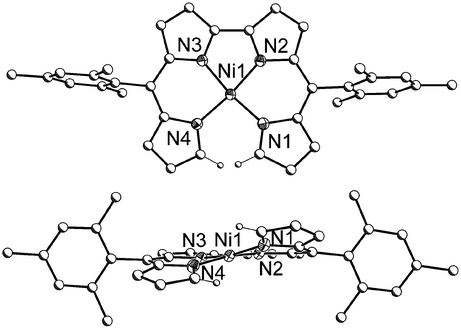 | ||
| Fig. 4 Solid-state structure of Ni-3b (40% ellipsoids). Carbon atoms are drawn with arbitrary radii; hydrogen atoms other than those on the α-carbons have been omitted for clarity. | ||
As depicted in Fig. 4, the solid-state structure of Ni-3b reveals a helical complex, consistent with the structures of other known Ni bidipyrrins.12 Interestingly, the degree of helicity, as described by the angle between the planes containing N(1)–Ni(1)–N(4) and N(2)–Ni(1)–N(3), is only 11.9° for Ni-3b. This interplanar angle is slightly smaller than the 13.0° measured for the only other α-unsubstituted bidipyrrinato Ni(II) complex reported and significantly smaller than the 19.9° measured for an α-methyl bidipyrrinato Ni(II) complex.12 The average Ni–N bond length of 1.873(2) Å is consistent with other bidipyrrinato Ni(II) complexes in the literature.12 The relatively short bond between the coupled α-carbons of 1.437(3) Å is indicative of its partial double-bond character, as can be envisioned from the resonance form placing the two imine nitrogens in one dipyrromethene moiety.
Electrochemical investigations of Ni-3b and H2-3b were undertaken to provide insight into the stability and electronic properties of these chromophores. Comparable to redox processes observed for numerous porphyrin species,13 the cyclic voltammogram of Ni-3b exhibits two reversible oxidations at 0.80 and 1.09 V, as well as two reversible reductions at 0.89 and 1.60 V (vs. Ag/AgCl). These observations are consistent with a series of measurements reported by Bröring and co-workers, which also show two reversible processes in both the oxidative and reductive directions for most compounds of this type.14 In addition to a reversible reduction at −1.05 V, H2-3b also undergoes an irreversible oxidation at 0.94 V versus Ag/AgCl, indicating that alkylation of the pyrrolic carbons is not required for stability over this broad potential range.
The electronic absorption spectra of these compounds change substantially on going from Ni-2 to Ni-3 to H2-3 in both series as illustrated by the spectra of the mesityl series shown in Fig. 5. Compounds Ni-2a and Ni-2b exhibit one broad absorption maximum around 471 and 478 nm, respectively, which are slightly higher in energy than transitions reported for other bisdipyrrinato Ni(II) complexes.8c,10 The absorption spectra become more complex after coupling: the main absorption bands of Ni-3a and Ni-3b are bathochromically shifted in comparison to their precursors, but they also exhibit electronic transitions at longer wavelengths (above 550 nm). Upon demetallation, an additional low-energy feature emerges in the electronic spectra of H2-3a and H2-3b. The metal-free compounds H2-3a and H2-3b show fluorescence emissions at 687 and 648 nm, with quantum yields of 1% and 5%, respectively. The higher value for H2-3b is consistent with the observation that rigid substituents such as mesityl groups at the meso-position in bisdipyrrinato Zn(II) complexes significantly increase the quantum yields for emission.15
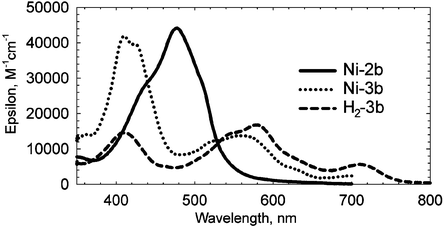 | ||
| Fig. 5 Electronic absorption spectra of Ni-2b, Ni-3b and H2-3b. | ||
In summary, the three-step, one-pot room temperature reaction of dipyrromethanes with DDQ, Et3N and NiCl2 described herein is a practical method to obtain meso-arylbisdipyrrinato Ni(II) complexes on the multigram scale. While the para-tolyl derivative, Ni-2a, showed sharp resonances in the 1H NMR spectrum, the mesityl analog, Ni-2b, exhibited broad signals that were shifted downfield, indicative of temperature-dependent paramagnetism. Furthermore, the oxidative coupling of two dipyrrins by employing Ni(II) as a template is a facile procedure for the preparation of previously unreported meso-aryl,-α,β-unsubstituted bidipyrrins and this methodology is compatible with aryl substituents bearing methyl groups at the ortho-positions. Demetallation using HCl results in the metal-free bidipyrrins and these chromophores are stable over a potential range of ∼2 V. Photophysical measurements of the dipyrrins reveal fluorescence in the low-energy region of the visible spectrum, with the mesityl derivative giving a higher quantum yield than the para-tolyl congener.
Experimental
General
1H NMR spectra were recorded on a Varian Mercury spectrometer at 300 MHz in CDCl3 and were referenced to the solvent residual peak. Absorption spectra were collected in CH2Cl2 on a Varian Cary 50 spectrophotometer. Reagents and solvents were used as received from Aldrich or Fisher Scientific. Dipyrromethanes 1a and 1b were prepared following modified literature procedures.7aFluorescence spectroscopy
Steady-state photoluminescence spectroscopy and quantum yield measurements were carried out using a SPEX Fluorolog 2 instrument. The standard used for quantum yield calculations was Ru(bipy)3Cl2 (Φ = 0.04). Samples H2-3a and H2-3b were dissolved in toluene and the concentration was adjusted so that their absorbance at the excitation wavelength (453 nm) matched that of a standard (0.2).Electrochemistry
Electrochemical measurements were made on an EG&G PAR VersaStatII potentiostat with a Pt disc working electrode, a Pt wire counter electrode, and an aqueous Ag/AgCl reference electrode (NaCl, 3 M) fitted with a Vicor frit as a salt bridge. In all cases dry, degassed C6H5CN was used as the solvent and tetra-N-butylammoniumhexafluorophosphate, at a concentration of 0.100 M, was used for the supporting electrolyte. A small portion of ferrocene (Fc) was added to the cell after each series of measurements to confirm the potential of the reference electrode. The E1/2 for the Fc/Fc+ couple remained constant at 0.46(1) V versus Ag/AgCl. The analyte concentration was 2.5(2) mM and all measurements were made at room temperature.X-Ray crystallography‡
Unit cell dimensions were obtained and intensity data collected on a Bruker CCD SMART diffractometer at low temperature, with monochromatic Mo-Kα radiation (λ = 0.710 73 Å). The data collections nominally covered over a hemisphere of reciprocal space by a combination of three sets of exposures; each set had a different ϕ angle for the crystal and each exposure covered 0.3° in ω. The crystal-to-detector distance was 5.0 cm. The data sets were corrected empirically for absorption using SADABS.16 The structure was solved and refined using the Bruker SHELXTL software package.17Syntheses
Acknowledgements
We thank the Ministère de l’Education Nationale et de la Recherche (support for F. Szydlo), the Research Corporation (Research Innovation Award), and the Alfred P. Sloan Foundation for providing financial support for this work. Support from the National Science Foundation is also gratefully acknowledged. We thank Prof. K. Schanze for assistance with photophysical measurements and Prof. M. Meisel and Mr. J.-H. Park for magnetic measurements.References
- (a) M. D. Watson, A. Fechtenkotter and K. Mullen, Chem. Rev., 2001, 101, 1267 CrossRef CAS; (b) A. Tsuda and A. Osuka, Science, 2001, 293, 79 CrossRef CAS; (c) R. B. Herbert, A. E. Kattah, A. J. Murtagh and P. W. Sheldrake, Tetrahedron Lett., 1995, 36, 5649 CAS.
- H. S. Gill, M. Harmjanz, J. Santamaria, I. Finger and M. J. Scott, Angew. Chem., Int. Ed., 2004, 43, 485 CrossRef CAS.
- A. W. Johnson and R. Price, J. Chem. Soc., 1960, 1649 RSC.
- Y. Zhang, A. Thompson, S. J. Rettig and D. Dolphin, J. Am. Chem. Soc., 1998, 120, 13537 CrossRef CAS.
- M. Bröring, Synthesis, 2000, 1291 CrossRef CAS.
- M. Bröring, D. Griebel, C. Hell and A. Pfister, J. Porphyrins Phthalocyanines, 2001, 5, 708 CrossRef CAS.
- (a) C. H. Lee and J. S. Lindsey, Tetrahedron, 1994, 50, 11427 CrossRef CAS; (b) J. S. Lindsey, in The Porphyrin Handbook, eds. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, Burlington, MA, 2000, vol. 1, p. 45 Search PubMed.
- (a) H. Fisher and H. Orth, Die Chemie des Pyrrols, Akademische Verlagsgesellschaft, Leipzig, 1st edn., 1940, vol. 2, p. 1 Search PubMed; (b) C. Brückner, V. Karunaratne, S. J. Rettig and D. Dolphin, Can. J. Chem., 1996, 74, 2182 CAS; (c) C. Brückner, Y. Zhang, S. J. Rettig and D. Dolphin, Inorg. Chim. Acta, 1997, 263, 279 CrossRef CAS; (d) S. M. Cohen and S. R. Halper, Inorg. Chim. Acta, 2002, 341, 12 CrossRef CAS; (e) L. Yu, K. Muthukumaran, I. V. Sazanovich, C. Kirmaier, E. Hindin, J. R. Diers, P. D. Boyle, D. F. Bocian, D. Holten and J. S. Lindsey, Inorg. Chem., 2003, 42, 6629 CrossRef CAS.
- (a) R. H. Holm and K. Swaminathan, Inorg. Chem., 1963, 2, 181 CrossRef CAS; (b) R. H. Holm, Acc. Chem. Res., 1969, 2, 307 CrossRef CAS; (c) A. La Cour, M. Findeisen, R. Hazell, L. Henning, C. E. Olsen and O. Simonsen, J. Chem. Soc., Dalton Trans., 1996, 3437 RSC.
- (a) J. E. Fergusson and C. A. Ramsey, J. Chem. Soc., A, 1965, 5222 Search PubMed; (b) Y. Murakami and K. Sakata, Inorg. Chem. Acta, 1968, 2, 273 CrossRef CAS; (c) F. A. Cotton, B. G. DeBoer and J. R. Pipal, Inorg. Chem., 1970, 9, 783 CrossRef CAS; (d) Y. Murakami, Y. Matsuda and K. Sakata, Inorg. Chem., 1971, 10, 1728 CrossRef CAS.
- A preliminary plot of the magnetic data is included in the supplementary material. Complete magnetic data will be presented in a future publication.
- M. Bröring and C. D. Brandt, Monatsh. Chem., 2002, 133, 623 CrossRef CAS.
- K. M. Kadish, G. Royal, E. V. Caemelbecke and L. Gueletti, in The Porphyrin Handbook, eds. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, Burlington, MA, 2000, vol. 9, p. 1 Search PubMed.
- M. Bröring, C. D. Brandt, J. Lex, H.-U. Humpf, J. Bley-Escrich and J.-P. Gisselbrecht, Eur. J. Inorg. Chem., 2001, 2549 CrossRef CAS.
- I. V. Sazanovich, C. Kirmaier, E. Hindin, L. Yu, D. F. Bocian, J. S. Lindsey and D. Holten, J. Am. Chem. Soc., 2004, 126, 2664 CrossRef.
- R. H. Blessing, Acta Crystallogr., Sect. A, 1995, 51, 33 CrossRef.
- G. M. Sheldrick, SHELXTL-6.12, Program for structure solution, refinement and presentation, BRUKER AXS Inc., Madison, WI, 2000 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H NMR spectra for all compounds and magnetic analysis of Ni-2b (χTvs.T plot). See http://www.rsc.org/suppdata/nj/b4/b412620c/ |
| ‡ CCDC reference numbers 254228 and 254229. See http://www.rsc.org/suppdata/nj/b4/b412620c/ for crystallographic data in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2005 |