Hydrogen bonds between ammonium ions and aromatic rings exist and have key consequences on solid-state and solution phase properties
Christos A.
Ilioudis
a,
Michael J.
Bearpark
b and
Jonathan W.
Steed
*c
aDepartment of Chemistry, King’s College London, Strand, London, WC2R 2LS, UK
bDepartment of Chemistry, Imperial College, Exhibition Road, London, SW7 2AZ, UK
cDepartment of Chemistry, University of Durham, South Road, Durham, DH1 3LE, UK. E-mail: jon.steed@durham.ac.uk
First published on 2nd December 2004
Abstract
A very small azaphane that exhibits high affinity for H+ has been synthesised. Included protons are stabilised by a charge-assisted NH-π interaction and hence the bound proton is effectively solvated by the aromatic ring. Exchange of H+ is slow on the 1H NMR time scale and the conformational characteristics of the macrobicycle are highly pH dependent.
Current understanding of the way in which biological and synthetic molecules fold and interact with one another is undergoing a transformation as the existence of new kinds of unconventional hydrogen bonds are recognized and allocated their correct place in the pantheon of intermolecular interactions.1–3 Structural, database and computational surveys have suggested that short contacts between NH groups and π-electron density are common in enzymes4 as well as a range of small molecules.5 It is not clear, however, whether such short distances are a result of enforced repulsive contacts or represent genuine attractive, directional interactions (calculated energies 0–2.9 kcal mol−1), although gas phase measurements indicate that HNMe3+ interacts approximately equally well with both benzene and a water molecule.6,7 We now report the properties of a system that shows that the postulated NH⋯π interaction has a major effect on solution behavior analogous to an NH⋯O hydrogen bond and represents solvation of the NH proton by an aromatic ring.
Structural and database evidence has been presented showing the existence of new hydrogen bonds from non-traditional hydrogen bond donors such as C–H groups to electronegative atoms2 and from donors such as OH and NH groups to surprising acceptors such as aromatic rings and alkynes.3 Structural studies can reveal geometrical characteristics but reveal little about the energy of such interactions, and are never decoupled from other crystal packing effects. Crucially, database studies provide no insight into whether new interactions have any observable consequences on molecular dynamics and solution properties such as acidity or basicity. Indeed, the debate over the question of the existence of various types of hydrogen bond has become fierce in recent years.8,9 Such questions can only be settled by careful study of the consequences of such interactions in systems in which they are, as much as possible, isolated from other dominating or interfering effects.
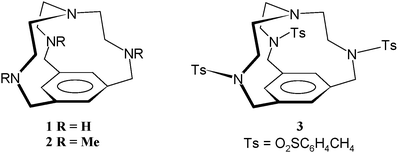
With reference to the NH⋯π interaction, we have designed the very small bicyclic cyclophane 1. The geometry of compound 1 is such that the lone pair of the tertiary bridgehead nitrogen atom is constrained to point inwards into the tiny central cavity. This cavity is much too small to bind to anions or metal cations and is suitable only for inclusion of a proton. This in geometry10 is conclusively established by both DFT and MP2 molecular modelling studies and by the X-ray crystal structure of the tritosylate (3). Small, rigid cyclophanes are known to experience unfavourable electron-electron repulsions between electron-rich groups forced into close proximity.11,12 In the case of 1 and 3 the molecules exhibit some strain as a result of the proximity of the tertiary amine lone pair and the aromatic ring. This strain is exhibited in the structure of 3 by the out-of-plane deformation of the benzylic carbon atoms C(7–9), which lie 0.260(11)–0.321(11) Å out of the aromatic ring mean plane.
The X-ray crystal structure of the protonated compound 1·4HCl·2H2O (Fig. 1) was obtained from a solution of 1 in aqueous HCl at pH < 2. The intra-cavity proton was located experimentally. The NH vector points directly at the aromatic ring centroid (N–H⋯centroid angle 179°), as shown in Fig. 1. The experimental H⋯centroid distance of 2.13 Å (the true distance is likely to be even shorter because of the underestimation of NH bond lengths by X-ray methods) is well below the sum of the van der Waals radii of H and C, while the N⋯centroid distance of 2.974(6) Å is at the lower end of the range seen for other structures postulated to contain an NH⋯π interaction5 and is marginally shorter than in 3, in agreement with the modelling studies. The attractive nature of the interaction is evidenced by the movement of the benzylic carbon atoms towards the aromatic ring plane [deviations in 1·4HCl 0.246(7)–0.258(8) Å].
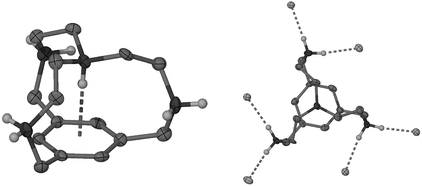 | ||
| Fig. 1 X-Ray crystal structure of the tetraprotonated cryptand in 1·4HCl·2H2O: (a) NH⋯π and (b) NH⋯Cl− interactions. | ||
Both DFT and MP2 calculations offer support to the conclusion that strain in the cyclophane is reduced upon protonation of the bridgehead nitrogen atom. DFT gives slightly closer agreement with the experimental bond lengths of 3 and 1·4HCl·2H2O. The DFT model of the neutral cryptand 1 indicates that the compound has approximate C3 symmetry with an endo bridgehead nitrogen atom exhibiting an N⋯centroid distance of 3.01 Å. The calculations indicate that upon protonation at this site the nitrogen atom moves very slightly but consistently closer to the aromatic ring. Gas phase DFT calculations [b3lyp/6-31+g(d,p)] suggest that the bridgehead is significantly the most favoured site for monoprotonation, by 14 kcal mol−1. If solvation effects are taken into account (iefpcm model, without geometry reoptimisation) this value is reduced to 7 kcal mol−1 but the inner-protonated form remains significantly more stable. It is well-known that while tertiary amines are more basic than secondary amines in the gas phase, this order is reversed in aqueous solution as a result of solvation effects.10 In this particular case the tethered aromatic ring in 1 is effectively playing the role of solvent for the intra-cavity proton. The results suggest that the stabilisation by the aryl ring is highly effective. Modelling of 1·4H+ suggests that the geometry changes little from 1·H+, highlighting the importance of the intra-cavity protonation and the preorganisation of 1.
It is noteworthy that thioether and sulfone analogues of 1, but bearing an apical CH group, have been reported by Pascal, Jr. et al.13,14 In the reported crystal structures, the CH group, which would not be expected to interact strongly with the aryl ring, is significantly further away from the aromatic ring centroid than in protonated 1 (Capical⋯centroid distances 3.206–3.314 Å). Remarkably, these workers were able to isolate a small amount of the hydrocarbon product of sulfone extrusion. While the material was not crystallographically characterised, the CH⋯centroid distance must be extremely short. The IR spectrum of the material shows evidence for extreme steric compression of the CH bond.14
Given the clear structural and theoretical evidence for the existence of a charge-assisted NH⋯π interaction in 1, we sought to examine its effects on the solution properties of the compound. The basicity of 1 was established by potentiometric titration with HCl in the presence of NaNO3, which gave log Ka values of 10.21(3), 8.50(4), 7.46(3) and 2.56(3). Changing the anion (Me4NCl) had little effect [log Ka = 10.12(6), 8.27(9), 7.57(5), 2.89(7)], suggesting that the basicity is not highly influenced by the hydrogen-bonding ability of the anion. The X-ray crystal structure of 1·4HCl shows the NH2+ groups all forming hydrogen bonds to Cl− anions with N⋯Cl distances in the range 3.047(4)–3.158(4) Å, while the shortest Nbridgehead⋯Cl− interaction is much longer at 4.372(4) Å. However, no evidence for anion chelation is noted. These log Ka values may be compared to values of 9.90, 9.35, 8.20 and 215 for the primary-amine-containing tertiary amine N(CH2CH2NH2)3, which is highly solvated on protonation at the termini, and 9.8, 9.17, 8.5, 7.21, 6.9, 6.70 and <2 for a large metacyclophane triethylenetriamine based macrobicycle containing two tertiary and six secondary amines that cannot engage in NH⋯π interactions.16 An important consequence of solvation effects in polar solution is that in triethylenetriamine based species protonation occurs initially at the primary or secondary amines and it can be very difficult to protonate the tertiary amine bridgehead nitrogen atom. However, there is ample evidence for protonation of macrobicyclic tertiary amines in cases where the proton is stabilised by conventional hydrogen bonds, particularly NH+⋯O, with tertiary amine basicity reaching values of 11.0 or, in one very small cryptand, 17.8!10
As a control, the basicity of the tri-N-methyl derivative 2 was determined in the presence of NaNO3, giving log Ka values of 10.43(5), 8.20(7), 7.14(5) and 3.12(6). The increase in the first log Ka value is consistent with intra-cavity protonation since inductive effects will render the cavity more electron-rich, while the decrease in the second and third values is consistent with decreased solvation of the tertiary amines compared to the secondary amine groups in 1. We sought to confirm the fact that initial protonation (as predicted by calculations) is at the bridgehead nitrogen by comparison of the 1H NMR spectra of the compound at a variety of pH values.
At pD 13 in D2O compound 1 shows three broad signals associated with the aliphatic CH2 groups, as well as a sharp signal associated with the aryl CH groups. Remarkably, however, at pD 11 an entirely separate, sharper set of signals begins to appear, consistent with the presence of a second species also possessing time-averaged C3v symmetry. The chemical shift of the benzylic protons (3.8 ppm) in this protonated species is similar to that of free 1 while the resonances assigned to the ethylenic backbone adjacent to the bridgehead are markedly shifted, suggesting bridgehead protonation. These spectra suggest that initial intra-cavity protonation occurs but that the resulting species is highly resistant to further protonation and exists in equilibrium with secondary amine protonated species as the pH is lowered. Raising the temperature to 90 °C results in the coalescence of the majority of the resonances into very broad signals. The high temperature indicates a relatively high, although NMR accessible, barrier to exchange between the two species. The presence of these two distinct sets of 1H NMR signals strongly suggests slow exchange between intra- and extra-cavity protons and indicates intra-cavity protonation at extremely high pH. Slow proton exchange has been noted before for noncyclophane cryptands in which the intra-cavity proton is stabilised by NH⋯O interactions.10,17 In other words, compound 1 is surprisingly basic as a result of the stabilisation of the intra-cavity proton by the cyclophane aromatic ring.
The 1H NMR spectrum of 1 reveals further interesting behaviour at pH 2 at room temperature. Under these conditions only a single fully protonated species is present, however, every one of the signals assigned to CH2 groups is doubled, indicating a lowering of the molecular symmetry from C3v to C3 and the freezing out of a chiral species by slowing of the propeller inversion motion. This conformation is consistent with the static, chiral structures observed crystallographically. The assignment of this fluxionality was confirmed by examination of the spectrum of neutral 1 in CDCl3 solution. At room temperature, the spectrum is broad. At +50 °C, the spectrum exhibits time averaged C3v symmetry, while at −50 °C a sharp spectrum is observed with geminal coupling constants of 12.1 Hz for the benzylic CH2 groups, for example (Fig. 2).
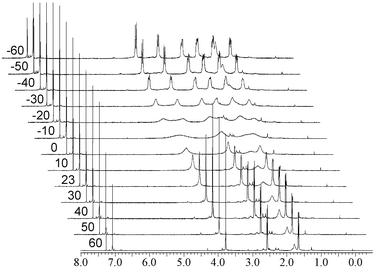 | ||
| Fig. 2 Variable-temperature 1H NMR spectra for compound 1 in CDCl3 solution. | ||
In conclusion, by rational molecular design we have constructed a model system that firmly establishes the ability of an aromatic ring to interact with an NH+ moiety. The protonation of 1 leads to a reduction in the strain in the compound and renders it more basic than unstrained cyclic and acyclic analogues. Exchange of the π-solvated proton with the surrounding medium is slow on the 1H NMR time scale in aqueous solution at temperatures up to 90 °C and the protonation of the cyclophane has a marked effect on its rate of propeller inversion.
Experimental
Syntheses
Potentiometric titration
All potentiometric titrations were performed at 25 °C under inert atmosphere, using carbonate-free NaOH. A Titrino model 736 GP was used. The protonation constants were determined from titrations of an approximately 10−3 M ligand solution containing an excess of HCl (0.01 M) in the presence of NaNO3 or Me4NCl to maintain the ionic strength at 0.1 M. The range of accurate pH measurements was considered to be 2–12. Stability constants were calculated with the program HYPERQUAD.Crystallography
Crystals of X-ray quality for 1 were grown by slow diffusion of acetone into a solution of 1 in diluted aqueous HCl (pH < 2). Crystals of X-ray quality for 3 were grown by slow diffusion of hexane into a solution of 3 in chloroform. Crystals were mounted on a thin glass fibre using silicon grease and cooled on the diffractometer to 120 K using an Oxford Cryostream low temperature attachment. Approximate unit cell dimensions were determined by the Nonius Collect program20 from 5 index frames of width 2° in ϕ using a Nonius Kappa CCD diffractometer, with a detector-to-crystal distance of 30 mm. The Collect program was then used to calculate a data collection strategy to 99.5% completeness for θ = 27.5° using a combination of 2° ϕ and ω scans of 10–120 s deg−1 exposure time (depending on crystal quality). Crystals were indexed using the DENZO-SMN package21 and positional data were refined along with diffractometer constants to give the final unit cell parameters. Integration and scaling (DENZO-SMN, Scalepack21) resulted in unique data sets corrected for Lorentz and polarisation effects and for the effects of crystal decay and absorption by a combination of averaging of equivalent reflections and an overall volume and scaling correction. Structures were solved using SHELXS-9722 and developed via alternating least-squares cycles and difference Fourier synthesis (SHELXL-9723) with the aid of the program XSeed.24 In general all non-hydrogen atoms were modelled anisotropically, while hydrogen atoms were assigned an isotropic thermal parameter 1.2 times that of the parent atom (1.5 for terminal atoms) and allowed to ride, except for acidic protons, which were located on the final difference Fourier map and refined freely. All calculations were carried out with either a Silicon Graphics Indy workstation or an IBM compatible PC.†Acknowledgements
We are especially grateful to Dr P. Gans (Leeds) for assistance with the potentiometric work and for the program Hyperquad. We thank King’s College London for a studentship (to CAI).References
- G. R. Desiraju and T. Steiner, The Weak Hydrogen Bond, OUP/IUPAC, Oxford, 1999 Search PubMed.
- G. R. Desiraju, Acc. Chem. Res., 1996, 29, 441 CrossRef CAS.
- F. H. Allen, J. A. K. Howard, V. J. Hoy, G. R. Desiraju, D. S. Reddy and C. C. Wilson, J. Am. Chem. Soc., 1996, 118, 4081 CrossRef CAS.
- S. K. Burley and G. A. Petsko, FEBS Lett., 1986, 203, 139 CrossRef CAS.
- C. S. Page and H. S. Rzepa, Electronic Conference on Trends in Organic Chemistry, 1995, www.ch.ic.ac.uk/etoc/papers/47/ Search PubMed.
- M. Meot-Ner (Mautner), J. Am. Chem. Soc., 1984, 106, 1265 CrossRef.
- C. A. Deakyne and M. Meot-Ner (Mautner), J. Am. Chem. Soc., 1985, 104, 474.
- T. Steiner, Chem. Commun., 1999, 313 Search PubMed.
- D. Braga, E. D’Oria, F. Grepioni, F. Mota, J. J. Novoa and C. Rovira, Chem.-Eur. J., 2002, 8, 1173 CrossRef CAS.
- A. Bencini, A. Bianchi, E. Garcia-Espana, M. Micheloni and J. A. Ramirez, Coord. Chem. Rev., 1999, 188, 97 CrossRef CAS.
- M. N. Jagadeesh, A. Makur and J. Chandrasekhar, J. Mol. Model., 2000, 6, 226 CrossRef CAS.
- G. P. Bartholomew and G. C. Bazan, Acc. Chem. Res., 2001, 34, 30 CrossRef CAS.
- R. A. Pascal, M. L. Carter, M. R. Johnson and D. M. Ho, Tetrahedron Lett., 1996, 37, 8125 CrossRef.
- R. A. Pascal, R. B. Grossman and D. Vanengen, J. Am. Chem. Soc., 1987, 109, 6878 CrossRef CAS.
- M. W. Hosseini and J. M. Lehn, Helv. Chim. Acta, 1988, 71, 749 CrossRef CAS.
- F. Arnaud-Neu, S. Fuangswasdi, B. Maubert, J. Nelson and V. McKee, Inorg. Chem., 2000, 39, 573 CrossRef CAS.
- P. B. Smith, J. L. Dye, J. Cheney and J. M. Lehn, J. Am. Chem. Soc., 1981, 103, 6044 CrossRef CAS.
- S. M. Dimick, S. C. Powell, S. A. McMahon, D. N. Moothoo, J. H. Naismith and E. J. Toone, J. Am. Chem. Soc., 1999, 121, 10286 CrossRef CAS.
- D. Chen, R. J. Motekaitis, I. Murase and A. E. Martell, Tetrahedron, 1995, 51, 77 CrossRef CAS.
- R. Hooft, Collect, Nonius, Delft, The Netherlands, 1998 Search PubMed.
- Z. Otwinowski and W. Minor, in Methods in Enzymology, eds. C. W. Carter and R. M. Sweet, Academic Press, London, 1997, vol. 276, pp. 307–326 Search PubMed.
- G. M. Sheldrick, SHELXS-97, Program for solution of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXL-97, Program for refinement of crystal structures, University of Göttingen, Germany, 1997 Search PubMed.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189 Search PubMed.
Footnote |
| † CCDC reference numbers 253845 and 253846. See http://www.rsc.org/suppdata/nj/b4/b415532g/ for crystallographic data in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2005 |