Zirconate complexes: multifaceted reagents
Jean-Pierre
Majoral
*a and
Maria
Zablocka
*b
aLaboratoire de Chimie de Coordination, CNRS, 205 route de Narbonne, 31077, Toulouse cedex 04, France. E-mail: majoral@lcc-toulouse.fr; Fax: +33 (0)5 61 55 30 03; Tel: +33 (0)5 61 33 31 23
bC.M.M.S, Polish Academy of Sciences, Sienkiewicza 112, 90363, Lodz, Poland. E-mail: zabloc@bilbo.cbmm.lodz.pl; Fax: +48 42 684-7126; Tel: +48 42 681-8952
First published on 16th December 2004
Abstract
Zirconium compounds play an important role in a large number of synthetic methods leading to simple or complex molecules for which straightforward preparations are not well-defined or appeared difficult to handle. Such a versatile behaviour of zirconium reagents can be explained by the fact that they can exhibit a metal centre with 14 to 18 electrons and a valence coordination number from 3 to 5. Among organozirconium species, zirconate complexes, not extensively studied up to now, seem very attractive species to deal with since convenient methods of preparation of these complexes are now available. The design, preparation, characterization and properties of these compounds are presented and discussed in this review.
Introduction
Over the past 20 years, interest in organozirconium chemistry has been rapidly increasing and a tremendous number of applications in synthetic chemistry starting from zirconocene complexes have been found.1 Recent contributions were highlighted in a special volume2 demonstrating the usefulness of such complexes for the development of modern synthetic methodologies.Special attention has been paid to the preparation and the properties of group 4 d° bis(cyclopentadienyl) complexes Cp2ZrXn (A, n = 1; B, n = 2; C, n = 3) with Zr–X bonds. They can exhibit metal centres with 14 to 18 electrons and valence coordination numbers from 3 to 5 (Fig. 1). Bent cationic bis(cyclopentadienyl) complexes A have received considerable attention as they have been identified as the active catalytic species in the homogeneous metallocene Ziegler–Natta process for the polymerization of olefins.3 The chemistry of B complexes has been also extensively explored, these compounds being used as reagents for the preparation of various new organic and organometallic compounds.
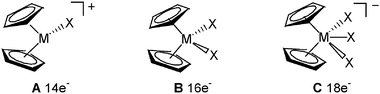 | ||
| Fig. 1 Group 4 d° metallocene complexes Cp2ZrXn (A, n = 1, B, n = 2; C, n = 3). | ||
An examination of the known chemistry of anionic C metallocenes shows that these five-coordinated derivatives are postulated as intermediates in a number of stoichiometric and catalytic reactions. However, only a very few of these zirconates C have been spectroscopically and structurally characterized. Such realistic assumptions concerning the transient generation of complexes of type C are, of course, very important because they offer plausible mechanistic explanations concerning the preparation of various organic derivatives, as it was demonstrated in a large number of papers. Despite this, there is always a need to have “in bottle” stable reagents, fully characterized, for use in a more comfortable way as starting materials for a diversified chemistry.
In order to take further steps towards these goals it is mandatory to have a rational view of the general principles governing the formation and the stabilization of such anionic metallocene derivatives C since no attempt has been made to provide a picture of the scope and limitations, characterization and properties of zirconate complexes.
It is not the purpose of this paper to review all the investigations done on this topic. Through selected examples we wish to show how it is possible to generate transient “ate” complexes or to form stable “ate” or zwitterionic “ate” complexes, thus providing the readers with tools for future developments in the field of the use of zirconium reagents in organic and heteroatomic chemistry, as well as in organometallic chemistry.
Anionic zirconocene complexes Cp2Zr(R)(R1)(R2).
As previously mentioned such zirconate complexes were found as key intermediates in a number of reactions.In 1989, Negishi and colleagues reported, in a pioneering work, the insertion of α-haloorganolithium reagents into acyclic zirconocene chlorides.4 Such a methodology is particularly useful as most organozirconocene complexes are electronically unsaturated (16 electrons), therefore the reaction may occur by formation and 1,2 -rearrangement of an 18-electron ate complex 1 (Scheme 1). The transient formation of an anionic zirconocene complex 2 was evoked to explain the “pair” selective and regioselective carbon–carbon bond forming reaction of zirconacyclopentane derivatives with Grignard reagents.5 It was shown that the Zr-catalyzed carbomagnesation of alkenes likely proceeds with formation of zirconacyclopentanes (Scheme 2). The anionic cyclic complex 3 was also implicated as an intermediate in similar reactions and in hydrogenation reactions.6,7
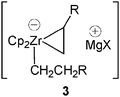
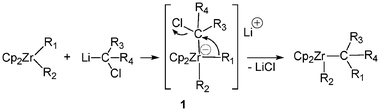 | ||
| Scheme 1 Transient formation of the 18-electron “ate” complex 1. | ||
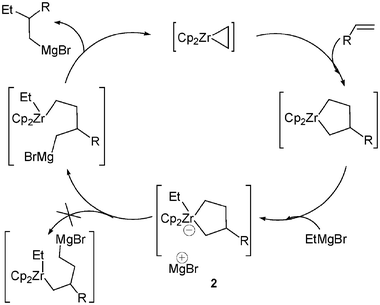 | ||
| Scheme 2 Zr-catalyzed carbomagnesation of alkenes. | ||
Whitby et al. proposed the generation, as intermediates, of zirconates 4 and 5 to explain the formation of complexes 6 and 7 arising in a first step from coordination of a lithiated reagent on zirconium (Scheme 3).8,9 This group has developed the insertion of a wide range of carbenoïds into organozirconocene chlorides derived by hydrozirconation to provide useful multi-component coupling methods.10,11 Ring expansion of zirconacyclo-pentanes and pentenes to afford 6-membered zirconacycles by insertion of a range of carbenoïds was also extensively reported.2,9
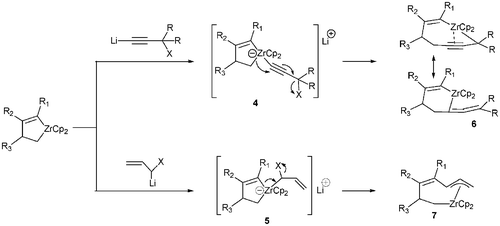 | ||
| Scheme 3 Transient generation of zirconocene-ate complexes 4 and 5. | ||
The zirconocene-promoted carbon-carbon bond formation via a 1,2-migration reaction of alkynylzirconium derivatives was proposed using the zirconate complex Li[Cp2Zr(C≡C–Ph)3] 8 (Scheme 4).12 Complex 8 was prepared by treatment of Cp2ZrCl2 with 3 equiv. of Li–C≡C–Ph in THF at −78 °C. Reaction of 9 with HCl produced (Z)-1,4-diphenyl-1-buten-3-yne, 10a, as a >96% isomerically pure compound. Similarly, the reaction of Cp2ZrCl2 with 3 equiv. of Li–C≡C–C(Me)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 followed by iodinolysis produced 10b in almost quantitatively yield. Zirconocene derivatives containing both alkynyl and aryl groups also undergo a similar reaction. Pre-formed Cp2Zr(C≡C–Ph)2 reacted with PhLi, providing upon quenching with HCl, (Z)-stilbene. Contrary to the previous zirconate complexes reported above, 8 was stable enough to be fully characterized by NMR. However, it appears that in the very large majority of cases the anionic zirconocene complexes are not stable enough to be characterized.
CH2 followed by iodinolysis produced 10b in almost quantitatively yield. Zirconocene derivatives containing both alkynyl and aryl groups also undergo a similar reaction. Pre-formed Cp2Zr(C≡C–Ph)2 reacted with PhLi, providing upon quenching with HCl, (Z)-stilbene. Contrary to the previous zirconate complexes reported above, 8 was stable enough to be fully characterized by NMR. However, it appears that in the very large majority of cases the anionic zirconocene complexes are not stable enough to be characterized.
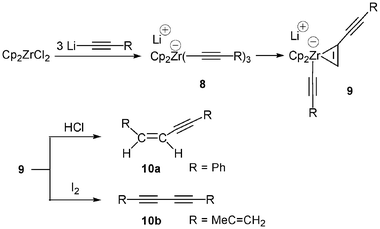 | ||
| Scheme 4 Carbon-carbon bond formation promoted by zirconate complexes. | ||
A remarkable exception to this behaviour was furnished by Stephan et al. who proposed synthetic pathways to several salts of the anion [Cp*2ZrH3]−X+, 11.13 It should be noted that early metal-based hydride anions analogous to the main group hydrides were unknown until this work.
Reactions of Cp*2ZrH2, prepared from [Cp*2Zr(N2)]2(μ-N2), with KH in THF afforded 11a (X = K). In a similar manner, addition of LiH gave [Cp*2ZrH3]Li, 11b. Another preparation involving the reaction of Cp*2ZrCl2 with 3 equiv. of n-BuLi under H2 affords [Cp*2ZrH3]Li·0.5LiCl–THF, 12. Alternatively, reaction of Cp*2ZrCl2 with LiAlH4 gave the species Cp*2ZrH(μ2-H2AlH2), which reacts with n-BuLi to give 11b in high yield (Scheme 5). Crystallographic studies of 11b and 12 confirm cation-anion pairing. Further NMR data affirm the formulation of the anion as a classical Zr(IV) trihydride formulation.
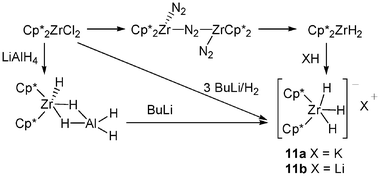 | ||
| Scheme 5 Synthesis of the anion Cp*2ZrH3−. | ||
The catalytic dehydro-oligomerization of 1,2-C6H4(PH2)2 with [Cp*2ZrH3][K(THF)2] results in deposition of the octamer (1,2-C6H4P2)8, 13, an unprecedented 16-membered ring of contiguous P atoms.14 In an effort to understand the process of formation of the macrocycle 13, the dimeric organophosphane species [C6H4(PH2)2], 14, was isolated, suggesting that 14 subsequently undergoes dehydro coupling to afford 13.
Zwitterionic (anionic zirconocene) complexes
Gell and Schwartz, in pioneering work, postulated the transient formation of a acyclic zwitterionic (phosphonium anionic zirconocene) complex during the synthesis of zirconium(IV) ylide hydride species as shown in Scheme 6.15 The strong nucleophile Me3PCH2 attacks the unsaturated zirconium(IV) alkyl hydride 15, producing an unstable 18-electron Zr(IV) species 16, which collapses to a Zr(II) intermediate 17 by reductive elimination of methylcyclohexane.15
A clean methylene-transfer reaction was observed when hydridozirconocene chloride 18 (Schwartz reagent) was treated with Ph3PCH2, leading to Cp2Zr(CH3)Cl and P(Ph)3via a 1,2-hydrogen migration from zirconium to carbon with transient generation of the zwitterion 1916 (Scheme 7). Later on the transient formation of another zwitterionic complex 20 was proposed in order to explain the formation of metallacyclic four-membered ring systems17 (Scheme 8).
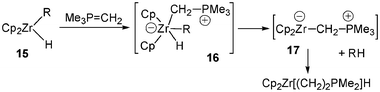 | ||
| Scheme 6 Synthesis of zirconium(IV) ylide hydride via generation of acyclic zwitterionic (phosphonium anionic zirconocene) complexes. | ||
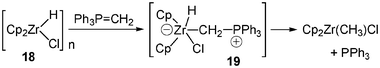 | ||
| Scheme 7 Methylene transfer reaction involving transient zwitterion 19. | ||
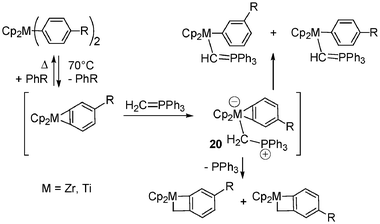 | ||
| Scheme 8 Formation of metallacyclic four-membered ring systems. | ||
However, even if the claims concerning the generation of highly unstable phosphonium anionic zirconocene complexes appeared to be reasonable, no real proof was furnished until the mid-nineties concerning the existence of such complexes.
Indeed, in 1996 two reactions allowed to isolate the fully characterized zirconated phosphonio dithiolate complexes 2218 (Scheme 9). The first one consists in the treatment of the adduct R3PCS221 (R = Bu, Me), obtained from carbon disulfide and the corresponding phosphine R3P, with the Schwartz reagent 18. Alternatively, carbon disulfide can be added first to the Schwartz reagent, the resulting mixture being treated with R3P. The complex 22a was isolated in high yield as a yellowish crystalline product. The nature of the substituents on phosphorus strongly affects the stability of these complexes: 22a decomposes in solution over 1 day but is stable as a powder for several weeks at −25 °C, while 22b has to be used as generated in situ. It should be noted that no 1,2-hydrogen shift, which would have resulted in the formation of complexes 23a or 23b, has been detected.
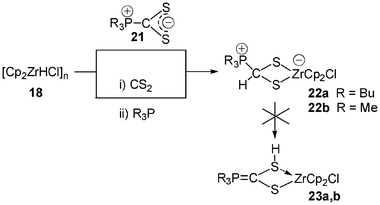 | ||
| Scheme 9 Synthesis of phosphonio dithiolate zirconate complexes 22. | ||
Complexes 22 present a diversified reactivity. 22a easily reacts with methyl iodide and benzyl bromide with formation in high yield of S,S′-dialkylated phosphonium salts 24a,b. Similarly, addition of 2 equiv. of acetylchloride to 22a affords the bisthioacylated phosphonium salt 25, while treatment of 22a with only 1 equiv. of acetylchloride gives rise to the other new methylene phosphonium salt 26 (Scheme 10).
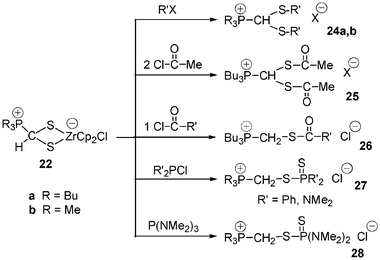 | ||
| Scheme 10 Reactivity of complexes 22. | ||
Remarkably, the reaction of 22a or 22b with chlorophosphines or aminophosphines led to the unprecedented methylene thiophosphorylated phosphonium salts 27 and 28. The mechanism of this unusual reaction has not been unequivocally established but can be regarded as a three-step process: (i) nucleophilic substitution at sulfur in 22a or 22b, (ii) then sulfurization on the tricoordinated phosphorus atom and (iii) hydrogen transfer in order to form the PCH2SR sequence.
A few other zwitterionic complexes bearing an anionic zirconocene moiety and a nitrogen cationic part were also reported. In an elegant work Whitby et al. described the synthesis of the triazenidoalkylzirconocene 29 (Scheme 11),19 which was found to be stable to air and water, surviving in aqueous workup intact and no change being observed after exposure of the crystals of 29 to light and air for 2 weeks. An explanation for this nonreactivity is that a bidendate coordination mode of the triazenido moiety gives an electronically saturated (18 electrons) zirconium centre.19 An X-ray structure determination of 29 confirmed the bidendate nature of the triazo ligand-metal interaction.
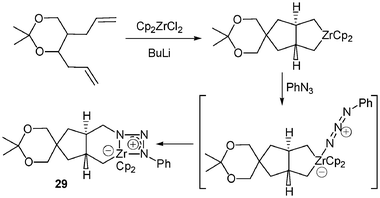 | ||
| Scheme 11 Synthesis of triazenidoalkylzirconocene 29. | ||
[3 + 2] cycloaddition reactions
X-Ray structure determinations of various zwitterionic (phosphonium anionic zirconocene) complexes appeared a little later, these unprecedented zwitterions being prepared via formal [3 + 2] cycloaddition reactions. Treatment of benzynezirconocene 30, generated by heating Cp2ZrPh2 in refluxing toluene, with alkynylphosphines 31 led to 2-phosphinozirconaindenes 32. Addition of methyl propiolate, for example, to 32 in toluene at room temperature afforded the complex 34 fully characterized by X-ray diffraction studies20 (Scheme 12). It was reasonably postulated that the first step of the reaction is the nucleophilic attack of phosphorus(III) on the unsubstituted acetylenic carbon with formation of the corresponding transient zwitterion 33. In the second step an intramolecular cyclization of the carbanionic centre of the betaïne species 33 on the zirconium metal fragment occurs to form the stable zirconate product 34.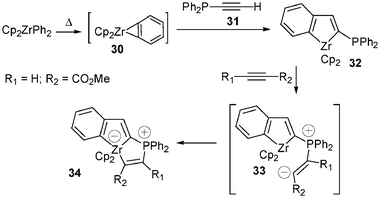 | ||
| Scheme 12 Synthesis of the zwitterion 34via treatment of the zirconaindene complex 32 with methyl propiolate. | ||
No reaction occurred on 32 with nonactivated terminal acetylenic systems such as tert-butyl, phenyl, and trimethylacetylene. Thus, zwitterionic organozirconocene-ate complexes are obtained with 32 when functional groups linked to the acetylenic moiety provide sufficient activation by conjugation with the carbon-carbon triple bond.
To expand the scope of the formation of anionic organozirconocene-ate complexes obtained from acetylenic compounds, investigations concerning the reaction of 32 with heteroacetylene reagents were performed. It is known through calculations and experimental studies that the regioselectivity of nucleophilic additions of tertiary phosphines on terminal heteroacetylene derivatives depends on the nature of the heteroatom directly bonded to the unsaturated system (Chart 1). The regioselective formation of 36 is in agreement with the nucleophilic attack of the phosphine moiety in 32 on the acetylenic carbon atom linked to the methoxy group to form the intermediate 35, which gives, after cyclization, complex 35’ (Scheme 13). The reaction of 32 with an acetylenic system directly substituted with a second-row element such as the phosphorus atom proceeds similarly but in much more drastic conditions (110 °C, 16 h, instead of room temperature, 3 h).21
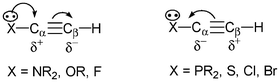 | ||
| Chart 1 | ||
![Synthesis of zwitterion 36via a [3 + 2] cycloaddition reaction.](/image/article/2005/NJ/b412439c/b412439c-s13.gif) | ||
| Scheme 13 Synthesis of zwitterion 36via a [3 + 2] cycloaddition reaction. | ||
Coupling reactions involving zirconocene complexes and propargylic systems lead generally to mixtures of products with low yields. In marked contrast, addition of heterosubstituted propargyl derivatives 36–38 to 32 gave regioselectively and quantitatively the anionic zirconocene complexes 39–41 (Scheme 14).
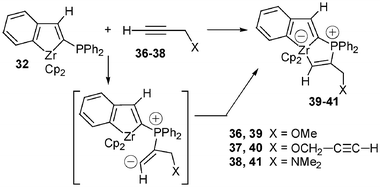 | ||
| Scheme 14 Reaction of propargylic systems with the zirconaindene 32. | ||
To summarize, [3 + 2] cycloadditions involving 32 and acetylenic systems lead to a large variety of stable anionic five-coordinate zirconocene-ate complexes. Nucleophilic reaction of the phosphine moiety 32 follows the proposed rule that a heteroelement in conjugation with the triple bond of a terminal acetylene orientates nucleophilic attack on Cα for first-row elements and on Cβ for elements of the second period (Chart 1).
To end up with such [3 + 2] cycloadditions using acetylenic systems it must be emphasized that the one-pot reaction of 1 equiv. of 30 with 2 equiv. of the acetylenic phosphine Ph2P–C≡C–H leads directly to the zwitterionic (phosphonium anionic zirconocene) complex 34a (Scheme 15).
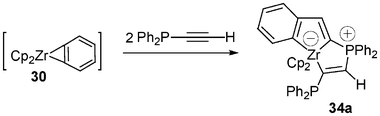 | ||
| Scheme 15 One-pot formation of the zwitterionic complex 34a. | ||
Such formal [3 + 2] cycloadditions reactions can be performed also with various aldehydes 42a–f22 (Scheme 16). Reactions are conducted in toluene for 1 h at either −78 °C or room temperature, depending on the aldehyde, and afforded the desired stable zwitterionic 18-electron d° anionic zirconocenes 43a–f in 53–73% yield after workup. It can be emphasized that these cycloadditions occur selectively on the carbonyl group, allowing therefore the preparation of diversely functionalized zwitterionic zirconocene complexes possessing either a free phosphino group (43b) or free alkenyl group (43c–e). NMR data of these complexes fit well with the proposed structures; one of them (43e) was fully identified by X-ray crystallographic studies, which confirm the formation of the suggested cycloadduct. Interestingly, the nucleophilic attack of the phosphine unit in 32 on Ph–C≡C–C(O)H took place selectively at the carbon atom of the aldehyde function to give quantitatively the zwitterion 44 after cyclization (Scheme 17).21
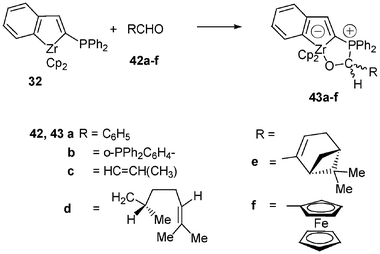 | ||
| Scheme 16 Reactivity of the zirconaindene 32 with aldehydes. | ||
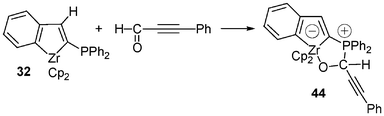 | ||
| Scheme 17 Reactivity of the zirconaindene 32 with Ph–C≡C–CHO. | ||
The ability of 32 to react with polyaldehydes was investigated: the same strategy was used to decorate the surface of a dendrimer of generation 4 (48 terminal aldehyde groups) and the surface of a dendrimer of generation 8 (768 aldehyde end groups) with respectively 48 and 768 anionic zirconocene moieties. These first zwitterionic metalladendrimers 46-G44 and 48-G88 with anionic early transition metal units grafted on the periphery were isolated in 70 and 76% yield, respectively (Scheme 18).
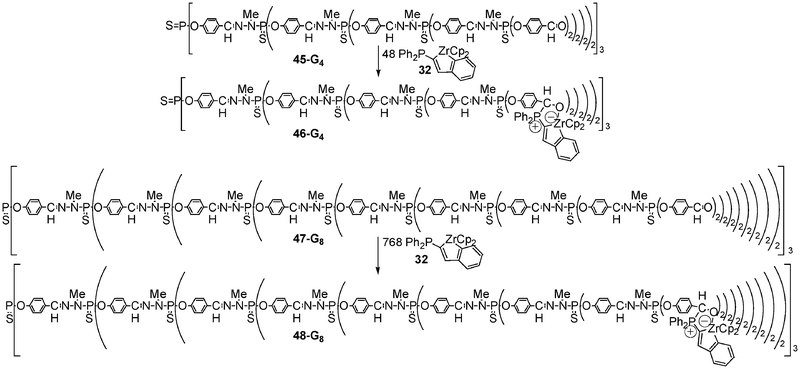 | ||
| Scheme 18 Synthesis of phosphorus dendrimers decorated with zwitterionic zirconate complexes. | ||
Remarkably, a controlled number of anionic zirconocene units can be selectively introduced into the internal layers of a polydendritic macromolecule. Indeed, treatment of the multidendritic system 49-G33-G22 constituted by a central dendrimer of generation 3 and by six internal dendrimers of generation 2 possessing 24 internal aldehyde groups, with 32 (excess) clearly leads to the polyzwitterionic zirconocene polydendritic structure 50-G33-G22 (88% yield),22 in which all the early transition metal anions are located within the cascade structure (Scheme 19).
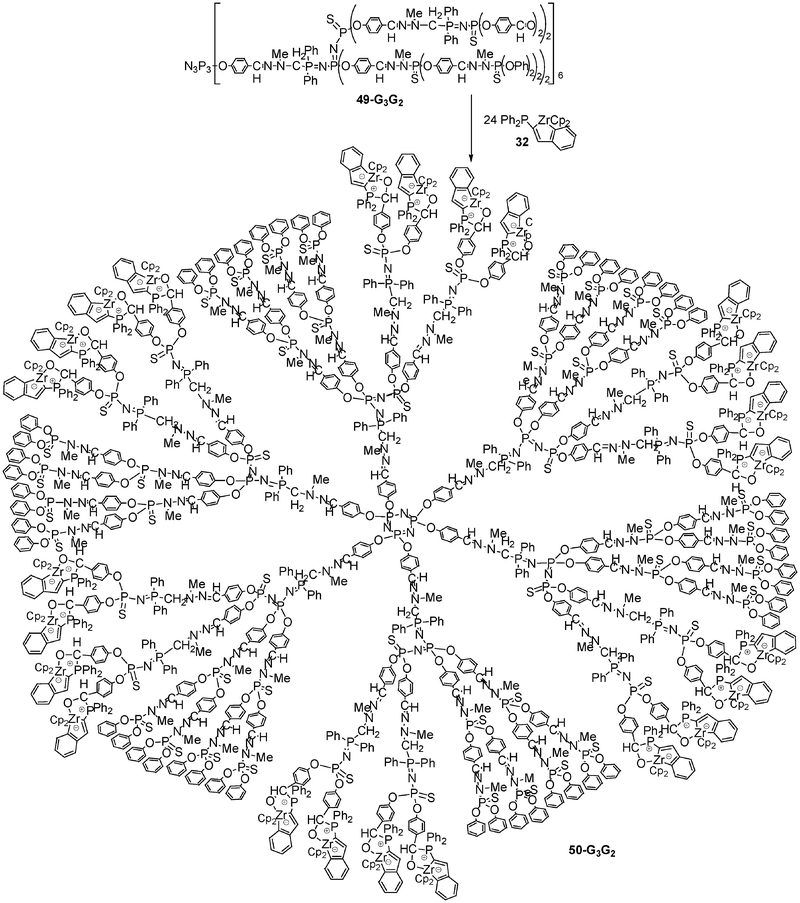 | ||
| Scheme 19 Incorporation of zwitterionic zirconate complexes within a multidendritic macromolecule. | ||
Such chemio- and regioselective cycloadditions were extended to the formation of complexes bearing zirconium–sulfur or zirconium–nitrogen bonds (Scheme 20).23 Indeed, the metallocene species 51–55 were isolated in good to excellent yields (53%–84%) by treatment of 32 with an equimolecular amount of the corresponding heterocumulene: CO2, CS2, Cy–NCN–Cy, R–NCS, R–NCO. Several X-ray diffraction studies were carried out to confirm the proposed zwitterionic structures. With isocyanates and isothiocyanates, the anionic charge is delocalized over the carbamoyl skeleton in I, but the preferred coordination site in these systems is the nitrogen atom. The η1-bonding mode of the carbamoyl group in complexes 54 and 55 is very strong since no competition between nitrogen and oxygen or between nitrogen and sulfur was observed either in the solid state or in solution.
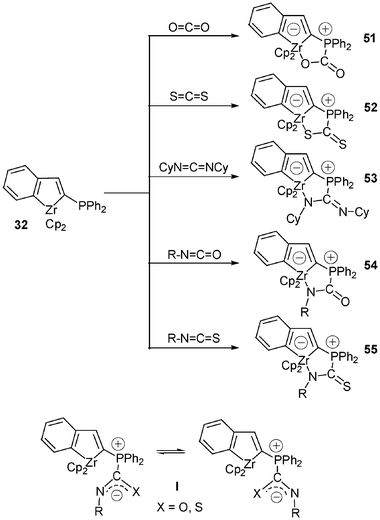 | ||
| Scheme 20 Formation of zwitterionic zirconate complexes bearing Zr–O, Zr–S or Zr–N bonds. | ||
Such a methodology leading to a large number of stable and well-characterized zwitterionic 18-electron d° anionic metallocene complexes can be illustrated with the reactivity of other 16-electron metallocene species, such as 56. Addition of phenyl isothiocyanate to 56 in toluene at −40 °C leads to the expected complex 57 isolated in 91% yield after workup. Surprisingly, the same reaction performed with 56 (1 equiv.) and carbon disulfide (0.5 equiv.) affords the unique “dimeric” species 58 arising from successive (or concomitant) cycloaddition reactions on the two carbon–sulfur double bonds of CS2. Under stoichiometric conditions, only half of the neutral complex 56 is transformed into the bis(zwitterionic) complex 58 (Scheme 21).23
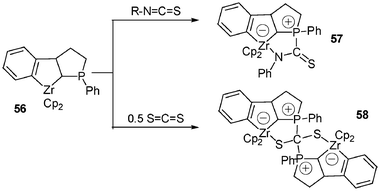 | ||
| Scheme 21 Reactivity of tricyclic zirconocene complexes with R–N=CS and CS2. | ||
[3 + 1] cycloaddition reactions
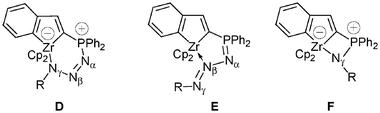
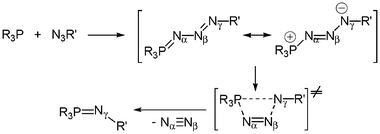
The chemistry of iminophosphorane compounds of general structure R3PN–R′, which incorporate a four-coordinate phosphorus and a formal double bond between the phosphorus and the nitrogen, is very well documented. Iminophosphoranes are employed in a number of useful reactions in organic chemistry, such as Aza-Wittig reactions24 or as very strong neutral bases.25 The Staudinger reaction of azides with tertiary phosphines is one of the two major routes in the preparation of iminophosphoranes.26 Such a reaction proceeds by nucleophilic attack of the phosphine on the terminal α-nitrogen atom of the azide to afford a linear phosphazide, rarely stable,27 which then dissociates to the iminophosphorane with elimination of dinitrogen (Scheme 22). Iminophosphoranes form complexes with a variety of metals by N-imino complexation (covalent or dative bonds).24 In marked contrast, only a few phosphazide complexes have been prepared.28 The unique seven-coordinate complex [WBr2(CO)3(Ar–NN–NPPh3)], 59a, was characterized by X-ray diffraction studies: the phosphazide ligand is bound to the tungsten metal fragment in a bidentate fashion through the α- and γ-nitrogen atoms.28b More recently, the cyclic (Z)-phosphazide 60 was found to act as a monodentate two-electron donor through the less sterically hindered β-nitrogen atom.28c Since in all the previous reactions involving [3 + 2] cycloaddition reactions and leading to zwitterionic phosphonium anionic zirconocene complexes the first step consists of a nucleophilic attack of the phosphine moiety, it was of interest to see if such a reaction can be observed with various azides and, for example, complexes 32 and 56. One can expect that a Staudinger reaction will take place with the formation of a transient phosphazide or the formation of the more stable iminophosphorane. The trapping of phosphazide through complexation of the γ-nitrogen atom to the zirconium centre may be anticipated because of the well-established zwitterionic character of the PN3R fragment (P+–NαNβ–N−γ–R)27 with formation of the zirconate species D. Complexation of phosphazide through the less-hindered nitrogen atom Nβ, which would lead to complexes of type E can be also envisaged. Otherwise, chelation of the iminophosphorane would lead to complexes of type F, whereby the zirconium counterpart plays the role of a Lewis acid in each case. Lastly, insertion of the azide into a Zr–C bond cannot be totally ruled out.29
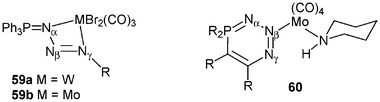
 | ||
| Scheme 22 Mechanism of the Staudinger reaction between phosphines and azides. | ||
Indeed, an unprecedented chelation of phosphazides, which involves a formal [3 + 1] cycloaddition that occurs exclusively with the α-nitrogen atom of the phosphazide moiety, was found with formation of a variety of new stable, polycyclic zwitterionic complexes.30 This was first observed when the α-phosphinozirconaindene 32 was reacted with a 4-fluoro-3-nitrophenyl azide (Scheme 23). No evolution of dinitrogen was detected and the structure of the resulting complex 61 was established by X-ray diffraction studies. Therefore, in marked contrast to the situation encountered for compounds 59 and 60, intramolecular donor-acceptor interactions only occur with the α-nitrogen atom, which suggests that this nitrogen atom is a better donor than Nγ and that the polarization of the phosphazide moiety is more correctly represented as –P+–N−–NN–R rather than P+–NN–N−–R, at least for the structure reported above.
![Synthesis of zwitterionic complexes 61 and 62–64 by [3 + 1] cycloaddition.](/image/article/2005/NJ/b412439c/b412439c-s23.gif) | ||
| Scheme 23 Synthesis of zwitterionic complexes 61 and 62–64 by [3 + 1] cycloaddition. | ||
The zwitterionic zirconocene-ate complex 61 is stable at room temperature and dinitrogen is only liberated on heating under reflux in toluene for 2 h to give rise to the new complex 62 (Scheme 23). This evolution of molecular nitrogen may imply a dissociation of the Zr–Nα bond. Two possible pathways to rationalize the formation of 62 may be proposed (Scheme 24). The first path involves the mechanism demonstrated for the Staudinger reaction with transient formation of a four-centred transition state, coordination of the γ-nitrogen atom to phosphorus, and elimination of the α- and β-nitrogen atoms (path a) to give compound 62. However, the transient formation of a six-membered ring (path b) cannot be totally ruled out. The X-ray structure analysis of 62 shows that the nitrogen atom is connected to zirconium [d(Zr–N) = 2.426(2) Å] and the phosphorus–nitrogen bond length [1.620 (2) Å] is in the normal range for such a bond. No structural change is observed for this derivative in comparison with 32; the fused tricyclic system is still planar (maximum deviation 0.062 Å).
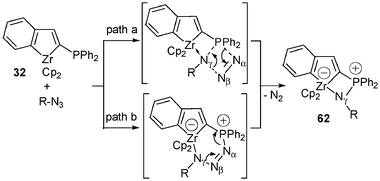 | ||
| Scheme 24 Proposed mechanism for the formation of complex 62. | ||
Interestingly, the reaction of 32 with the azide N3P(O)(OPh)2 in toluene at room temperature for 1 h gave the zwitterionic derivative 63 directly (Scheme 23); the transient formation of a phosphazide complex was not detected in this case. A similar result was observed when a toluene solution of 32 was treated for 1 h with trimethylsilylazide: the complex 64 was the only product formed and was isolated in 61% yield (Scheme 23). However, this reaction did not proceed at room temperature; it was necessary to reflux for 1 h in order for the reaction to go to completion.
Such a formal [3 + 1] cycloaddition involving azides and α-zirconated phosphines can be extended to tricyclic systems such as 56 (Scheme 25). Indeed, when a solution of 56 and N3P(O)(OPh2)2 in toluene was stirred at room temperature for 1 h, in contrast to the reaction that led to 63, the formation of a transient phosphazide complex 65 was detected by 31P NMR spectroscopy.
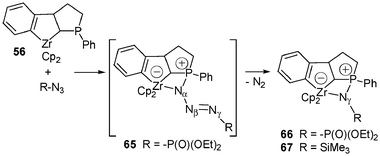 | ||
| Scheme 25 Synthesis of zwitterionic zirconaphosphazide complexes 66 and 67. | ||
Lastly, as in the reaction of trimethylsilylazide with 32, the iminophosphorane complex 67 was directly formed when the tricyclic system 56 was treated with trimethylsilylazide at room temperature. Remarkably, only one isomer of each complex 66 and 67 was formed, as indicated by NMR spectroscopy.
Therefore, it appears that the lifetime of transient phosphazide complexes is greatly dependent on the nature of both the starting α-zirconated phosphine and the azide used. As previously shown,24 phosphazides are thermodynamically stabilized by the presence of electron-withdrawing groups on nitrogen and of electron-donating groups on phosphorus. This was also demonstrated in this work. Concomitant [3 + 1] and [3 + 2] cycloaddition reactions were observed when α-phosphinozirconaindene 32 was treated with difunctional reagents such as 4-azidotetrafluorobenzaldehyde.30 The bisphosphonium species 68 was isolated and fully characterized when the reaction is performed at room temperature for 30 min. When 68 was heated in refluxing toluene, dinitrogen was liberated and adduct 69 was formed (Scheme 26). A similar reactivity was found when compound 32 was treated with 4-azidophenyl isocyanate at room temperature: the stable polycyclic bis(zwitterionic) adduct 70 containing four- and five-membered rings with Zr–C–P–N and Zr–N–C–P–C backbones is formed in near quantitative yield. As for all the reactions reported above, the phosphazide complex can be easily transformed into an iminophosphorane-like complex 71 by heating in refluxing toluene (Scheme 27).
![The [3 + 1] and [3 + 2] cycloadditions between 4-azidotetrafluorobenzaldehyde and 32.](/image/article/2005/NJ/b412439c/b412439c-s26.gif) | ||
| Scheme 26 The [3 + 1] and [3 + 2] cycloadditions between 4-azidotetrafluorobenzaldehyde and 32. | ||
![The [3 + 1] and [3 + 2] cycloadditions between 4-azidophenyl isothiocyanate and 32.](/image/article/2005/NJ/b412439c/b412439c-s27.gif) | ||
| Scheme 27 The [3 + 1] and [3 + 2] cycloadditions between 4-azidophenyl isothiocyanate and 32. | ||
Analogous reactions conducted with the tricyclic system 56 instead of 32 and with 4-azidotetrafluorobenzaldehyde or with 4-azidophenyl isocyanate allowed the formation of the bis(zwitterionic) complexes 72 and 73 (Scheme 28). Contrary to what was seen with the other phosphazide complexes, 72 and 73 do not cleanly lose dinitrogen when refluxed in toluene; decomposition of these complexes occurs with formation of numerous unidentified derivatives.
![The [3 + 1] and [3 + 2] cycloadditions between 56 and 4-azidotetrafluorobenzaldehyde or 4-azidodiphenyl isothiocyanate.](/image/article/2005/NJ/b412439c/b412439c-s28.gif) | ||
| Scheme 28 The [3 + 1] and [3 + 2] cycloadditions between 56 and 4-azidotetrafluorobenzaldehyde or 4-azidodiphenyl isothiocyanate. | ||
Miscellaneous ways of synthesis of zwitterionic complexes
To finish up with the formation of zwitterionic (phosphonium anionic zirconocene) complexes, two other ways of formally obtaining these species can be emphasized: the first one involves the sulfuration of 32 leading to the complex 74. X-Ray structure analysis of 74 revealed an interaction between zirconium and sulfur: the P–S bond length was found to be slightly longer [2.005 (1) Å] than those generally observed for thiophosphoryl groups (1.93–1.95 Å). Indeed the P–S bond length lies in between those of single and double bonds, suggesting a zwitterionic character for 7431 (Scheme 29). The second method deals with an exchange reaction between the zircona complex 56 and trichlorophosphineimine 75, leading to a metallaazaspirophosphane 76 (Scheme 30). The solid state structure of 76 has been corroborated by a single-crystal X-ray diffraction study32 showing a slight zwitterionic character for this complex, the P–N bond length [1.615(2) Å] being in between that of single and double bonds. A similar reaction performed with the complex 77 and Cl3PN–t-Bu affords the zirconabisazaspirophosphane 78.
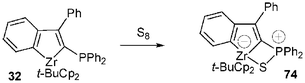 | ||
| Scheme 29 Synthesis of a zwitterionic complex via sulfuration of the phosphinozirconaindene 32. | ||
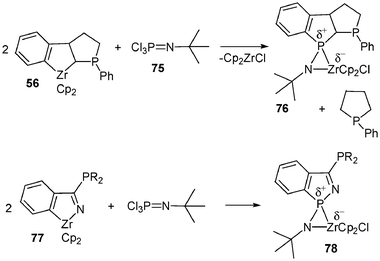 | ||
| Scheme 30 Synthesis of zirconabisazaspirophosphanes 76 and 78. | ||
If the synthesis of zwitterionic (phosphonium anionic zirconocene) complexes is now well-documented, that of the corresponding ammonium species is quite rare19 and it is only very recently that the preparation and X-ray characterization of a bis-sulfonium zirconocene-ate dimer was proposed.33 A toluene solution of diphenylphosphinoacetylene and a zirconocene thioaldehyde equivalent, Cp2Zr(Me)SCH2Ph, prepared from dimethylzirconocene and phenylmethanethiol, led to such a complex isolated as dimer 79. Remarkably, the cleavage of such a binuclear thiazirconacycle with borane afforded the monomeric thiazirconacyclic complexes 80, thus pointing out a weak association through bridging sulfur atoms in dimer 79 (Scheme 31).
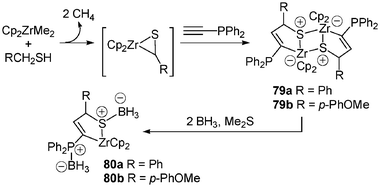 | ||
| Scheme 31 Preparation of bis-sulfonium zirconocene-ate dimer. | ||
Reactivity of zwitterionic (phosphonium anionic zirconocene) complexes
In marked contrast with the work done with unstable zirconate complexes such as those mentioned in the first part of this report, a few reactions were described dealing with the corresponding stable zwitterionic complexes. Addition of HCl to 34a leads to the cleavage of the three Zr–C bonds to give the divinyl phosphonium compound 81 as the major product. Addition of MeI to the same zwitterionic complex affords the bisphosphonium zirconate 82 (Scheme 32). Even in the presence of an excess of MeI no electrophilic cleavage of one of the three Zr–C bonds was observed.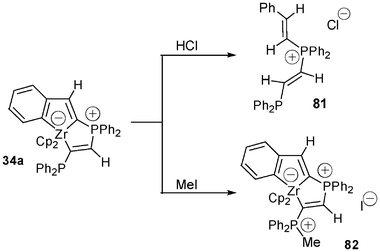 | ||
| Scheme 32 Reactivity of the anionic five-coordinate zirconocene complex 34a. | ||
Concluding remarks
Throughout this manuscript is reported either the transient generation of anionic zirconocene complexes or the isolation and full characterization of stable zwitterionic [phosphonium (sulfonium or “ammonium”) anionic zirconocene] complexes. Transient zirconate complexes were postulated in a number of reactions leading to the formation of linear, cyclic or polycyclic derivatives, but because of their “inherent” instability, it is often difficult to orientate the reactions that they are initiating. Indeed, their role and behaviour are often explained a posteriori. In marked contrast the corresponding zwitterionic species are remarkably stable and they can be easily manipulated, but the major drawback of these complexes is their lack of reactivity. However, the zirconated phosphino dithiolate complexes 22, owing to their Zr–S bonds, represent a good compromise between reactivity and stability since they easily offer the possibility to prepare a variety of phosphonium salts, for example, difficult or even impossible to prepare via classical methods. Similarly, bis-sulfonium zirconocene-ate dimers seems to be also versatile reagents.It is clear that studies concerning the design of new zirconates with controllable activity is highly desirable not only for creating new and efficient tools in organic and organometallic chemistry but also for expanding the number of useful applications that were already proposed with complexes of type A and B. As an example the fascinating potential of both zirconium on the one hand and phosphorus sulfur or nitrogen chemistries on the another hand will help chemists to further develop this stimulating and still lively field of challenging research.
References
- (a) For recent reviews, see: H. G. Alt and A. Köppl, Chem. Rev., 2000, 100, 1205 Search PubMed; (b) G. Erker, Acc. Chem. Res., 2001, 34, 309 CrossRef CAS; (c) E.-I. Negishi, Pure Appl. Chem., 2001, 73, 239 CrossRef CAS; (d) P. Wipf and C. Kendall, Chem.-Eur. J., 2002, 8, 1778 CrossRef.
- K. Suzuki and P. Wipf, Tetrahedron, 2004, 60, 1267 CrossRef CAS.
- (a) R. F. Jordan, J. Mol. Catal. A: Chem., 1998, 128, 1 CrossRef; (b) W. E. Piers, Chem.-Eur. J., 1998, 4, 13 CrossRef CAS and references therein.
- E.-I. Negishi, K. Akiyoshi, B. O’Connor, K. Takagi and G. Wu, J. Am. Chem. Soc., 1989, 111, 3089 CrossRef CAS.
- T. Takahashi, T. Seki, Y. Nitto, M. Sabura, C. J. Rousset and E.-I. Negishi, J. Am. Chem. Soc., 1991, 113, 6266 CrossRef CAS.
- (a) A. H. Hoveyda, J. P. Morken, A. F. Houri and Z. Hu, J. Am. Chem. Soc., 1992, 114, 6692 CrossRef CAS and references therein; (b) T. Takahashi, N. Suzuki, M. Kageyana, Y. Nitto, M. Saburi and E.-I. Negishi, Chem. Lett., 1991, 1579 CAS.
- (a) K. S. Knight and R. M. Waymouth, J. Am. Chem. Soc., 1991, 113, 6268 CrossRef CAS; (b) D. P. Lewis, P. M. Muller, R. J. Whitby and R. U. H. Jones, Tetrahedron Lett., 1991, 32, 6797 CrossRef CAS; (c) C. J. Rousset, E.-I. Negishi, N. Suzuki and T. Takahashi, Tetrahedron Lett., 1992, 33, 1965 CrossRef CAS.
- G. J. Gordon and R. J. Whitby, Chem. Commun., 1997, 1321 RSC.
- S. F. Fillery, G. J. Gordon, T. Luker and R. J. Whitby, Pure Appl. Chem., 1997, 69, 633 CrossRef CAS.
- (a) A. N. Kasatkin and R. J. Whitby, Tetrahedron Lett., 1997, 38, 4857 CrossRef CAS; (b) A. Kasatkin and R. J. Whitby, J. Am. Chem. Soc., 1999, 121, 7039 CrossRef CAS.
- (a) A. N. Kasatkin and R. J. Whitby, Tetrahedron Lett., 1999, 40, 9353 CrossRef CAS; (b) A. N. Kasatkin and R. J. Whitby, Tetrahedron Lett., 2000, 41, 6211 CrossRef CAS; (c) A. N. Kasatkin and R. J. Whitby, Tetrahedron Lett., 2000, 41, 6201 CrossRef CAS; (d) A. N. Kasatkin and R. J. Whitby, Tetrahedron, 2003, 59, 9857 CrossRef CAS.
- (a) K. Takagi, C. J. Rousset and E.-I. Negishi, J. Am. Chem. Soc., 1991, 113, 1440 CrossRef CAS; (b) Complex 9 has been characterized by X-ray diffraction: R. Choukroun, J. Zhao, C. Lorber, P. Cassoux and B. Donnadieu, Chem. Commun., 2000, 1511 Search PubMed.
- N. Etkin, A. J. Hoskin and D. W. Stephan, J. Am. Chem. Soc., 1997, 119, 11420 CrossRef CAS.
- N. Etkin, M. C. Fermin and D. W. Stephan, J. Am. Chem. Soc., 1997, 119, 2954 CrossRef CAS.
- K. I. Gell and J. Schwartz, Inorg. Chem., 1980, 19, 3206.
- G. Erker, P. Czisch, R. Mynott, Y. H. Tsay and C. Krüger, Organometallics, 1985, 4, 1310 CrossRef CAS.
- H. J. R. de Boer, O. S. Akkermann, F. Bickelhaupt, G. Erker, P. C. Czisch, R. Mynott, J. M. Wallis and C. Krüger, Angew. Chem., Int. Ed. Engl., 1986, 25, 639 CrossRef.
- A. Gudina, A. Igau, B. Donnadieu and J. P. Majoral, J. Org. Chem., 1996, 61, 9585 CrossRef.
- T. Luker, R. J. Whitby and M. Webster, J. Organomet. Chem., 1995, 492, 53 CrossRef CAS.
- Y. Miquel, A. Igau, B. Donnadieu, J. P. Majoral, N. Pirio and P. Meunier, J. Am. Chem. Soc., 1998, 120, 3504 CrossRef CAS.
- Y. Miquel, V. Cadierno, B. Donnadieu, A. Igau and J. P. Majoral, Organometallics, 2000, 19, 54 CrossRef CAS.
- V. Cadierno, A. Igau, B. Donnadieu, A. M. Caminade and J. P. Majoral, Organometallics, 1999, 18, 1580 CrossRef CAS.
- V. Cadierno, M. Zablocka, B. Donnadieu, A. Igau and J. P. Majoral, Organometallics, 1999, 18, 1882 CrossRef CAS.
- For a review, see: (a) A. W. Johnson, Ylides and Imines of Phosphorus, Wiley, New York, 1993 Search PubMed; (b) For recent examples, see: M. T. Reetz, E. Bohres and R. Goddard, Chem. Commun., 1998, 935 Search PubMed; (c) R. W. Reed, B. Santarsiero and R. G. Cavell, Inorg. Chem., 1996, 35, 4292 CrossRef CAS.
- See, for example: (a) R. Schwesinger and H. Schlemper, Angew. Chem., Int. Ed. Engl., 1987, 26, 1167 CrossRef; (b) R. Link and R. Schwesinger, Angew. Chem., Int. Ed. Engl., 1992, 31, 850 CrossRef and references therein.
- The second major route to the iminophosphoranes is the Kirsanov reaction; see ref. 24.
- For X-ray structures of phosphazides see: (a) M. Alajarin, P. Molina, A. Lopez-Lazaro, C. Foces-Foces and C. Fernandez-Castano, Tetrahedron, 1996, 52, 9629 CrossRef CAS; (b) J. R. Goerlich, M. Farkens, A. Fischer, P. G. Jones and R. Schmutzler, Z. Anorg. Allg. Chem., 1994, 620, 707 CAS; (c) A. N. Chernega, M. Y. Antipin, Y. T. Struchkov, M. P. Ponomarchuk, L. F. Kasukhin and V. P. Kukhar, Zh. Obshch. Khim., 1992, 62, 2675; (d) C. G. Ghidester, J. Smuszkovicz, D. J. Duchamp, L. G. Laurian and J. P. Freeman, Acta Crystallogr., Sect. C, 1988, 44, 1080 CrossRef; (e) A. N. Chernega, M. Y. Antipin, Y. T. Struchkov, I. E. Boldeskul, M. P. Ponomarchuk, L. F. Kasukhin and V. P. Kukhar, Zh. Obshch. Khim., 1984, 54, 1979 CAS.
- (a) G. L. Hilhouse and B. L. Haymore, J. Organomet. Chem., 1978, 162, C23 CrossRef CAS; (b) G. L. Hilhouse, G. V. Goeden and B. L. Haymore, Inorg. Chem., 1982, 21, 2064 CrossRef CAS; (c) K. Bieger, G. Bouhadir, R. Réau, F. Dahan and G. Bertrand, J. Am. Chem. Soc., 1996, 118, 1038 CrossRef CAS.
- (a) K. W. Chiu, G. Wilkinson, M. Thornton-Pett and M. Hursthouse, Polyhedron, 1984, 3, 79 CrossRef; (b) T. Luker, R. J. Whitby and M. Webster, J. Organomet. Chem., 1995, 492, 53 CrossRef CAS; (c) Recently, it was also demonstrated that azides add smoothly, and without loss of N2, to a polarized metal–metal bond in an early/late heterobinuclear complex, with formation of a bridging imido complex: T. A. Hanna, A. M. Baranger and R. G. Bergman, Angew. Chem., Int. Ed. Engl., 1996, 35, 653 Search PubMed.
- V. Cadierno, M. Zablocka, B. Donnadieu, A. Igau, J. P. Majoral and A. Skowronska, Chem.-Eur. J., 2000, 6, 346 CrossRef CAS.
- Y. Miquel, A. Igau, B. Donnadieu, J. P. Majoral, L. Dupuis, N. Pirio and P. Meunier, Chem. Commun., 1997, 279 RSC.
- K. Owsianik, M. Zablocka, B. Donnadieu and J. P. Majoral, Angew. Chem., Int. Ed., 2003, 42, 2176 CrossRef CAS.
- E. Ortega, N. Pirio, P. Meunier and B. Donnadieu, Chem. Commun., 2004, 678 RSC.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2005 |