Luminescent chemosensors: from molecules to nanoparticles
Luca
Prodi
Dipartimento di Chimica “G. Ciamician”, Università degli Studi di Bologna, Via Selmi 2, I-40126, Bologna, Italy. E-mail: luca.prodi@unibo.it; Fax: +39-051-2099456; Tel: +39-051-2099481
First published on 10th December 2004
Abstract
The need to develop sensors for different target analytes is well-recognised and confirmed by the considerable research efforts spent for the preparation of more and more efficient sensory devices. In our laboratories we have been working on this subject for the past few years and in this contribution some selected examples of the systems we have studied will be presented. Through the discussed compounds, the reader will be able to follow our step-by-step approach towards this fascinating research field. The first essential move for a fruitful strategy in the development of efficient macroscopic sensory devices is a thorough understanding of the systems responsible for the binding event and of its consequent transduction into a detectable signal. These species are often molecular or supramolecular structures that behave as molecular-level sensory devices, which are usually referred to as chemosensors. Among them, fluorescence-based chemosensors present many interesting features, such as high sensitivity and versatility. Starting from this first and fundamental step our aims and future perspectives will be presented. There is still a great demand for ever more efficient chemosensors; nanostructured systems such as nanoparticles, in which signal amplification can be obtained, will certainly play a very important role in this field. For this reason this contribution concludes with a section on the results obtained on nanoparticles, which are the natural carry-over of our research work.
Introduction
The need for chemical sensors
The development of chemical sensors is dramatically changing the potentialities of chemical analysis. Following the definition given by IUPAC, a chemical sensor is a device that transforms chemical information, ranging form the concentration of a specific sample component to total composition analysis, into an analytically useful signal.1 Classical methodologies require collection, transportation, eventual pretreating of the sample, and, in many cases, expensive instrumentation to be used only by trained staff. Chemical sensory devices have been conceived to bypass these restrictions since they are cheap and user-friendly analytical tools. In addition, if properly designed they allow to monitor in situ analyte concentrations in real time and, eventually, in real space.2–13 Such characteristics explain why chemical sensors have already found wide application in many fields, such as environmental monitoring, process control, food and beverage analysis, medical diagnosis, and, lately, in monitoring toxic gases and explosives for security reasons.2–13 It is evident that all these fields are of great importance from a social and economic point-of-view, and this makes the demand for the development of sensory devices even more urgent.The power of luminescence spectroscopy
Among the different chemical sensors, fluorescence-based ones present many advantages: fluorescence measurements are usually very sensitive (even single molecule detection is possible, although only under special conditions), of low cost, easily performed, and versatile, offering subnanometer spatial resolution with submicron visualisation and submillisecond temporal resolution.2,7,13 The versatility of fluorescence-based sensors originates also from the wide number of parameters that can be tuned in order to optimise the convenient signal. Even very complex analytical problems can be indeed overcome by controlling the excitation and emission wavelengths, the time window of signal collection, and the polarisation of the excitation beam or of the emitted light. In most cases luminescence intensity changes represent the most directly detectable response to target recognition; more recently, however, other properties such as excited state lifetime and fluorescence anisotropy have also been preferred as diagnostic parameters, since they are less affected by the environmental and experimental conditions.14When designing the structure of a luminescent chemosensor, one has to take into account some photophysical parameters in order to achieve the best sensitivity. First of all, the system should present, in order to obtain the highest luminescence signal, a very high molar absorption coefficient (ε) and a very high luminescence quantum yield (Φ). The luminescence intensity, in very diluted solutions, is in fact directly proportional to these quantities. In photoluminescence analysis, autofluorescence and light scattering are possible sources of relevant interferences, particularly when biological samples are involved. In order to increase the signal-to-noise ratio, it is then important to eliminate as much as possible these phenomena; this can be done using three distinct approaches. The first one is based on the development and use of red and near-infrared (NIR) dyes, which show absorption and luminescence bands in the 600–1100 nm region. These dyes offer minimal background as a result of reduced scattering (due to the inverse 4th power dependence on the wavelength) and of the absence of natural fluorescence of biomolecules in this spectral range. In addition, NIR systems are also very attractive because bright light sources such as diode lasers working in this spectral region are available at low cost. The possible drawback of this approach is that the fluorescence efficiency tends to decrease on diminishing the energy of the excited state.15 The second approach is based on the use of phosphorescent dyes with long lifetimes at room temperature. In this case, the background light is excluded by the use of time-resolved spectroscopy, since the scattered light and the fluorescence from natural fluorophores decay much faster than phosphorescence and can therefore be eliminated by delaying the measurement. Typical commonly used dyes are complexes of Eu(III) and Tb(III)16 and polypyridine complexes of Ru(II), Os(II) and Re(I).17 Finally, a large Stokes shift can also be of value, since it helps to reduce the interferences from the Rayleigh–Thyndall and Raman bands. Among the different strategies to obtain this feature there are the use of dyes whose lowest excited state is of charge transfer character, or the use of a combination of two different dyes in which efficient fluorescence resonance energy transfer (FRET) occurs.14
Furthermore, when designing a luminescent chemosensor, one can take great advantage of the variety of possible ways for modulating the photophysical properties of a dye. Typical examples are the introduction of proton-, energy- and electron-transfer processes, the presence of heavy atom effects, changes of electronic density, and the destabilisation of a non-emissive nπ* excited state. The knowledge of the rules governing these processes is of great importance to obtain an efficient signal transduction mechanism.
Sensory devices and chemosensors: the role of chemists
It is evident from the discussion above that the design of efficient chemical sensory devices requires a multidisciplinary approach: it is in fact essential that chemists, biologists, physicists, material scientists and engineers collaborate in order to obtain the best system for the desired application. The main role of chemists in this multidisciplinary team consists in the design and development of the interface between the device itself and the matrix to be analysed, that is the part that directly interacts with the analyte. This moiety is responsible for the affinity and selectivity of the whole device, and, in many cases, also of the transduction process, determining, as a consequence, the sensitivity of the system.A very fruitful approach recently followed by chemists for the design of new efficient chemical sensors is based on the principles of supramolecular chemistry.13,18–20 This bottom-up approach starts the design from the molecular level, thus ensuring a resolution that cannot be achieved with a top-down approach. The molecular or, more often, supramolecular species that carry out these functions are conventionally referred to as chemosensors, mostly to distinguish them from chemical sensors, that usually are intended as macroscopic devices.21–23 It is, however, important to note that chemosensors and probes also respond to the definition of chemical sensor given by IUPAC, if the concept of sensors is extended also to molecular-level devices.24
As already discussed, luminescent chemosensors can and do already find use in many disciplines.2–13 In biochemistry, clinical and medical sciences, and cell biology, freely mobile sensor molecules are employed extensively in microscopy, allowing to draw maps of the concentration of a given target analyte in a sample in real time.25 For this kind of application, chemosensors should be sufficiently soluble in water and offer suitable selectivities and affinities in this solvent.
For many other applications in analytical chemistry and environmental sciences, however, only the immobilisation of luminescent chemosensors to yield insoluble sensitive materials, and the consequent development of a sensory device, can allow continuous measurement of analyte concentrations. In this case, the water solubility of the chemosensor is not a fundamental requirement; rather, when it is not covalently linked to a suitable support, its solubility could be a problem since the chemosensor would be washed out while performing the analysis in flow. However, the system, when immobilized, should present the requested selectivities and affinities for the analytes in water solutions.
The characteristics of the supporting material and the methods of immobilisation are in this context crucial for the efficiency of the sensory devices, and they are again a special task for chemists,6,13,26 but their discussion is outside the scope of this paper.
The term selectivity deserves at this stage a final comment. The conventional approach to chemical sensing is related to the design of specific receptors able to efficiently bind the analyte of interest. This approach requires the synthesis of separate, highly selective chemosensors, one for each target compound, and therefore it is appropriate only when a few analytes have to be identified in matrices with controlled background and interferences. An alternative strategy that finds a wide range of practical applications is based on the use of sensor arrays, taking inspiration from the mammalian senses of taste and smell. In these kinds of artificial arrays, called electronic noses and tongues (if they are built to analyse gaseous and liquid matrices, respectively), each sensing element does not need to be selective towards a specific analyte. In this approach, the identification of target species cannot be derived from the response of a single element; rather, it is the pattern of responses produced over the whole collection of sensors that allows the identification of the matrix under analysis.12
This paper will focus on the research performed in our laboratory, with the intention of showing the approach lying behind our research activity and the future perspectives we are aiming to.
From molecules…
As outlined so far, the development of new chemosensors is the first essential step for the design of efficient sensors, and, as many others in this field, we started from the study of simple molecular and supramolecular systems.The first, large, series of chemosensors I would like to discuss here is the one obtained by linking together crown ether receptors and 8-hydroxyquinoline derivatives. Their design27 is based on the idea to combine the ability of 8-hydroxyquinoline derivatives to modulate their fluorescence properties when complexed to suitable metal ions28 with the additional tool of controlling the selectivity towards the target ion provided by the receptor moiety.
The first component of this large family to be studied in our laboratory was compound 1, bearing a chloride as substituent on the 8-hydroxyquinoline. Interestingly, it proved to be a very efficient luminescent chemosensor for Mg2+ ions.29 The uncomplexed species shows in MeOH–H2O (1∶1) a weak luminescence band centred at 540 nm. Its very low quantum yield (Φ < 5 × 10−5) is consistent with the luminescence properties of the parent neutral 8-hydroxyquinoline in protic solvents, where intra- and intermolecular photoinduced proton transfer (PPT) processes are responsible for the high efficiency of the radiationless deactivation to the ground state.30 In addition, photoinduced electron transfer (PET) processes involving the nitrogen atom of the crown can offer an additional nonradiative deactivation process to the ground state. Addition to a solution of this species of an increasing amount of magnesium ions leads to a large enhancement of the fluorescence band (Fig. 1, complex quantum yield 0.042) allowing quantitative detection of this species in solution with high sensitivity.
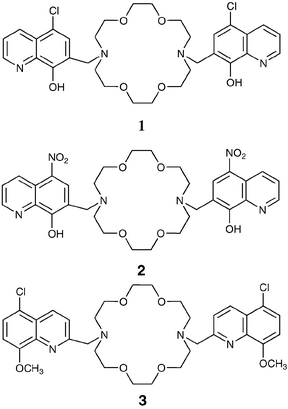
 | ||
| Fig. 1 Fluorescence spectra of 1 (2.5 × 10−5 M, λexc = 360 nm) in methanol–water (1∶1 v/v, pH 7.0) and upon addition of an increasing amount of Mg2+ ions. Inset: fluorescence intensity (λexc = 360 nm, λem = 520 nm) vs. equivalents of Mg2+ ions. | ||
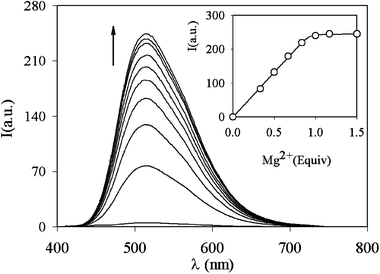
The fluorescence enhancement is caused by a process involving the deprotonation of the hydroxy groups induced by the complexation of the metal ions (Scheme 1); this process, indeed, strongly depends on the pH conditions. This dependence is depicted in Fig. 2, which shows the absorbance of 1 as a function of pH. In the absence of metal ions, it can be observed that the deprotonation of the hydroxy groups of the ligand occurs at pH above 9. If Mg2+ ions are present, it takes place at pH below 7, making 1 able to indicate the presence of these ions also in physiological conditions. Under these conditions the other earth alkali metal ions form less stable and nonluminescent complexes, since they are able to induce deprotonation of the hydroxy groups only at pH above 9. All these features, which are particularly favourable, suggested to us the use of this and very similar compounds as fluorescent probes for the intracellular detection of magnesium ions. The preliminary results obtained so far show that 1 and similar species indeed cross cellular membranes and become fluorescent because of the high intracellular concentration of magnesium (Fig. 3).
 | ||
| Scheme 1 Schematic representation of the complexation process for 1. | ||
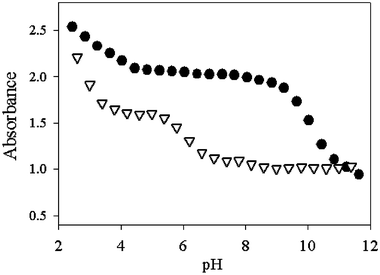 | ||
| Fig. 2 Absorbance of 1 (1 × 10−5 M) alone (circles) or in the presence of Mg2+ (2.7 × 10−3 M, triangles) at various pH values. | ||
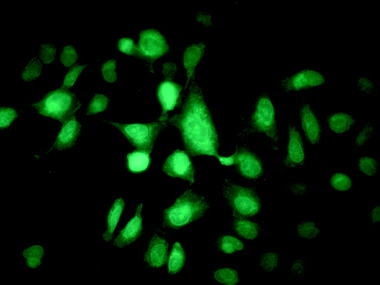 | ||
| Fig. 3 U2OS (human osteosarcoma) cells, grown on a glass cover slip and stained with 1 (50 µM). | ||
We have investigated the uptake of these probes by different kinds of cells containing different amounts of magnesium; a good correlation between the data obtained using fluorescence spectroscopy and atomic absorption spectroscopy has been obtained in all cases. Moreover, addition of EDTA in excess causes, as expected, the quenching of fluorescence intensity.31 A complete investigation on the behaviour of this probe for the in vivo monitoring of magnesium, including the possibility to follow its cellular compartmentalisation, are now in progress and will be published in due course. This chemosensor could represent a great improvement for this kind of analysis, considering that commercially available probes for these ions typically show lower association constants for Mg2+ and also a stronger selectivity for Ca2+ over Mg2+.
Starting from the knowledge that the deprotonation process was crucial both for the complexation and for the switching on of the fluorescence, we studied 2, which is very similar to 1, having in the 5 position a nitro group instead of the chlorine one.32 The substitution with the nitro group was intended to change significantly the acid–base properties of the pyridine nitrogen and of the hydroxy group. Interestingly, 2 forms particularly stable complexes with Hg2+ ions. The severe risks for human health and the environment posed by mercury ions have attracted the interest of many groups worldwide for the development of highly selective sensors for this cation.7,10,33 In methanol–water (1∶1 v/v, pH 7.0) the absorption spectrum of the free ligand, which is dominated in the visible region by a charge-transfer band involving the electron-withdrawing nitro groups, shows noticeable changes upon Hg2+ complexation, as can be seen in Fig. 4. The uncomplexed species presents a luminescence band centred at ca. 470 nm and its very low quantum yield (Φ = 5 × 10−5) is consistent with the already discussed luminescence properties of the parent chromophore; also in this case PET processes cannot be ruled out. Upon complexation with Hg2+ and excitation at 420 nm, the fluorescence intensity of 2 increases by a factor of 12 (Fig. 5), and the association constant was estimated to be greater than 1 × 108 M−1. The fluorescence intensity reached its maximum value when one equivalent of the metal ion was added, indicating that the stoichiometry of the complex is 1∶1.
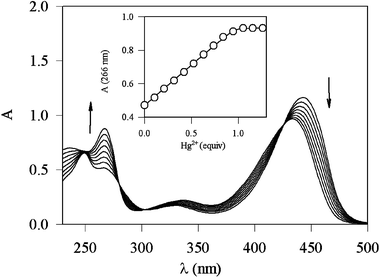 | ||
| Fig. 4 Absorption spectra of 2 in methanol–water (1∶1 v/v, pH 7.0) and upon addition of an increasing amount of Hg2+ ions. | ||
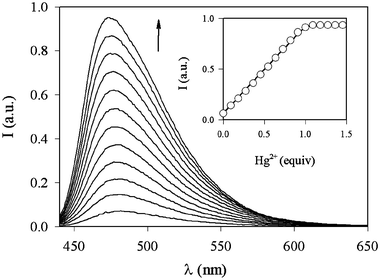 | ||
| Fig. 5 Fluorescence spectra (λexc = 430 nm) of 2 in methanol–water (1∶1 v/v, pH 7.0) and upon addition of an increasing amount of Hg2+ ions. Inset: fluorescence intensity (λexc = 430 nm, λem = 480 nm) vs. equivalents of Hg2+ ions. | ||
The fluorescence enhancement can again be observed only when the deprotonation of the hydroxy group occurs, in this case at pH above 3. From the point-of-view of possible applications, it is evident that a 12-fold intensity enhancement is not high enough to ensure the sensitivity needed for the detection of mercury in highly diluted conditions. However, this is a rare example of fluorescent enhancement upon mercury complexation33 since this metal ion typically causes luminescence quenching, usually because of the heavy atom effect. For this reason, we believe that this system can be a good starting point for the design of more appropriate sensors.
Another possible way to tune the sensitivity and selectivity of this class of compounds was found to be the substitution of the hydroxy group with a methoxy function as in 3.34 In this case, the dependence of the photophysical properties on pH is obviously less dramatic for two reasons: (i) the chromophore has lost one acid centre and (ii) intramolecular PPT processes in this system are no longer possible. On the other hand, in protic solvents intermolecular proton transfer is still active, together with PET processes involving the nitrogen of the crown moiety. 3 is selective for Cd2+ ions and its complexation caused the highest fluorescence enhancement among all the transition metal ions studied. The quantum yield of the complex is remarkably high (0.66 in methanol solution, Fig. 6) and it is accompanied by high molar absorption coefficients (95 000 M−1 cm−1 at 252 nm), although in the UV region. These interesting features make 3 a promising candidate as a chemosensor for the detection of Cd2+, which has well-established deleterious effects on human health.35 For this reason, studies are in progress for its immobilisation on quartz or silica surfaces, a fundamental step for practical applications.
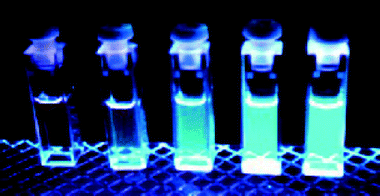 | ||
| Fig. 6 Methanol solutions of 3 (1 × 10−4 M) with an increasing amount (from left to right, 0, 2, 4, 6, 8 × 10−5 M) of Cd2+ ions irradiated with UV light. | ||
To understand the reason for the so different behaviour shown by the closely related chemosensors 1–3, we investigated also many other species containing different crown ether receptors and 8-hydroxyquinoline derivatives.36 In particular, we were able to observe a notable feature: the macrocyclic rings are not always directly involved in complex formation. In particular, in the complexes of 2 with Hg2+, Zn2+ and Mg2+, the crown ethers are not chelated to the metal ions; rather, the macrocyclic rings behave as a scaffold for the quinolines to fold around the metal ions. In the case of Hg2+, a pseudotetrahedral coordination sphere is generated, while in the cases of Zn2+ and Mg2+ octahedral coordination spheres are formed with water molecules at the apical positions.
Upon closer inspection, it becomes evident that there is also a key role for the amine groups in the macrocyclic ring. These amine groups can act as general bases and serve to deprotonate the hydroxyl group in the metal ion complexes. This deprotonation inhibits both PPT and PET processes and yields fluorescent complexes. In the 2-Zn2+ and 2-Mg2+ structures the protons were indeed found on the nitrogen atoms of the amine groups in the macrocycle.
The extensive amount of data obtained so far for this class of chemosensors has required much effort; however, we believe that this will be of great value for the design of new and more efficient species. In particular, we are aiming to synthesise new derivatives that could offer: (i) even greater selectivity, by changing the nature of the crown ether and the substituents on the chromophores, and (ii) higher sensitivity, by introducing substituents that could lead to greater absorptivity and fluorescence quantum yields in the red part of the spectrum, without influencing the selectivity. In addition, new quinoline derivatives will be also synthesised in order to optimise membrane crossing and cellular compartmentalisation.
Another modification we introduced and investigated was the replacement of each hydroxy group with a benzene sulfonamido group,37 yielding 4 and 5. These two species contain the same chromophores as the acyclic 6-methoxy-8-p-toluensulfonamidoquinoline (TSQ)38 and its ester ether derivative Zinquin,39 respectively, that are currently the most widely used zinc probes. It is important to stress that several chemosensors for the detection of Zn2+ ions has been recently reported, especially for fluorescent imaging, which has been proven to be the most suitable technique for in vivo monitoring of zinc ions.40 The responses of these two ligands to the addition of metal ions are dramatic. In particular, addition of Zn2+ ions to a solution of 4 or 5 leads to strong changes of the absorption spectra until one equivalent of metal ion has been added, while no changes are observed upon further additions. In contrast, a different pattern is observed for the fluorescence spectra. The fluorescence intensity, in fact, increases in an almost linear way up to the addition of 2 equiv. of zinc. All these findings can be rationalised in terms of the formation in solution of two different complexes with 1∶1 and 1∶2 (ligand∶metal) stoichiometries, respectively, as supported by X-ray data. Qualitatively, the changes observed in the absorption and luminescence spectra are very similar to those reported for Zinquin and TSQ at the same pH. At neutral pH, the complexation process is associated with the deprotonation of the sulfonamido group. From the changes in the absorption spectra, it is evident that the inclusion of the first metal ion leads to the deprotonation of both the chromophoric groups, while the formation of the binuclear complexes causes only minor changes. The change of the charge density close to the two fluorescent moieties is, however, sufficient to further increase the luminescence band, which, having a charge-transfer character, is very sensitive to the polarity of the environment. The increase in the excited state lifetime observed on going from the mononuclear to the binuclear complex lends further support to this interpretation.
In our opinion 4 and 5 could also be of interest for applications in the field of cell biology, as already discussed for 1, and for this reason we are currently investigating in this direction.
Continuing to profit from the favourable properties of hydroxyquinoline derivatives and of our experience in the photophysics of calix[4]arene receptors,41 we designed and studied 6 and 7.42 Contrary to what was observed with quinoline-functionalised crown ethers, no changes in the absorption or emission spectra were observed for either ligands 6 or 7 with Hg2+, Mg2+ and Zn2+ metal ions. In addition, it is noted that their fluorescence is relatively high compared to that of compounds 1–3. In these systems, in fact, two of the main mechanisms for the nonradiative deactivation of the excited state present in the azacrown derivatised systems, namely PET and intramolecular PPT, cannot be active. Alkali and alkaline earth metal ion complexes of 6 usually show a bathochromic shift and a quenching of the fluorescence band with respect to the free ligand; on the contrary, the corresponding complexes of 7 present hypsochromic shifts and an increase of fluorescence intensities for alkaline earth cation complexes. NMR spectroscopic data and semi-empirical calculations support the hypothesis that this difference in the photophysical properties is due to a different mode of coordination of the quinoline part to the metal ion. For 6, complexation involves both the quinoline N and O atoms, while in 7 the N atom is too far from the metal ion. These results were very useful for understanding the nature of the interactions among metal ions and hydroxyquinoline ligands, and they supplement the data obtained from the systems with the same chromophoric units but with crown ethers as receptors.
It should be noted at this point how delicate is the balance among all the factors influencing the metal ion coordination ability of the receptor and the consequent photophysical response of the fluorophore. This makes it sometimes very difficult to predict the performances of a designed chemosensor. Only the knowledge coming from a detailed structural and spectroscopic characterisation of many and varied systems of this kind can yield precious suggestions for the realisation of future devices.
It is at this stage even more evident that one has to thoroughly understand the widest possible number of chromophores and receptors, together with their mutual interactions, in order to design species responding to the different demands in sensor technology. In this context, another system of particular interest for us is 8.43 Although amidic groups are usually only poorly efficient in metal ion coordination, their binding tendency can be widely enforced when they are inserted in a receptor structure in which other anchoring centres co-operatively take part in the complexation. Fabbrizzi et al. have shown that a well-known category of ligands, dioxotetraamines (and in particular dioxo-2,3,2-tet, 1,4,8,11-tetraazaundecane-5,7-dione) can be successfully employed for signalling the presence of Ni2+ and Cu2+ if connected to anthracene.44 The substitution of this chromophore with the well-known Ru(bipy)32+ luminophore in 8 has been considered, taking into account that it is water soluble at a reasonable concentration (up to 10−2 M), can be excited with visible light, and possesses a relatively long-lived excited state (ca. 1 µs in deaerated solution), thus allowing lifetime-based sensing.17a The complexation mechanism of 8 involves the deprotonation of the two amide groups. This very endoergonic process can take place only with metal ions that benefit from a large ligand field stabilisation, that is Ni2+ and Cu2+ only. In addition, the different extent of ligand field stabilisation, higher for Cu2+ with respect to Ni2+, allows also to distinguish between these ions, making these ligands extremely selective. In this regard, it has to be noted that in the past years much effort has been expended for the detection of copper ions due to their biological and environmental significance.45
In particular, the complexation properties of 8 towards transition metal cations have been examined by comparing the changes in its fluorescence intensity (If) and lifetime (τ) as a function of pH, in the presence and absence of transition metal cations. When no metal cations are added to solutions containing 8, If remains constant in the 2 < pH < 12 range. In this pH range, in fact, its luminescence quantum yield (0.030) and lifetime (440 ns) in aerated water solutions are very similar to those observed for the parent Ru(bpy)32+ chromophore under the same conditions. This indicates that the dioxo-2,3,2-tet fragment does not substantially perturb the excited state properties of the Ru moiety. On the other hand, when Ni2+ or Cu2+ are added in a 1∶1 molar ratio with respect to 8, the Ifvs. pH plot shows a typical sigmoidal profile (Fig. 7), which indicates that the binding of metal ions by the dioxo-2,3,2-tet fragment takes place in the narrow pH range of the steeply descending portion of the sigmoid.
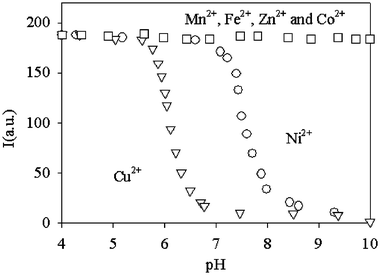 | ||
| Fig. 7 Luminescence intensity (λexc = 450 nm, λem = 610 nm) vs. pH for solutions containing ligand 8 and a metal cation in 1∶1 stoichiometry. The metal species referring to each curve is indicated in the plot. | ||
Fig. 7 shows that complexation by 8 (1 × 10−5 M) begins at pH 5.8 and it is complete at pH 6.8 for Cu2+, while for Ni2+ it begins at pH 7.5, being complete at pH 8.5. In the descending Ifvs. pH plot portion one can also observe the appearance in the excited state decay profile of a second component with a much shorter lifetime (11 and 15 ns for Ni2+ and Cu2+, respectively), clearly indicating an intramolecular quenching process; this component becomes the only one present at higher pH values.
From flash photolysis experiments, no evidence of the presence of RuI species could be detected, the only transient absorbing species being due to the excited state of the RuII chromophore. Furthermore, steady state experiments performed at 77 K showed that the quenching process was very fast also in frozen medium at low temperature. All these findings suggest that an energy transfer (ET) mechanism is the most likely explanation for the luminescence quenching of 8.
The selectivity of this system is proven by the negligible fluorescence intensity changes in the Ifvs. pH plot in the presence of other metal centres (1∶1 molar ratio), such as Mn2+, Fe2+, Zn2+ and Co2+, indicating that no complexation takes place at the dioxo-2,3,2-tet fragment with these ions. In addition, working on a solution of 8, buffered at pH = 7.0, no changes in If are observed upon addition of up to 2 equiv. of Ni2+ (or other divalent first-row transition metal cations), while subsequent addition of Cu2+ causes the expected fluorescence quenching, demonstrating that selectivity for Cu2+ over Ni2+ can be obtained by choosing the correct pH value (see Fig. 8). Finally, solutions buffered at pH 7.0 and containing 8 at concentrations as low as 10−7 M revealed an easily detectable variation of If on addition of 1 equiv. of Cu2+, indicating that 8 is a suitable sensor for copper cations under analytically relevant conditions.
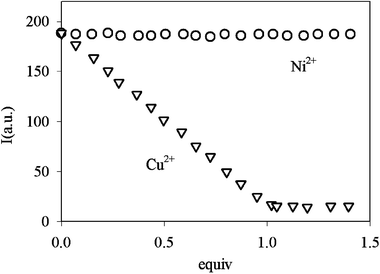 | ||
| Fig. 8 Luminescence intensity vs. equivalents of added Ni2+ and Cu2+ for 8, in a solution buffered at pH 7.0. | ||
The last series of chemosensors for metal ions presented here (946 is their archetype) is based on the dansyl chromophore, which is well-known to show an intense and large luminescence band in the 400–600 nm region.47 This band is very sensitive to the polarity of the solvent, having a considerable charge-transfer character, caused by the mixing of the 1La and 1Lb states of naphthalene with a charge-transfer state arising from the promotion of a lone-pair electron on the amino group into an antibonding orbital of the naphthalene ring. The fluorescence of 9 does not depend on the pH conditions in the 3–11 pH range, while strong changes in its absorption and luminescence properties (see Fig. 9) were observed upon addition of Cu2+, Co2+, Zn2+ and Cd2+ at pH 9.5. In particular, for the former two metal ions, a strong quenching (Irel
= 1%) of the luminescence intensity was observed, while for Zn2+ and Cd2+ a blue shift accompanied by an enhancement of the fluorescence quantum yield could be detected. The complexation mechanism involves, for all these metal ions, the deprotonation of the sulfonamide groups. For Zn2+ and Cd2+ ions, the observed blue shift has been attributed to an increase of the electronic density on the naphthalene ring due to the deprotonation/complexation process, which moves towards higher energy the amine-to-naphthalene charge-transfer state present in the dansyl chromophore. Cu2+ and Co2+ ions, together with this effect, introduce energetically lower lying excited states centred on the metal, which deactivate the luminescent excited state centred on the dansyl chromophore. Interestingly, addition to 5 × 10−5 M solutions of 9 of up to 100 molar equivalents of Na+, K+, Ca2+, Sr2+, Ba2+, Eu3+, Mn2+, Pb2+, Fe2+, Fe3+, Cr3+, Ni2+ and Hg2+ did not lead to any changes in the absorption and emission spectra of the dansyl chromophore over the whole pH range examined (3–11). It is noteworthy that the complexation equilibrium is pH dependent since it involves the deprotonation of the sulfonamide groups.
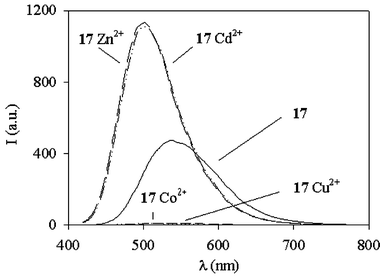 | ||
| Fig. 9 Fluorescence spectra (λexc = 338 nm) of 9 (9.2 × 10−5 M) and of its complexes with Cu2+, Co2+, Zn2+ and Cd2+ in acetonitrile–water (1∶1 v/v) solution at pH 9.5. | ||
It was observed that, while the complexation with copper ions is complete at pH 6, the complexation with the other metal ions requires higher pH values, which makes 9 very selective for Cu2+ at physiological pH. A very similar behaviour as a chemosensor was observed for the ligand in which only one amine of the tren moiety was reacted with a dansyl chromophore, the only difference being the formation of metal complexes at more acidic pH. It has to be said that, although the dansyl chromophore is widely used in fluorescence-based analysis, for applications in very diluted solutions this dye suffers in terms of sensitivity from its relatively low molar absorptivity. For this reason, 9 and related systems have not been directly tested in practical applications; their interesting properties have, however, suggested to us their use in the derivatisation of nanoparticles. The signal amplification obtained in these systems gives the possibility to overcome this problem of low absorptivity (see below).
The dansyl moiety was also used in 10, a chemosensor designed for chiral recognition, in which this chromophore is appended to a β-cyclodextrin via a spacer containing a chiral phenylalanine unit.48 I would like to stress here that enantioselectivity is one of the most important goals for the sensing of organic substances, since biological activity is strictly correlated with stereochemistry.49,50 The isomer of 10 containing L-phenylalanine (10a) was found to have a predominant conformation in which the dansyl group is self-included in the cyclodextrin cavity, while in the one containing D-phenylalanine (10b) the conformation in which the dansyl group lies outside the cavity predominated. As a consequence, the two isomers were found to bind Cu2+ with different affinities, as revealed by fluorescence quenching in competitive binding measurements. Interestingly, addition of D- or L-amino acids to a solution containing the copper complexes of either 10a or 10b induced an increase in fluorescence intensity, which was dependent on the amino acid used and, in some cases, on its absolute configuration. 10a was found to be more enantioselective than 10b, suggesting that the self-inclusion in the cyclodextrin cavity strongly increases the chiral discrimination ability of the copper(II) complex. Using 10a, particularly good enantioselectivities were obtained for Pro, Val, Leu, Ser, Phe and PhGly. The enantioselectivity in fluorescence response was found to be due to the formation, with different association constants, of diastereomeric ternary complexes. These results suggest that 10a can be used for analytical purposes, at least when fast screening procedures are required since, contrary to many other proposed methods for parallel analysis of enantiomers, only a simple mixing procedure, which can easily be automated, is required. Of course, this is a very appealing characteristic for possible applications.
As outlined in the Introduction, great interest is currently devoted to luminophores with very long excited state lifetimes. Among this class of fluorophores, complexes of Eu3+ and Tb3+ ions, which typically possess luminescence lifetimes in the microsecond–millisecond range, are particularly studied for the development of labels and sensors.16 In this context, we have investigated the complexes of 11 with Tb3+ and Eu3+51 as possible chemosensors for anions. Undoubtedly, the development and improvement of highly selective ionophores that would allow quantitative detection of anions remains a central issue in analytical chemistry and many groups are very active in this field, also using lanthanide complexes.5211 consists of one P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O fragment and two methylene linked bipy subunits. Our choice of this particular ligand was based on the expectation that the PO moiety would provide relatively stable complexes due to the strong coordination ability of this group and that the bipy fragments coordinated to the Ln would effectively act as photon antenna able to transfer energy to the Eu3+ and Tb3+ ions with high efficiency. Furthermore, the ligand does not complete the coordination sphere of the Ln metal ions, leaving some positions easily accessible. The ligand and the Ln complexes are sketched in Scheme 2.
O fragment and two methylene linked bipy subunits. Our choice of this particular ligand was based on the expectation that the PO moiety would provide relatively stable complexes due to the strong coordination ability of this group and that the bipy fragments coordinated to the Ln would effectively act as photon antenna able to transfer energy to the Eu3+ and Tb3+ ions with high efficiency. Furthermore, the ligand does not complete the coordination sphere of the Ln metal ions, leaving some positions easily accessible. The ligand and the Ln complexes are sketched in Scheme 2.
![Structure of the complexes [Ln·11]3+.](/image/article/2005/NJ/b411758a/b411758a-s2.gif) | ||
| Scheme 2 Structure of the complexes [Ln·11]3+. | ||
The results obtained showed that the photophysical properties of these complexes depend drastically on the presence of anions. In particular, addition of nitrate anions to an acetonitrile solution containing the [Ln·11]3+ complex as triflate salt results in an up to seven-fold increase of the luminescence intensity for the Tb3+ complex and an eleven-fold increase for the Eu3+ complex. This increase is caused by the displacement of solvent molecules from the first coordination shell of the Ln cations by the competing nitrate anions and by concomitant important changes in the coordination features of the bipy moieties. In particular, absorption, emission and excitation spectra suggest that when two nitrate anions are coordinated to the metal ion, 11 assumes a different conformation, in which the energy transfer from the bipy subunits to the metal ion is more efficient, so that a more intense metal-centred luminescence is observed. Quantum mechanical calculations support this conclusion, suggesting that upon coordination of the second anion one bipy arm moves closer to the lanthanide ion, thus increasing their mutual electronic interaction. Similar results were obtained with another family of Eu3+ complexes,53 whose ligands were designed with the same strategy followed for 11, but replacing the two bipy units with a terpy moiety. It is important to point out that, since these species can signal the presence of anions only in aprotic solvents, they cannot be used as such for anion detection in real samples. However, these systems are of interest not only because chemosensors for anions are relatively rare, but also because they suggest that the idea of leaving some position(s) easily accessible can offer an interesting signal transduction mechanism. The next synthetic step will be to design new ligands, based on the same strategy, able to form stable complexes also in water solutions. Furthermore, we have observed in preliminary experiments that these complexes, when deposited in thin films, are able to bind coordinating species, such as amines, in the gaseous phase with a concomitant quenching of the luminescence intensity, suggesting their possible use in electronic noses with optical transduction.
… to nanoparticles
So far we have discussed molecular systems in which one receptor unit is usually linked to up to three chromophoric moieties. We have seen that systems of this kind, described by many laboratories worldwide, have interesting properties and that some of them can also be of use in practical applications. Of course, all the systems presented so far can be improved, and we are devoting much effort on this research since we believe that progress in this direction is still of great value. On the other hand, we are also convinced that many new approaches to improve the performance of chemical sensing are extremely challenging. As an example, the introduction of combinatorial chemistry12d,54 is attracting the attention of many chemists for the development of new specific receptors. When we were thinking about the possibility to undertake new strategies, we were attracted by the importance that engineers working in sensor technology were giving to a particular keyword, specifically signal amplification. It was evident to us that this would mean a significant improvement in the detection limits; however, it was much less evident how to translate this keyword into ‘chemical language’. We found an interesting solution to this problem in the possibility that the coordination of a single molecule of the target analyte could change the properties of a large number of active units at the same time. This approach is particularly useful with luminescent sensors, since even very weak electronic interactions can be sufficient for inducing energy- and electron-transfer processes over a relatively large scale. In this way, every receptor unit of the system can be ‘electronically wired’ to a large number of dyes around it, so that the luminescence properties of all of them can be tuned by the state of a single receptor. A typical class of compounds that is able to show this effect is dendrimers;55 their drawback is, however, that their synthetic procedures require considerable efforts.Our aim then was to synthesise new structures in which a large number of dyes could interact with each other and with suitable receptors by means of energy- and/or electron-transfer processes. Generally speaking, the organisation of photophysically active units in structures such as derivatised surfaces and conjugated polymers typically gives rise to collective effects that can be exploited for the design of new functional materials.26,56 Among the different possibilities, we chose the modification of the surface of nanoparticles as suitable path to constrain a set of fluorescent units into an organised network. Fluorescent nanoparticles are, in fact, very promising for the design of labels and sensors due to the relative ease of their synthesis and because of their peculiar properties. Among the different kinds of nanoparticles, we have addressed our attention to the ones having a gold or silica nucleus. As far as the former ones are concerned, since the metal core can give rise to electron- and energy-transfer processes, they are often not (or almost not) luminescent, even when luminophores are attached to their surface.57 In some cases, however, a large fluorescence enhancement has been found for fluorophores deposited near metal particles under carefully optimised conditions58 and this effect deserves particular attention.
As far as silica nanoparticles are concerned, they can be prepared in a very straightforward way and their surface easily modified by means of alkoxysilane derivatives. This versatility, in our opinion, makes silica nanoparticles a good choice as a scaffolding structure for a network of dye moieties, especially considering that these materials are transparent to visible light and inert as far as energy- and electron-transfer processes are concerned. Therefore, dye-coated silica nanoparticles constitute a suitable system to characterise intermolecular photophysical processes at their surface, avoiding any interference from the particle nucleus.
In our first attempt59 to synthesise dye-coated silica nanoparticles, we chose the dansyl chromophore, because of the peculiar response of its photophysical properties to the protonation of the amino group. As I have already discussed for 9 and related compounds, the lowest excited state of dansyl has, in fact, a charge-transfer character. In the protonated unit, this charge-transfer state is destabilised and, as a consequence, the π-π* transition localised on the naphthalene becomes the lowest in energy. This leads to a strong blue shift of both absorption and luminescence bands that allows, in partially protonated polydansylated systems, to selectively excite and detect the outcoming fluorescence of the two different units. Hence, a partially protonated polydansylated system represents a good example of a network composed of two different kinds of chromophores (Dns and DnsH+) whose relative composition can be controlled by changing the degree of protonation (Fig. 10).
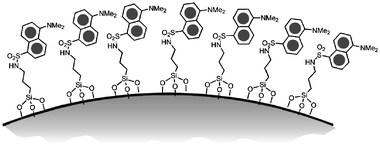 | ||
| Fig. 10 Schematic representation of dansyl covered silica nanoparticles. | ||
We prepared the fluorescent nanoparticles by covering preformed silica colloids with silane derivatives (Fig. 11); each nanoparticle (mean diameter 18 nm) bears an average number of about 4000 dansyl units. The main goal of the photophysical investigation was to analyse the cooperative effects that arise from the organisation of several chromophoric units into a high density pattern such as at the surface of these nanoparticles. Their photophysical characterisation revealed that they respond to protonation in acetonitrile solutions with an amplified quenching of the dansyl luminescence (up to five quenched units per added proton) due to multicomponent charge-transfer processes. On the contrary, when protonated in pure chloroform they behave as an efficient antenna system, where the energy absorbed by the DnsH+ is efficiently transferred to the luminescent Dns chromophore. The different behaviour observed in the two solvents can be conveniently explained by the role of environmental polarity on the delicate balance among several different photophysical processes, such as fluorescence emission and energy- and electron-transfer processes. All these results are due to the possible occurrence in an organised network of photophysical processes involving a large number of chromophores, leading to large signal amplification, even upon small chemical inputs. The possibility to tune the photophysical properties of these systems makes them extremely interesting for the design of new photoactive devices.
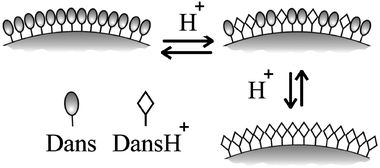 | ||
| Fig. 11 Schematic representation of the effect of protonation on dansyl covered silica nanoparticles. | ||
Afterwards, taking advantage of the knowledge and experience gained, we have also proposed60 an alternative synthetic strategy to obtain nanoparticles derivatised with fluorophores presenting energy transfer between them, specifically to introduce a fluorescent alkoxysilane directly during the formation of the colloids. Inclusion of dyes in silica nanoparticles usually takes advantages of inverse microemulsion techniques that, unfortunately, do not lead to covalent grafting on the polymeric matrix, with the consequent risk of dye leaching. The introduction of a triethoxysilane group directly on the dye molecule allows, instead, its covalent anchoring during the TEOS polymerisation. It is interesting to note that this synthetic strategy, contrary to the one described before, leaves the surface of the colloids almost unmodified and therefore available for further modification.
To prepare the nanoparticles, we modified the Stoeber procedure,61 adding a 1% (mol/mol with respect to tetraethoxysilane, TEOS) of a fluorescein derivatised triethoxysilane (Fig. 12) during the polymerisation process. The formation of the colloids was confirmed by TEM images that were graphically elaborated to evaluate the average size of the particles (23 ± 3 nm). Flow field-flow fractionation62 (FlFFF) experiments gave definitive evidence that the fluorophores were bound to the silica matrix.
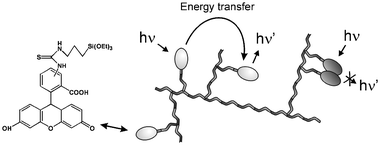 | ||
| Fig. 12 Schematic representation of fluorescein containing silica nanoparticles. | ||
The absorption and luminescence bands of the dye in these nanoparticles did not undergo noticeable spectral shifts, as instead observed in more highly doped systems.63 For all the solutions examined the fluorescence intensity at 520 nm upon excitation at 480 nm was directly proportional to the absorbance (A < 0.05) at the excitation wavelength. This proportionality indicates that in the nanoparticles the average quantum yield of fluorescein is the same, independent of the size of the colloids. This average quantum yield was about 50% with respect to that of free fluorescein in solution, while the excited state lifetime of the fluorescein in the nanoparticles was the same as that one found in solution. It has to be noted that multiexponential decays have been observed in other materials, when highly doped with fluorescein.64 In addition, the luminescence of fluorescein in the nanoparticles is almost completely depolarised (P < 0.02). This was an unexpected result, since the rotational mobility of the fluorophores in the colloids is strongly reduced, so that a different mechanism for depolarisation had to be considered. Two kinds of processes that involve the fluorescein units in nanoparticles have been, therefore, taken into account to explain the photophysical results. The first one leads to the complete quenching of some fluorophores and it is known to be due to short-range interactions between fluorescein molecules.58 The second one leads to fluorescence depolarisation and involves transfer of the excitation energy between the different dye units in the nanoparticles. It has to be noted that the Forster distance for fluorescein-fluorescein energy transfer is about 47 Å.58 These two processes are schematically depicted in Fig. 12. The possibility of having different kinds of interactions depending on intermolecular distance makes fluorescein a good probe to test the structure of the fluorophore network.
The results obtained have shown that the local density of the dye molecules is not homogeneous in the nanoparticles. In the more dense regions they are close enough to give self-quenching, causing a decrease of the overall average fluorescence quantum yield. On the other hand, the observation that no fast components are present in the decay of the fluorescence suggests, together with the observed depolarisation, that the recorded emission is due to dye molecules close enough to take part to energy-transfer processes but too far apart to engage in self-quenching. Such a discontinuous behaviour implies the presence of groups of fluorophores completely quenched and others whose fluorescence efficiency is unmodified, suggesting some degree of pre-organisation of the fluorophores during the formation of nanoparticles. The very low degree of polarisation of the fluorescence rules out the presence of “isolated” fluorophore molecules too far away to transfer their excitation energy to other fluorescein moieties. We would like to stress again that the possibility to have long-distance energy migration among the chromophores inside the nanoparticles is a key step for having an efficient signal amplification; for this reason these results open up, in our opinion, new perspectives in the design of functional nanosystems with high potential in applications, in particular in the field of sensors and labels. In the described systems the communication between the photoactive units is very efficient and it should be recalled that this is an essential condition for obtaining the desired signal amplification effect.
The next step we have undertaken, aiming to more and more efficient chemosensors, is the synthesis, following the same strategy, of nanoparticles containing species that present a dansyl group and a linear polyamine receptor unit covalently linked together (Fig. 13).65 Interestingly, upon addition of small amounts of Cu2+, Co2+ and Ni2+ in water–ethanol solutions, the typical fluorescence of dansyl is quenched: in particular, after the addition of only 2% (respect to the number of chromophoric units) of copper ions (10−8 M), a more than 20% decrease of the luminescence intensity can be observed. This result is evidence that the amplification process in these systems does indeed occur. I would like to stress that this system could be considered an implement of 9, since the signal amplification overcomes the problems related to the relatively low absorptivity of dansyl derivatives. Interestingly, Brasola et al. obtained similar results with nanoparticles derivatised with chromophores and receptors that are not covalently linked one to another,66 demonstrating again the validity of this strategy. Of course, better designed systems could be appended to the nanoparticles in order to increase the selectivity but, to our opinion, these results clearly indicate that silica nanoparticles, which are typically made with cheap material and simple synthetic procedures, are very promising systems for the development of chemical sensor technologies.
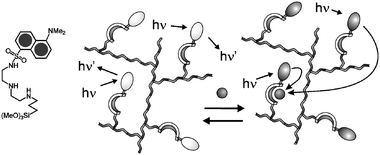 | ||
| Fig. 13 Schematic representation of the quenching processes occurring in silica nanoparticles upon copper complexation. | ||
Conclusions and future perspectives
The great interest devoted by the scientific community to chemical sensors can be easily understood considering that these species can offer the possibility to monitor in situ, in real time and real space, the concentrations of analytes for many different purposes. They can have, indeed, important applications in environmental monitoring, process control, food and beverage analysis, medical diagnosis, and toxic gas and explosive detection, fields of crucial importance in our society. A fruitful approach to the design and synthesis of efficient sensory devices is the development of supramolecular entities, referred to as chemosensors, able to recognise the target analyte with the desired affinity and selectivity, and presenting an efficient signal transduction mechanism. I have reported here some examples of luminescent chemosensors studied in our laboratory, trying to make clear the principal strategies followed for translating analyte recognition into luminescence signal modulation. A deep understanding of this aspect is essential in the development of efficient macroscopic sensory devices. This is, in my opinion, a very good reason to explain the efforts spent up to now on this research area. In order to address practical needs, however, better selectivities and sensitivities have often still to be reached. Several approaches are possible to solve this problem. The first one is, of course, to keep on working on the well-established receptor-spacer-luminophore scheme, trying to optimise all the steps that lead to the chemosensor response. Nowadays this approach is becoming rather mature, but I think that there is still a lot of room for innovative research in this direction. The ingenuity of chemists is, however, suggesting also more innovative approaches, often taking advantage of a multidisciplinary environment. For example, engineers pay a lot of attention to the concept of signal amplification, an approach that chemists have borrowed and in which we strongly believe. This can be obtained, in chemistry, when a single binding event with the target analyte induces changes in the properties of a large number of active units at the same time. Photophysical processes, such as energy and electron transfer, are able to induce such an effect, even if very weak electronic interactions are present among the different components. This makes luminescent sensors even more appealing. In this context, we strongly believe that the use of nanoparticles, in which energy- and electron-transfer processes can occur over a relatively large number of units, can be a fruitful strategy for improving sensor performances.To conclude, I think that what Czarnik67 stated a decade ago: “there is still a great demand for more and more efficient chemosensors”, is today even more valid: a wide horizon is in fact still open to further investigations. The ingenuity of chemists is suggesting new developments; among them, I believe that nanoparticles performing signal amplification can play a crucial role.
Acknowledgements
It is an honour for me to dedicate this paper to Dr Roberto Ballardini and to Prof. Maria Teresa Gandolfi, both recently retired, who had a key role in my first steps in the wonderful world of photochemistry, and to Marco Montalti and Nelsi Zaccheroni, without whose irreplaceable collaboration all the work described here would have never appeared.Financial support from the Ministero dell’Istruzione, dell’Università e della Ricerca (FISR, project SAIA) and the University of Bologna (funds for Selected Topics) is gratefully acknowledged.
References
- A. Hulanicki, S. Glab and F. Ingman, Pure Appl. Chem., 1991, 63, 1247 CrossRef.
- Fluorescent Chemosensors of Ion and Molecule Recognition, eds. J.-P. Desvergne and A.W. Czarnik, NATO-ASI Series, Kluwer Academic Publishers, Dordrecht, 1996 Search PubMed.
- U. E. Spichiger-Keller, Chemical Sensors and Biosensors for Medical and Biological Applications, Wiley–VCH, Weinheim, 1998 Search PubMed.
- Fluorescent Chemosensors for Ion and Molecule Recognition, ed. A. W. Czarnik, American Chemical Society, Washington, D.C., 1992 Search PubMed.
- See, for example: Coord. Chem. Rev., 2000, 205, 1–232, special issue on luminescent sensors, guest editor L. Fabbrizzi Search PubMed.
- See, for example: Chem. Rev., 2000, 100, 2477–2738, special issue on chemical sensors, guest editors A. B. Ellis and D. R. Walt Search PubMed.
- (a) A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515 CrossRef; (b) A. P. de Silva, G. D. McClean, T. S. Moody and S. M. Weir, in Handbook of Photochemistry and Photobiology, ed. H. S. Nalwa, American Institute of Physics, Stevenson Ranch, CA, USA, 2003, vol. 3, p. 217 Search PubMed.
- L. Fabbrizzi, M. Licchelli and A. Taglietti, J. Chem. Soc., Dalton Trans., 2003, 3471 RSC.
- K. Rurack, Spectrochim. Acta, Part A, 2001, 57, 2161 CrossRef CAS.
- O. S. Wolfbeis, in Biomedical Optical Instrumentation and Laser-Assisted Biotechnology, ed. A. M. Verga Scheggi, Kluwer Academic Publishers, Dordrecht, The Netherlands, 1996, p. 327 Search PubMed.
- R. Martinez-Manez and F. Sancenon, Chem. Rev., 2003, 103, 4419 CrossRef CAS.
- (a) K. J. Albert, N. S. Lewis, C. L. Schauer, G. A. Sotzing, S. E. Stitzel, T. M. Vaid and D. R. Walt, Chem. Rev., 2000, 100, 2595 CrossRef CAS; (b) W. Göpel, Sens. Actuators B, 1998, 52, 125 CrossRef; (c) U. Weimar and W. Göpel, Sens. Actuators B, 1998, 52, 143 CrossRef; (d) J. J. Lavigne and E. V. Anslyn, Angew. Chem., Int. Ed., 2001, 40, 3118 CrossRef; (e) A. D’Amico, C. Di Natale and R. Paolesse, Sens. Actuators B, 2000, 68, 324 CrossRef; (f) S. S. Schiffman and R. Gutierrez-Osuna, IEEE Spectrum, 1998, 1, 22.
- (a) L. Prodi, F. Bolletta, M. Montalti and N. Zaccheroni, Coord. Chem. Rev., 2000, 205, 59 CrossRef CAS; (b) C. Bargossi, M. C. Fiorini, M. Montalti, L. Prodi and N. Zaccheroni, Coord. Chem. Rev., 2000, 208, 17 CrossRef CAS; (c) M. Montalti, L. Prodi and N. Zaccheroni, in Handbook of Photochemistry and Photobiology, ed. H. S. Nalwa, American Institute of Physics, Stevenson Ranch, CA, USA, 2003, vol. 3, p. 271 Search PubMed; (d) L. Prodi, M. Montalti, N. Zaccheroni and L. S. Dolci, in Topics in Fluorescence Spectroscopy, eds. C. D. Geddes and J. R. Lakowicz, Springer, vol. 10, in press Search PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, 2nd edn., Kluwer Academic/Plenum Publishers, New York, 1999 Search PubMed.
- Near-Infrared Dyes for High Technology Applications, eds. S. Daehne, U. Resch-Genger and O. S. Wolfbeis, NATO-ASI Series, Kluwer Academic Publishers, Dordrecht, 1998 Search PubMed.
- (a) Applications of Fluorescence in Immunoassays, ed. I. A. Hemmilä, Wiley, New York, 2nd edn., 1991 Search PubMed; (b) I. Hemmilä and R. Harju, in Bioanalytical Applications of Labelling Technologies, eds. I. Hemmilä, T. Stählberg and P. Mottram, Wallacoy and EG&G Cie Pl, 1995, p. 124 Search PubMed; (c) N. Sabbatini, M. Guardigli and I. Manet, in Advances in Photochemistry, eds. D. C. Neckers, D. H. Volman and G. von Bünau, John Wiley & Sons, New York, 1997, vol. 23, p. 213 Search PubMed; (d) D. Parker, Coord. Chem. Rev., 2000, 205, 109 CrossRef CAS; (e) L. Prodi, S. Pivari, F. Bolletta, M. Hissler and R. Ziessel, Eur. J. Inorg. Chem., 1998, 1959 CrossRef CAS.
- (a) A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. Von Zelewsky, Coord. Chem. Rev., 1988, 84, 85 CrossRef CAS; (b) X.-Q. Guo, F. N. Castellano, L. Li, H. Szmacinski and J. R. Lakowicz, Anal. Biochem., 1997, 254, 179 CrossRef CAS; (c) X.-Q. Guo, F. N. Castellano, L. Li and J. R. Lakowicz, Anal. Chem., 1998, 70, 632 CrossRef CAS; (d) J. M. Kurner, I. Klimant, C. Krause, H. Preu, W. Kunz and O. S. Wolfbeis, Bioconjugate Chem., 2001, 12, 883 CrossRef CAS.
- R. Pinalli, F. F. Nachtigall, F. Ugozzoli and E. Dalcanale, Angew. Chem., Int. Ed., 1999, 38, 2377 CrossRef CAS.
- K. D. Schierbaum, T. Weiss, E. U. Thoden van Velzen, J. F. J. Engbersen, D. N. Reinhoudt and W. Göpel, Science, 1994, 265, 1413 CrossRef.
- F. C. J. M. Van Veggel, in Comprehensive Supramolecular Chemistry, eds. J. L. Atwood, J. E. D. Davies, D. D. MacNicol, F. Vögtle and D. N. Reinhoudt, Pergamon, Oxford, 1996, vol. 10, pp. 171–185 Search PubMed.
- J. Janata and M. Josowicz, Anal. Chem., 1998, 70, 179R CrossRef CAS.
- E. Bakker, P. Bühlmann and E. Pretsch, Chem. Rev., 1997, 97, 3083 CrossRef CAS.
- P. Bühlmann, E. Pretsch and E. Bakker, Chem. Rev., 1998, 98, 1593 CrossRef.
- V. Balzani, M. Venturi and A. Credi, Molecular Devices and Machines, Wiley–VCH, Weinheim, 2003 Search PubMed.
- R. Y. Tsien, Annu. Rev. Neurosci., 1989, 12, 227 CrossRef CAS.
- (a) L. A. J. Chrisstoffels, A. Adronov and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2000, 39, 2163 CrossRef CAS; (b) N. J. van der Veen, S. Flink, M. A. Deij, R. J. M. Egberink, F. C. J. M. van Veggel and D. N. Reinhoudt, J. Am. Chem. Soc., 2000, 122, 6112 CrossRef CAS; (c) O. S. Wolfbeis, Anal. Chem., 2000, 72, 81R CrossRef CAS; (d) J. M. Kürner, O. S. Wolfbeis and I. Klimant, Anal. Chem., 2002, 74, 2151 CrossRef CAS; (e) P. Yang, G. Wirnsberger, H. C. Huang, S. R. Cordero, B. Scott, M. D. McGehee, T. Deng, G. M. Whitesides, B. F. Chmelka, S. K. Buratto and G. D. Stucky, Science, 2000, 287, 465 CrossRef CAS; (f) B. J. Scott, M. H. Bartl, G. Wirnsberger and G. D. Stucky, J. Phys. Chem. A, 2003, 107, 5499 CrossRef CAS; (g) C. E. Fowler, D. Khushalani, B. Lebeau and S. Mann, Adv. Mater., 2001, 13, 649 CrossRef CAS.
- A. V. Bordunov, J. S. Bradshaw, X. X. Zhang, N. K. Dalley, X. Kou and R. M. Izatt, Inorg. Chem., 1996, 35, 7229 CrossRef CAS.
- (a) Molecular Luminescence Spectroscopy, ed. S. G. Schulman, John Wiley & Sons, Inc., London, 1985 Search PubMed; (b) F. E. Lytle, D. R. Storey and M. E. Juricich, Spectrocim. Acta, Part A, 1973, 29, 1357 Search PubMed; (c) K. Hiraki, K. Morishige and Y. Nishikawa, Anal. Chim. Acta, 1978, 97, 121 CrossRef CAS.
- L. Prodi, F. Bolletta, M. Montalti, N. Zaccheroni, J. S. Bradshaw, P. B. Savage and R. M. Izatt, Tetrahedron Lett., 1998, 39, 5451 CrossRef CAS.
- E. Bardez, I. Devol, B. Larrey and B. Valeur, J. Phys. Chem., 1997, 101, 7786 Search PubMed and references therein.
- G. Farruggia and L. Masotti, unpublished results.
- L. Prodi, C. Bargossi, M. Montalti, N. Zaccheroni, N. Su, J. S. Bradshaw, R. M. Izatt and P. B. Savage, J. Am. Chem. Soc., 2000, 122, 6769 CrossRef CAS.
- (a) E. M. Nolan and S. J. Lippard, J. Am. Chem. Soc., 2003, 125, 14270 CrossRef CAS; (b) K. Rurack, M. Kollmannsberger, U. Resch-Genger and J. Daub, J. Am. Chem. Soc., 2000, 122, 968 CrossRef CAS; (c) A. B. Descalzo, R. Martínez-Máñez, R. Radeglia, K. Rurack and J. Soto, J. Am. Chem. Soc., 2003, 125, 3418 CrossRef CAS and references therein.
- L. Prodi, M. Montalti, N. Zaccheroni, J. S. Bradshaw, R. M. Izatt and P. B. Savage, Tetrahedron Lett., 2001, 42, 2941 CrossRef CAS.
- S. Dobson, Cadmium–Environmental Aspects, World Health Organization, Geneva, 1992 Search PubMed.
- (a) L. Prodi, M. Montalti, J. S. Bradshaw, R. M. Izatt and P. B. Savage, J. Inclusion Phenom. Macrocycl.. Chem., 2001, 41, 123 CrossRef CAS; (b) R. T. Bronson, M. Montalti, L. Prodi, N. Zaccheroni, R. D. Lamb, N. K. Dalley, R. M. Izatt, J. S. Bradshaw and P. B. Savage, Tetrahedron, 2004, 60, 11139 CrossRef CAS; (c) G.-P. Xue, J. S. Bradshaw, N. K. Dalley, P. B. Savage, K. E. Krakowiak, R. M. Izatt, L. Prodi, M. Montalti and N. Zaccheroni, Tetrahedron, 2001, 57, 7623 CrossRef CAS.
- G. Xue, J. S. Bradshaw, N. K. Dalley, P. B. Savage, R. M. Izatt, L. Prodi, M. Montalti and N. Zaccheroni, Tetrahedron, 2002, 58, 4809 CrossRef CAS.
- (a) C. J. Frederickson, E. J. Krasarskis, D. Ringo and R. E. Frederickson, J. Neurosci. Methods, 1987, 20, 91 CrossRef CAS; (b) P. D. Zalewski, I. J. Forbes and W. H. Betts, J. Biochem., 1993, 296, 403 CAS.
- C. J. Fahrni and T. V. O’Halloran, J. Am. Chem. Soc., 1999, 121, 11448 CrossRef CAS.
- (a) K. Kikuchi, K. Komatsu and T. Nagano, Curr. Opin. Chem. Biol., 2004, 8, 182 CrossRef CAS; (b) P. Jiang and Z. Guo, Coord. Chem. Rev., 2004, 248, 205 CrossRef CAS; (c) C. J. Chang, E. M. Nolan, J. Jaworski, K.-I. Okamoto, Y. Hayashi, M. Sheng and S. J. Lippard, Inorg. Chem., 2004, 43, 6774 CrossRef; (d) D. A. Pearce, N. Jotterand, I. S. Carrico and B. Imperiali, J. Am. Chem. Soc., 2001, 123, 5160 CrossRef CAS; (e) H. A. Godwin and J. M. Berg, J. Am. Chem. Soc., 1996, 118, 6514 CrossRef.
- (a) L. Prodi, F. Bolletta, M. Montalti, N. Zaccheroni, A. Casnati, F. Sansone and R. Ungaro, New J. Chem., 2000, 24, 155 RSC; (b) A. Casnati, F. Giunta, F. Sansone, R. Ungaro, F. Bolletta, M. Montalti, L. Prodi and N. Zaccheroni, Supramol. Chem., 2001, 13, 419 CAS.
- A. Casnati, F. Sansone, A. Sartori, L. Prodi, M. Montalti, N. Zaccheroni, F. Ugozzoli and R. Ungaro, Eur. J. Org. Chem., 2003, 1475 CrossRef CAS.
- I. Costa, L. Fabbrizzi, M. Licchelli, P. Pallavicini, L. Parodi, L. Prodi, F. Bolletta, M. Montalti and N. Zaccheroni, J. Chem. Soc., Dalton Trans., 1999, 1381 RSC.
- L. Fabbrizzi, M. Licchelli, P. Pallavicini, A. Perotti and D. Sacchi, Angew. Chem., Int. Ed. Engl., 1994, 33, 1975 CrossRef.
- (a) Y. Zheng, J. Orbulescu, X. Ji, F. M. Andreopoulos, S. M. Pham and R. M. Leblanc, J. Am. Chem. Soc., 2003, 125, 2680 CrossRef CAS; (b) M. Berton, F. Mancin, G. Stocchero, P. Tecilla and U. Tonnellato, Langmuir, 2001, 17, 7521 CrossRef CAS; (c) R. Kramer, Angew. Chem., Int. Ed., 1998, 37, 772 CrossRef CAS; (d) A. Torrado, G. K. Walkup and B. Imperiali, J. Am. Chem. Soc., 1998, 120, 609 CrossRef CAS.
- (a) L. Prodi, F. Bolletta, M. Montalti and N. Zaccheroni, Eur. J. Inorg. Chem., 1999, 455 CrossRef CAS; (b) L. Prodi, M. Montalti, N. Zaccheroni, F. Dallavalle, G. Folesani, M. Lanfranchi, R. Corradini, S. Pagliari and R. Marchelli, Helv. Chim. Acta, 2001, 84, 690 CrossRef CAS.
- Y.-H. Li, L.-M. Chan, L. Tyer, R. T. Moody, C. H. Hirmel and D. M. Hercules, J. Am. Chem. Soc., 1975, 97, 3118 CrossRef CAS.
- S. Pagliari, R. Corradini, G. Galaverna, S. Sforza, A. Dossena, M. Montalti, L. Prodi, N. Zaccheroni and R. Marchelli, Chem.-Eur. J., 2004, 10, 2749 CrossRef CAS.
- F. Votgle and P. Knops, Angew. Chem., Int. Ed. Engl., 1991, 30, 658.
- (a) L. Prodi, F. Bolletta, M. Montalti, N. Zaccheroni, P. Huszthy, E. Samu and B. Vermes, New J. Chem., 2000, 24, 781 RSC; (b) L. S. Dolci, P. Huszthy, E. Samu, M. Montalti, L. Prodi and N. Zaccheroni, Collect. Czech. Chem. Commun., 2004, 69, 885 CrossRef CAS.
- (a) M. Montalti, L. Prodi, N. Zaccheroni, L. J. Charbonnière, L. Douce and R. Ziessel, J. Am. Chem. Soc., 2001, 123, 12694 CrossRef CAS; (b) L. J. Charbonnière, R. Ziessel, M. Montalti, L. Prodi, N. Zaccheroni, C. Boehme and G. Wipff, J. Am. Chem. Soc., 2002, 124, 7779 CrossRef CAS.
- (a) M. D. Best and E. V. Anslyn, Chem.-Eur. J., 2003, 9, 51 CrossRef CAS; (b) J. I. Bruce, R. S. Dickins, L. J. Govenlock, T. Gunnlaugsson, S. Lopinski, M. P. Lowe, D. Parker, R. D. Peacock, J. J. B. Perry, S. Aime and M. Botta, J. Am. Chem. Soc., 2000, 122, 9674 CrossRef CAS; (c) J. Lisowski, J. L. Sessler and T. D. Mody, Inorg. Chem., 1995, 34, 4336 CrossRef CAS; (d) J. Lisowski, J. L. Sessler, V. Lynch and T. D. Mody, J. Am. Chem. Soc., 1995, 117, 2273 CrossRef CAS.
- L. Prodi, M. Montalti, N. Zaccheroni, G. Pickaert, L. Charbonnière and R. Ziessel, New J. Chem., 2003, 27, 134 RSC.
- (a) P. Schuster, Proc. Natl. Acad. Sci. USA, 2000, 97, 7678 CrossRef CAS; (b) J. Drews, Science, 2000, 287, 1960 CrossRef CAS; (c) D. R. Liu and P. G. Schultz, Angew. Chem., Int. Ed., 1999, 38, 37 CAS; (d) J. K. M. Sanders, Phil. Trans. R. Soc. London, Ser. A, 2004, 362, 1239 CrossRef CAS.
- (a) V. Balzani, P. Ceroni, S. Gestermann, C. Kauffmann, M. Gorka and F. Vögtle, Chem. Commun., 2000, 853 RSC; (b) F. Vögtle, S. Gestermann, C. Kauffmann, P. Ceroni, V. Vicinelli and V. Balzani, J. Am. Chem. Soc., 2000, 122, 10398 CrossRef; (c) V. Balzani, P. Ceroni, S. Gestermann, M. Gorka, C. Kauffmann and F. Vögtle, J. Chem. Soc., Dalton Trans., 2000, 3765 RSC.
- (a) T. M. Swager, Acc. Chem. Res., 1998, 31, 201 CrossRef CAS; (b) D. T. McQuade, A. E. Pullen and T. M. Swager, Chem. Rev., 2000, 100, 2537 CrossRef CAS; (c) J. Kim, D. T. McQuade, A. Rose, Z. Zhu and T. M. Swager, J. Am. Chem. Soc., 2001, 123, 11488 CrossRef CAS; (d) C. N. Fleming, K. A. Maxwell, J. M. De Simone, T. J. Meyer and J. M. Papanikolas, J. Am. Chem. Soc., 2001, 123, 10336 CrossRef CAS.
- (a) M. Montalti, L. Prodi, N. Zaccheroni, R. Baxter, G. Teobaldi and F. Zerbetto, Langmuir, 2003, 19, 5172 CrossRef CAS; (b) M. Marcaccio, M. Margotti, M. Montalti, F. Paolucci, L. Prodi, S. Roffia and N. Zaccheroni, Collect. Czech. Chem. Commun., 2003, 68, 1395 CrossRef CAS; (c) M. Montalti, L. Prodi, N. Zaccheroni and G. Battistini, Langmuir, 2004, 20, 7884 CrossRef CAS.
- J. R. Lakowicz, C. D. Geddes, I. Gryczynski, J. Malicka, Z. Gryczynski, K. Aslan, J. Lukomaska, E. Matveeva, J. Zhang, R. Badugu and J. Huang, J. Fluorescence, 2004, 14, 425 CrossRef CAS and references therein.
- M. Montalti, L. Prodi, N. Zaccheroni and G. Falini, J. Am. Chem. Soc., 2002, 124, 13540 CrossRef CAS.
- M. Montalti, L. Prodi, N. Zaccheroni, A. Zattoni, P. Reschiglian and G. Falini, Langmuir, 2004, 20, 2989 CrossRef CAS.
- W. Stoeber, A. Fink and E. Bohn, J. Colloids Interface Sci., 1968, 26, 62 CrossRef.
- J. C. Giddings, Science, 1993, 260, 1456 CrossRef CAS.
- N. A. M. Verhaegh and A. van Blaaderen, Langmuir, 1994, 10, 1427 CrossRef CAS.
- See, for example: J. R. Lakowicz, J. Mailcka, J. Huang, Z. Gryczynski and I. Gryczynski, Biopolymers, 2004, 74, 467 Search PubMed.
- M. Montalti, L. Prodi and N. Zaccheroni, unpublished results.
- E. Brasola, F. Mancin, E. Rampazzo, P. Tecilla and U. Tonellato, Chem. Commun., 2003, 3026 RSC.
- A. W. Czarnik, Chem. Biol., 1995, 2, 631 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2005 |