The metathesis reactions: from a historical perspective to recent developments
Didier Astruc
Nanosciences and Catalysis Group, LCOO (CNRS UMR 5802), Université Bordeaux I, 351 Cours de la Libération, 33405, Talence cedex, France. E-mail: d.astruc@lcoo.u-bordeaux1.fr
First published on 21st December 2004
Abstract
Metathesis is one of the most spectacular recent improvements in synthetic strategies for organic synthesis and polymer science. The historical aspects and modern developments of the metathesis reactions are summarized here. In particular, emphasis is placed on the leading role played by the mechanistic work and proposals of Yves Chauvin and on the history of the efficient catalysts discovered by the groups of R. R. Schrock and R. H. Grubbs. It is pointed out how the Chauvin metathesis mechanism, with formation of a metallacyclobutane, has been generalized to many organometallic reactions that also involve square intermediates comprising a metal atom. Subsequently, the progressive development of ideas by Schrock and Grubbs during the last three decades has brought the field to the forefront of synthetic chemistry. The quest for efficient metathesis catalysts is a success story, starting from organometallic mechanisms, that has now invaded the worlds of organic synthesis and polymer science. Indeed, the Schrock’ and Grubbs’ catalysts and their derivatives are now the most efficient catalysts compatible with functional groups for the metathesis reactions. They considerably shorten synthetic schemes by affording new routes and therefore have changed the way chemists think about synthesis.
Introduction
Metathesis,1–9 with its multiple aspects, has become one of the most important chemical reactions and is now extremely useful. This area has gone beyond the research stage in inorganic and organometallic chemistry to develop in organic, medicinal, polymer and materials chemistry to such an extent that it has now become a familiar tool for the specialists of these fields. The goal of the present review article is to delineate the history of olefin metathesis while showing how pioneers have investigated the mechanism and catalysts, and to highlight the most recent aspects and implications. In particular, the unique disclosure by Yves Chauvin of the metathesis mechanism and the little known implication of his metallo-squares in most organometallic catalysis mechanisms (including alkane and alkyne metathesis) is emphasized. Then, how the considerable breakthroughs by Richard Schrock and Robert Grubbs in terms of catalyst discoveries and uses was made possible by initial syntheses and fundamental studies of transition-metal-alkylidene and alkylidyne complexes is illustrated in its historical context. Meanwhile, we wish to underline the role of the development of ideas and research efforts to lead to a success story in the advancement of chemistry and its applications. Finally, the review includes the most recent developments, applications and perspectives of metathesis with emphasis on catalyst design.Since the discovery of the Wilkinson–Osborn catalyst that allows the hydrogenation of olefins10a and its efficient asymmetric version by Kagan,10b,c considerable hope has been engaged in homogeneous catalysis because of its high selectivity, the perfect knowledge of the molecular mechanism leading to improvements and optimization, and the numerous medicinal applications. Among the many homogeneous catalytic reactions, those involving the formation of carbon-carbon bonds are of course essential in organic chemistry, as well as in polymer and materials science. Among these reactions, metathesis occupies a central role because it shortens many multi-step synthetic schemes and directly leads to valuable polymers.
Metathesis reactions: reaching an understanding
Metathesis: fragments changing place
The etymology of the word metathesis comes from the Greek μεταθεσιζ (metathesis) that means transposition. Thus, metathesis is invoked when, for instance, ions are exchanged in a solution containing two ion pairs in order to produce the most stable ion pairs [eqn. (1)].11 In the same way, two carbenes of an olefin can be exchanged to give, if they are different, another recombination leading to the two symmetrical olefins [eqn. (2)] or the two carbynes of an alkyne to give the two symmetrical alkynes [eqn. (3)].| A+B− + C+D− ⇌ A+D− + C+B− | (1) |
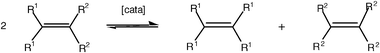 | (2) |
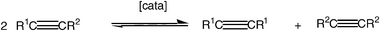 | (3) |
 | ||
| Fig. 1 History of the olefin metathesis reactions. | ||
The metathesis reactions are in principle under thermodynamic control, that is they are equilibrated, which clearly is an inconvenience. In fine chemical synthesis, this problem can be circumvented by choosing to carry out metathesis of a terminal alkene or alkyne. The formation of ethylene (respectively acetylene) displaces the reaction towards the product. This strategy also applies to olefins terminated by an ethylidene group, because metathesis then produces 2-butenes whose volatility also displaces the reaction towards the products. Operating under reduced pressure insures elimination of the volatile olefin in order to displace the metathesis reaction [eqns. (4) and (5)]. In fact, many metathesis reactions are under kinetic control. Note in passing that alkene metathesis is most often complicated by the formation of both Z and E isomers [eqn. (4)], whereas this problem does not exist in alkyne metathesis [eqn. (5)], disclosed for the first time by Blanchard, Mortreux and coworkers.14 The latter reaction with terminal bis-alkynes can thus selectively lead to cycloalkynes that selectively give Z-cycloalkenes after partial hydrogenation. Yet, alkyne metathesis is presently much less developed than alkene metathesis.5f
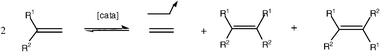 | (4) |
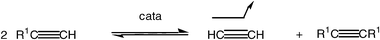 | (5) |
The Chauvin mechanism
At the end of the 1960’s, the metathesis reaction was very mysterious. Catalytic systems were either oxides such as WO3/SiO2 used in industry for the transformation of propene to ethylene and butenes or Ziegler–Natta derived systems such as WCl6 (or MoCl5) + AlXnR3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) −n or (SnR4). Mechanistic ideas had appeared, but they did not match the results of some metathesis experiments. For instance, Calderon had suggested a mechanism involving an intermediate π-cyclobutane-metal species,12 but the metathesis reaction does not give cyclobutane and metathesis catalysts do not lead to olefins by reaction with cyclobutane derivatives (Fig. 2).
−n or (SnR4). Mechanistic ideas had appeared, but they did not match the results of some metathesis experiments. For instance, Calderon had suggested a mechanism involving an intermediate π-cyclobutane-metal species,12 but the metathesis reaction does not give cyclobutane and metathesis catalysts do not lead to olefins by reaction with cyclobutane derivatives (Fig. 2).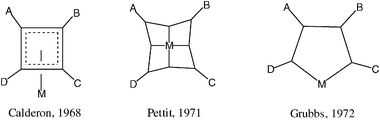 | ||
| Fig. 2 Representative proposals of intermediates for olefin metathesis that were later disproved. | ||
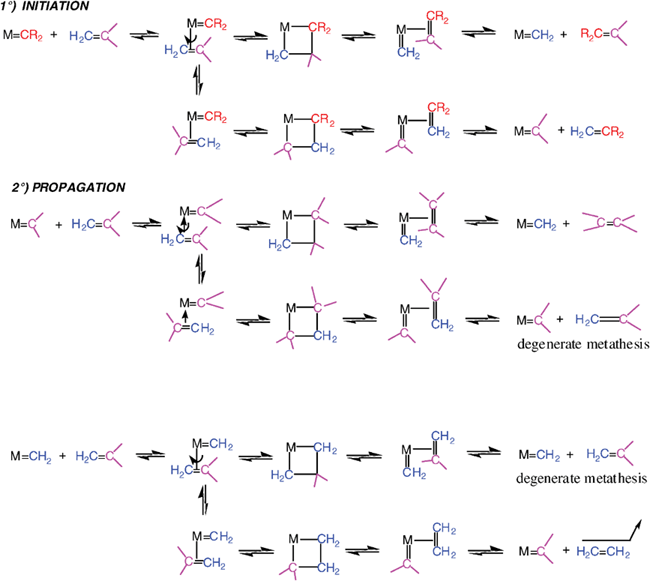
In the process of thinking about the metathesis mechanism, Yves Chauvin from the Institut Français du Pétrole, had taken into account the report of E. O. Fischer on the synthesis of a tungsten-carbene complex,15 that of Natta on the polymerization of cyclopentene by ring opening catalyzed by a mixture of WCl6 and AlEt316 and that of R. L. Banks and G. C. Bailey on the formation of ethylene and 2-butene from propene catalyzed by [W(CO)6] on alumina.12,13 Consequently, Yves Chauvin and his student Jean-Louis Hérisson published their proposition of metathesis mechanism in 1971 (Scheme 1).17
 | ||
| Scheme 1 Chauvin’s mechanism, proposed in 1971, for the catalyzed olefin metathesis involving metal alkylidene and metallacyclobutane intermediates. | ||
The latter involves a metal-carbene species (or more precisely metal-alkylidene), the coordination of the olefin onto the metal atom of this species, followed by the shift of the coordinated olefin to form the metallocyclobutane intermediate, and finally the topologically identical shift of the new coordinated olefin in the metallocyclobutane in a direction perpendicular to the initial olefin shift. This forms a metal-alkylidene to which the new olefin is coordinated, then liberated. This new olefin contains a carbene from the catalyst and the other carbene from the starting olefin. The new metal-alkylidene contains one of the two carbenes of the starting olefin and it can re-enter into a catalytic cycle of the same type as the first one. In fact, depending on the orientation of the coordinated olefin, the new catalytic cycle can give two different metallacyclobutenes, one leading to the symmetrical olefin and the other one leading the starting olefin. This latter cycle is said to be degenerate olefin metathesis. Thus, the catalytic cycles alternatively involve both metal-alkylidene species resulting from the combination of the metal with each of the two carbenes of the starting olefin.
Chauvin and Hérisson not only suggested the metallacyclobutane mechanism, but also published several experiments to confirm it. For instance, the reaction of a mixture of cyclopentene and 2-pentene led to C-9, C-10 and C-11 dienes in the ratio 1∶2∶1. Also, the reaction of a mixture of cyclooctene and 2-pentene led almost exclusively to the C-13 product. The latter reaction, but not the first one, was compatible with Calderon’s mechanism. In 1973, Chauvin published other results showing that the WCl6 + MeLi mixture catalyzes the formation of propene by reaction of 2-butene, which was proposed to proceed via methylation of tungsten, followed by the α-elimination in the tungsten-carbon bond of W–CH3 to form W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 (H), then metathesis.18
CH2 (H), then metathesis.18
The importance of the Chauvin mechanism for overall organometallic catalysis
Chauvin’s mechanism introduced several new ideas. First, it proposed the implication of a metal-carbene complex to initiate the catalysis of the metathesis reaction. This idea first suggested that one could just synthesize metal-alkylidene complexes and let them react as catalysts with olefins to carry out the metathesis reaction. Of course, many authors later engaged in such research directions, first delineated by Chauvin. The induction time was very long, however. Relatively few chemists became interested in such a route in the first half of the decade following Chauvin’s proposal. On the contrary, other mechanistic hypotheses were proposed well after Chauvin’s publication. It is true that only Fischer-type transition-metal-carbene complexes stabilized by the presence of an heteroatom on the carbenic carbon atom such as [LnWC–OR(R′)] were known at that time.15 This type of carbene complex often does not catalyze the metathesis reaction as is now known, but gives the cyclopropanation of olefins by reductive elimination within the metallacyclobutane intermediate. Several years after Chauvin’s publications, some authors showed that tungsten-carbene complexes also stabilized by heteroatoms are active in olefin metathesis. The first result of this kind was published by Casey and Burkhardt who showed that [W(CO)5(CPh2)] reacts with isobutene to form 1,1′-diphenylethylene [eqns. (6) and (7)]20a, and Chauvin himself reported in 1976 that even some Fischer-type carbene could promote metathesis.18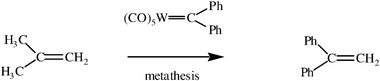 | (6) |
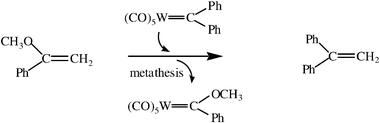 | (7) |
Another very important aspect of the Chauvin mechanism concerns the intermediacy of the metallacyclobutane. Such metallocyclobutane complexes are sometimes stable, and some stable metallacyclobutenes have indeed been shown to be involved in metathesis. Elegant studies by Grubbs’ group showed that Tebbe’s complex [Cp2Ti(CH2)(ClAlMe2)]19a reacts with olefins in the presence of dimethylaminopyridine to give titanacyclobutanes that slowly catalyze metathesis and could be used to identify all the intermediates in olefin metathesis (Scheme 2).19
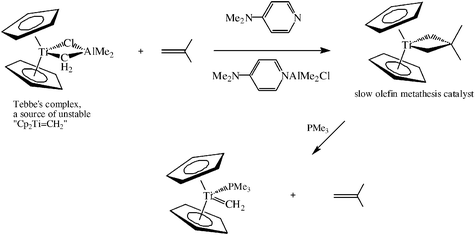 | ||
| Scheme 2 Grubbs’ studies on Tebbe’s complex, a source of TiCH2 species, showing the formation of titanacyclobutanes. These metallocycles are slow metathesis catalysts that allowed the observation of the intermediates in olefin metathesis. | ||
The square containing the transition metal and formed by the shift of the olefin coordinated to the metal in the metal-alkylidene species is not only involved in alkene metathesis, but also in many other catalytic organometallic mechanisms. Indeed, the metathesis of alkynes and the metathesis polymerization of cycloalkenes and alkynes formulated by Katz are completely analogous.20b–d Moreover, it is possible to represent by a metallo-square scheme the mechanisms of σ-bond metathesis and β-elimination. Scheme 3 gathers together the different organometallic reactions involving a metallo-square.
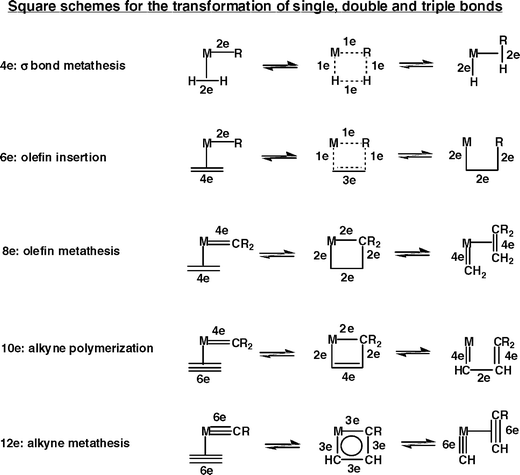 | ||
| Scheme 3 Various organometallic reactions involving the intermediacy of Chauvin-type metallo-square schemes. | ||
From Wilkinson’s metal-alkyl complexes to Schrock’s alkylidene and alkylidyne complexes
From the middle of the XIXth century to the middle of the XXth century, chemists believed that metal-alkyl compounds were intrinsically unstable, because of the supposedly too low energy of the metal-carbon bond. Geoffrey Wilkinson then synthesized stable binary metal-alkyl complexes that did not contain β-hydrogen, showing that this instability was in fact kinetic, due to β-elimination, because chemists had been trying to make binary metal-ethyl complexes.21Organometallic chemists could then synthesize a whole series of thermally stable binary (and other) metal-alkyl complexes with alkyl groups lacking β-hydrogens, such as methyl, benzyl, neopentyl, trimethylsilylmethyl and mesityl, even if the metal had necessarily (for binary compounds) less than 18 valence electrons in the valence shell, in conflict with Sidgwick’s 18-electron rule (Fig. 3).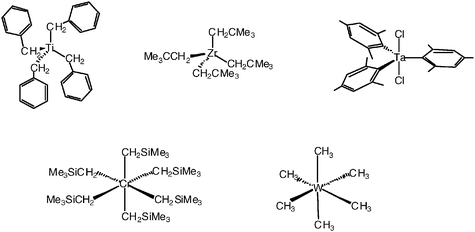 | ||
| Fig. 3 Transition-metal-alkyl complexes owing their stability to the lack of β-hydrogens, despite numbers of metal valence electrons that are much lower than 18. | ||
Richard Schrock was, at Harvard, a Ph.D. student of John Osborn, who himself had been a Ph.D. student of Geoffrey Wilkinson, who was at Imperial College, London, after Harvard had turned down his promotion for tenure. All chemists know how influential scientific filiations are for the determination of one’s scientific area and ideas. Indeed, the influence of Wilkinson on his scientific grandson Dick Schrock turned out to be noteworthy: Schrock, then at Du Pont, tried to synthesize [Ta(CH2CMe3)5], which would not contain β-hydrogens and thus, according to this principle, should have been stable. Good luck smiles only on good scientists, and the expected compound did not form. An α-elimination reaction by σ-bond metathesis occurred while he was attempting to coordinate the fifth neopentyl group, which produced one mole of neopentane and led to the isolation of the first stable metal-alkylidene complex, [Ta(CH2CMe3)3(CHCMe3)], which has the high oxidation state of V (Scheme 4).
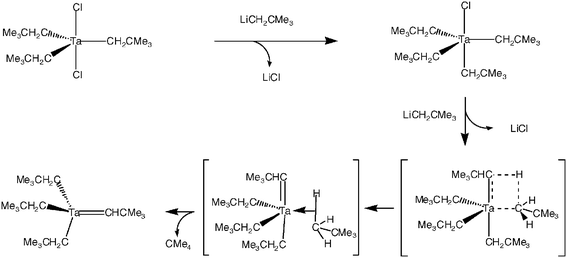 | ||
| Scheme 4 Mechanism involving σ-bond metathesis for the α-elimination observed by Schrock in the first synthesis of a stable metal-alkylidene complex. | ||
Schrock then synthesized other high oxidation state niobium- and tantalum-alkylidene and -alkylidyne complexes by various routes, including the first methylene complex, [TaCp2(CH3)(CH2)], and characterized all these complexes by X-ray crystal structures and the very low-field proton and carbon NMR signals of the carbene and carbyne ligands.22
We were then in the mid-1970’s and yet the metathesis reaction had to wait, because none of these alkylidene complexes catalyzed the metathesis of olefins. In fact, the metallocyclobutanes that did form upon reaction of the alkylidene complexes with olefins followed the ordinary β-elimination pathway of metal-alkyl complexes that had less than 18 valence electrons on the metal and contained β-hydrogens. The site liberation, however, would produce catalytically active species and these alkylidene complexes were shown by Schrock to be catalysts for the dimerization of olefins. Two equivalent olefins coordinate to the unsaturated metal center, giving a metallocyclopentane whose β-elimination gives metal butenyl hydride intermediates that reductively eliminate to form 1-butene (Schemes 5 and 6).24 Note that, in the same vein, Yves Chauvin had discovered extremely efficient and selective titanium olefin dimerization catalysts.25
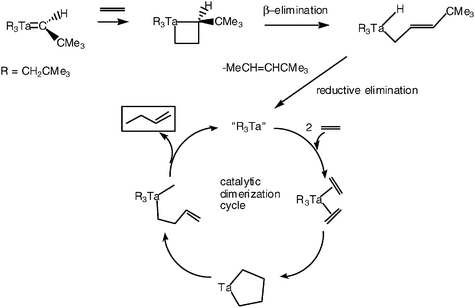 | ||
| Scheme 5 Catalytic dimerization disclosed by Schrock subsequent to the reaction of a tantalum-alkylidene complex with an olefin.24 See also Chauvin’s work for a more efficient titanium catalyst for olefin dimerization proceeding by the same mechanism.25 | ||
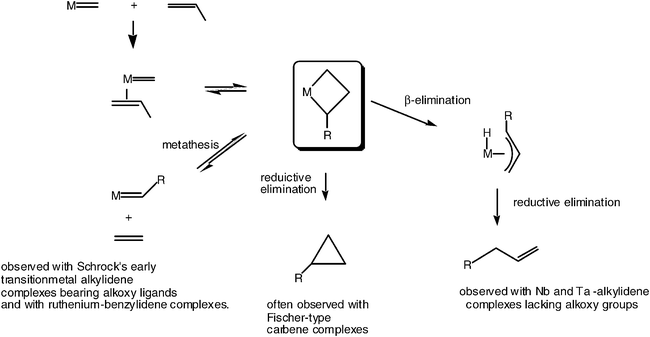 | ||
| Scheme 6 The three modes of evolution of a metallacyclobutane complex formed by reaction of a transition-metal-alkylidene species and an olefin. | ||
It was in 1980 that Dick Schrock’s group at MIT reported a tantalum-alkylidene complex, [Ta(CH–t-Bu)Cl(PMe3)(O–t-Bu)2], which catalyzed the metathesis of cis-2-pentene.22e This provided the very first proof for Chauvin’s mechanism of olefin metathesis with a well-defined high oxidation state alkylidene complex, almost a decade after Chauvin’s proposal. The reason that this complex catalyzes the metathesis reaction, whereas the other members of the family of niobium- and tantalum-alkylidene complexes failed to do so, was the presence of alkoxide ligands. Earlier, other catalysis experiments had been carried out with [W(CO)5(CPh2)], but as we know that almost any tungsten-containing molecular compound catalyzes alkene metathesis, eventually after decomposition of the precursor, these experiments cannot be considered as really significant. Molybdenum and tungsten, however, were obviously the most active metals in alkene metathesis and, around 1980, Schrock and his group considerably increased their efforts in the search for stable molecular alkylidene and alkylidyne complexes of these metals that would catalyze the metathesis of unsaturated hydrocarbons. This search was successful22f and eventually produced a whole family of molybdenum- and tungsten-alkylidene complexes of the general formula [M(CHCMe2Ph)(N–Ar)(OR2], R being bulky groups. These compounds presently are the most active alkene metathesis catalysts. (Fig. 4).23
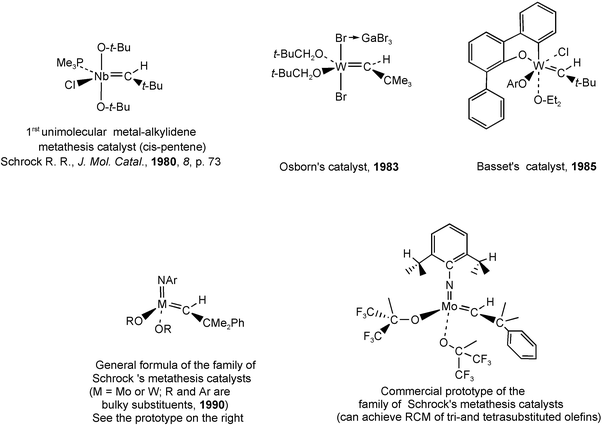 | ||
| Fig. 4 First Nb molecular catalyst and main families of molecular Mo and W metathesis catalysts. | ||
By 1980, Schrock’s group had also reported an active tungsten-alkylidyne catalyst for alkyne metathesis.22f Other chemists such as John Osborn in Strasbourg26 and Jean-Marie Basset27 in Lyon played an important role in the history of metathesis by reporting tungsten complexes that were active as olefin metathesis catalysts in the 1980’s (Fig. 4). Osborn reported early well-defined W(VI) alkylidene metathesis catalysts (Fig. 4) and showed the living character of the polymerization system and the intermediacy of a tungstacyclobutane by 1H NMR spectroscopy. Basset reported aryloxoalkoxoalkylidene W(VI) catalysts and one of the first examples of Lewis-acid-free initiators that allowed the polymerization of substituted norbornenes following the ROMP mechanism.
The advantage of Schrock’s catalysts, whose most efficient members were reported in 1990, was that even though they are extremely active, they are molecular (without additives) and also provided a commercial catalyst and chiral versions for the first examples of asymmetric metathesis catalysis.28
Schrock’s closely related Mo-alkylidyne complexes do not react with olefins, but they selectively and efficiently metathesize alkynes without the need for a co-catalyst. For instance, the prototype [W(CCMe3)(O–t-Bu)3] effects several hundred turnovers per minute under mild conditions. Some reactions even proceed at 25 °C. Here again, the alkoxide ligands are crucial for metathesis.
Grubbs’ pragmatic, then efficient mechanistic and synthetic approach to useful metathesis catalysts
Two classes of metal-alkylidene complexes are usually distinguished: those containing a nucleophilic carbene of the Schrock type and those with an electrophilic carbene of the Pettit type.29 This other talented American chemist was interested in methylene complexes, because they would be organometallic models for the implication of methylene intermediates in the Fischer–Tropsch process. Thus, in 1966, he reported the generation of the elusive species [FeCp(CO)2(CH2)]+, although characterization of this species only included its reactivity.30a Later, the replacement of Cp (η5-C5H5) by Cp* (η5-C5Me5) allowed us to characterize the rotation barrier about the FeCH2 bond by NMR.30b A related Fischer-type ruthenium complex, [RuCp{C(Me)OMe}(CO)(PCy3)][PF6], stabilized by a methoxy group on the carbene carbon, was synthesized by Malcolm Green’s group at Oxford in 1971. This was the first reported ruthenium-carbene complex.31 The reactivity of this type of iron and ruthenium complexes towards olefins is again cyclopropanation, because of the strongly electrophilic character of the carbene ligand due to the positive charge, further increased by the electron-withdrawing carbonyl ligands. The success of Grubbs’ approach to stable benzylidene complexes containing the electrophilic benzylidene ligand may appear, by comparison, somewhat surprising, but it is due to the neutrality of the complexes, thus affording a considerably reduced electrophilicity of the carbene ligand, and the great versatility of ruthenium, a magic metal in inorganic and organometallic chemistry.Grubbs had been interested for a long time in the metathesis reaction, as indicated by his mechanistic proposal of a metallocyclopentane intermediate early on.32 He had noticed Natta’s 1965 publication on the catalysis by RuCl3 of the polymerization of cyclobutene and 3-methylcyclobutene by ring opening.33 This process (in butanol) had been developed by Norsorex. In this context, the Ziegler–Natta polymerization of olefins under mild conditions obviously had a considerable impact on polymer chemistry. The delineation of a new polymerization mechanism, however, was not a simple task. Well-inspired by this approach, Grubbs published in 1988 the polymerization of 7-oxanorbornene into a high molecular weight monodisperse polymer (Mw = 1.3 × 106 g mol−1; Mw/Mn = 1.2) by RuCl3 or [Ru(H2O)6] (OTs)2 (OTs = toluene sulfonate). This catalysis was all the more remarkable as it was conducted in water.34a Shortly afterwards, he could show, in the course of the same reaction, the formation of a Ru-alkylidene intermediate, then the polymerization of cyclooctene, an olefin with little constraints, when the alkylidene ligand source was ethyl diazoacetate added to the aqueous solution of [Ru(H2O)6] (OTs)2.34
Consecutively and according to the same logic, a great step forward was accomplished by Grubbs in 1992. He reported the first molecularly well-defined ruthenium-carbene complex that promoted the ROMP of low-strain olefins as well as the catalytic RCM of functionalized dienes. Grubbs showed that these vinylidene complexes, [RuCl2(PR3)(CH–CHCPh2)] (R = Ph or Cy), were efficient molecular catalysts for these polymerization reactions (Scheme 7) and other metathesis reactions such as those involving ring closing of terminal diolefins.34b–d
 | ||
| Scheme 7 Metathesis mechanism for the ring-opening metathesis polymerization (ROMP) of a cyclic olefin. | ||
Interestingly, Noels’ group reported, also in 1992, the Ru-catalyzed ROMP of cycloolefins initiated by diazoesters.34e In 1995, this group showed that addition of such diazoesters to [Ru(η6-cymene)PR3] (R = Cy or t-Bu) produces very active arene-free ruthenium-carbene catalysts in which the carbene proton could be observed by 1H NMR, shedding light onto the catalyst structure.34f In 1995, the new molecularly well-defined catalysts [Ru(CHPh)Cl2(PR3)2], R = Ph or Cy, whose structures are closely related to the vinylidene ones published three years earlier, appeared and were commercialized with R = Cy. [Ru(CHPh)Cl2(PCy3)2] is now known as the first-generation Grubbs catalyst and is still today the most used metathesis catalyst by organic chemists, because of its stability to air and compatibility with a large variety of functional groups (except for amines and nitriles and basic media).34d The best organometallic research groups also offered astute alternative syntheses to Grubbs catalysts.35
Fine mechanistic studies with this catalyst led Grubbs’ group to conclude that the mechanism first involved the dissociation of one phosphine to generate the reactive 14-electron ruthenium intermediate. In order to accelerate this dissociative step, Grubbs introduced, in place of one phosphine, one of Arduengo’s cyclic bis-amino carbene ligands that are relatively stable, even in the free forms obtained by deprotonation of the corresponding imidazolium cation.36 These ligands are excellent σ-donors without π-acceptor properties and have been known for several decades, but they only recently have become very popular in organometallic chemistry and catalysis.36a It is Herrmann’s group that first synthesized ruthenium complexes with two such carbene ligands in the context of the catalysis of olefin metathesis, but their catalytic activity was shown to be modest.36d In Grubbs’ first generation catalysts containing only one such ligand, they increase the electron density at the ruthenium center, however, and their trans effect labilizes the ruthenium-phosphine bond, favoring phosphine dissociation. Thus, the second generation of “Grubbs catalysts” [RuCl2{C(N(mesityl)CH2)2}(PCy3)(CHPh)] and its catalytic activity in metathesis were successively proposed within a few months by the groups of Nolan,37c Grubbs,37b,d–h and Fürstner and Herrmann.37c It is presently the most used catalyst for efficient cross-metathesis reactions, although it is not tolerant to amines and nitriles (for instance, with acrylonitrile, Schrock’s catalyst is efficient, in contrast to the ruthenium catalysts). On the contrary, this new, commercially available, catalyst is even more active although it is also more thermally stable than the first one (Fig. 5).
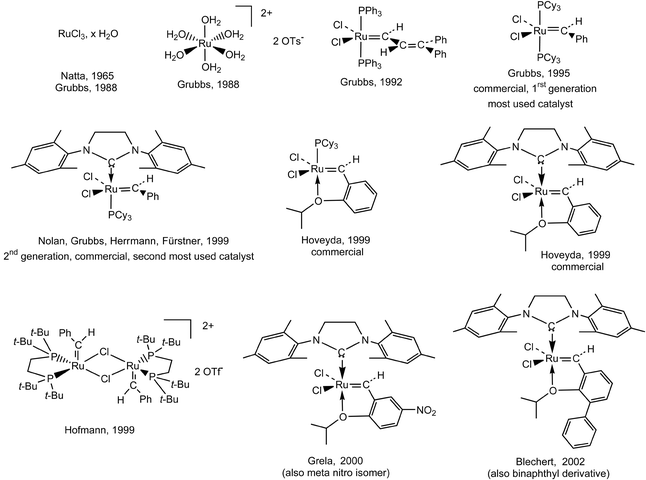 | ||
| Fig. 5 Grubbs-type (or derived) ruthenium metathesis catalysts, air-stable and compatible with most functional groups. | ||
Along this line, Hoveyda,38 Hofmann,41 Grela14i and Blechert39 reported other related, very active, stable and functional-group tolerant ruthenium metathesis catalysts. The first Hoveyda metathesis catalyst is derived from Grubbs’ first generation catalysts. It bears only one phosphine and a chelating carbene ligand. The second one bears, in addition, Arduengo’s carbene instead of the phosphine. Both catalysts are now commercially available, although expensive. Grela recently reported variations of the Hoveyda catalyst with increased efficiency (active even at 0 °C) when the aryl group of the benzylidene ligand bears a nitro group in the meta or para positions or two methoxy substituents (Fig. 5). Grela’s successful idea was to destabilize the Ru–O(ether) bond in order to favor the ether decoordination that generates the catalytically active 14-electron species.38b The family of Hoveyda catalysts, whose activity compares with that of the second generation Grubbs catalyst, are especially useful for difficult cases of metathesis of polysubstituted olefins and selective cross metathesis (CM) in which homo-coupling needs be avoided.38 The most successful variation of these Ru-benzylidene catalysts so far was reported by Blechert39 whose strategy to sterically destabilize the Ru–O(ether) bond was to introduce an aryl (phenyl or naphthyl) substituent on the benzylidene aryl in the ortho position relative to the O(ether). The catalytic efficiency and stability of Blechert complexes surpasses those of all the other Ru catalysts, although it has been shown several times that the catalytic efficiency depends on the type of metathesis reaction examined and the tolerance towards the required functional group (Fig. 5).39b
As an example, Janine Cossy demonstrated that the CM between allylsilanes and α,β-unsaturated carbonyl compounds is catalyzed by Hoveyda’s second catalyst with excellent stereoselectivity.44b,c Peter Hofmann’s catalyst, also a very active one, was obtained by chloride abstraction, providing a dicationic dimer from a ruthenium analog bearing a cis-diphosphine.41 Indeed, Grubbs’ success story has encouraged the search for ruthenium metathesis catalysts and other ruthenium structures or variations have been published,1,37–43 including ruthenium catalysts containing other ligands,39–49 water-soluble ones,46 reactions carried out in ionic liquids,47,48 on solid supports7b,38c and dendrimers.40,45,49
With the dendrimer strategy, the challenge is to locate ruthenium catalysts that are at the same time robust enough to form stable metallodendrimers and yet active in metathesis catalysis. Such a compromise could be successfully achieved with chelating phosphines that strongly attach ruthenium to dendrimers of generations 1 to 4 (with 4, 8, 16 and 32 dendritic branches). The solubility of the last generation complex is weak because of the steric bulk at the periphery, but that of the first free generations is good in common solvents. Interestingly, the rate of the ring-opening-metathesis polymerization of norbornene using these metallodendritic catalysts is much higher for the metallodendrimers than for the monometallic model complex, probably because of the more facile phosphine decoordination in the dendrimer than in the model, consistent with relative stabilities and DFT calculations, but the dendritic effect is then negative in terms of rates because of the inhibiting steric effect as the metallodendrimer generation increases (Scheme 8).
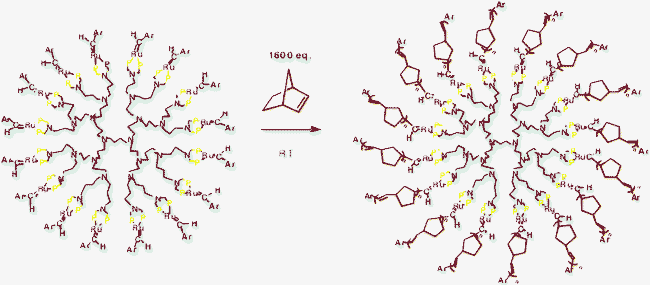 | ||
| Scheme 8 Formation of dendritic stars by ROMP of generations 1 to 3 metallodendritic ruthenium benzylidene complexes (generation 3 shown here; the two chloride ligands on the Ru atoms are omitted for clarity; the ROMP is faster with the metallodendrimers than with a monometallic model).45 | ||
The multiple applications of olefin metathesis
Olefin metathesis is very useful in industry given the large quantities of hydrocarbon handled and the need for propylene, produced by metathesis from ethylene and 2-butene, is enormous. Unimolecular, sophisticated functional-group-tolerant catalysts are not necessary, however. Industry still uses heterogeneous metathesis catalysts for propylene production. Likewise, complex homogeneous mixtures that are catalytic active for polymerization are used, but not the molecular Schrock and Grubbs catalysts.The great popularity of the Schrock and Grubbs catalysts that has led to their widespread use in organic chemistry is due to their tolerance of a large variety of functional groups, combined with their efficiency and, for Grubbs catalysts, their ease of handling in air. Six types of metathesis reactions are known and all of them can be catalyzed by Schrock and Grubbs metathesis catalysts. They can facilitate extensive organic transformations, including the synthesis of low-dispersity polymers (Scheme 9).
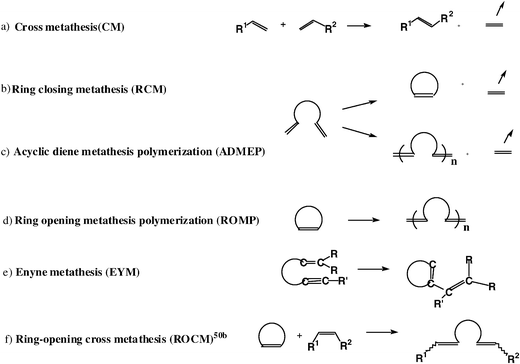 | ||
| Scheme 9 Different types of olefin metathesis, all proceeding according to the Chauvin mechanism and catalyzed by Schrock-type or Grubbs-type metathesis catalysts. Tandem, domino and cascade metathesis reactions couple several of these reactions (in particular ROMP + RCM). | ||
The most popular reaction among organic chemists is the ring-closing metathesis (RCM) of terminal diolefins, which can be achieved under ambient conditions in air using the first generation Grubbs catalyst. The easiest reaction of this kind is the formation of five-membered rings such as cyclopentenes and heterocyclic analogs, but it is also relatively easy to form large rings if terminal diolefins are used as precursors,50 for instance, for the synthesis of Sauvage’s molecular knots [eqn. (8)].50b
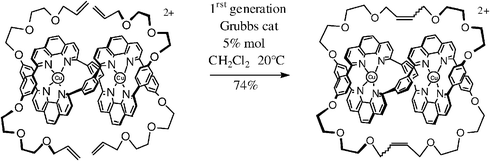 | (8) |
A recent application, only possible using the second generation Grubbs catalyst, disclosed by our group was the synthesis of organobimetallic and organic cyclophane capsules in two steps from the simple mesitylene sandwich complex [FeCp(η6-mesitylene)][PF6]. First, the CpFe+-induced nona-allylation under ambient conditions using KOH and allyl bromide yields [FeCp{η6-1,3,5-C6H3(C(allyl)3)3}][PF6]. Metathesis of this complex or its free arene ligand yields the capsules in one pot,51 resulting from nine metathesis reactions including six RCM and three CM reactions (Scheme 10).51a
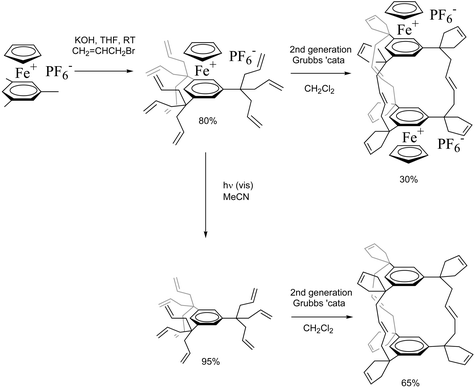 | ||
| Scheme 10 Formation of organic and organometallic cyclophane capsules by triple RCM + triple CM. | ||
Superb examples of RCM in the metal coordination sphere have been reported by Gladysz and his research group.51b
Enantioselective metathesis catalysis is presently another major challenge. The first example of a chiral metathesis catalyst was reported by Schrock in a 1993 reaction and the first example of very efficient enantioselective ROMP was published by Shrock’s and Hoveyda’s groups using a chiral molybdenum catalyst in 1998;2b it was later followed by another example reported by Grubbs’ group. Examples of enantioselective RCM reaction are becoming numerous [see, for instance, the synthesis of (+)-brevomicin in eqn. (9)52]. Since 1998, Schrock and Hoveyda have published several other examples, in particular cascade or domino enantioselective reactions by ROMP followed by RCM [eqn. (10)].28
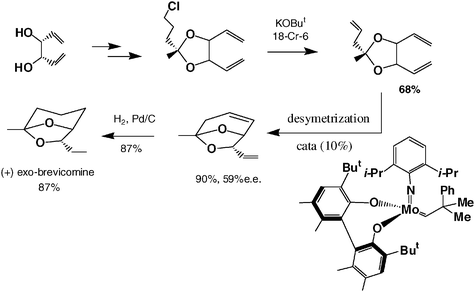 | (9) |
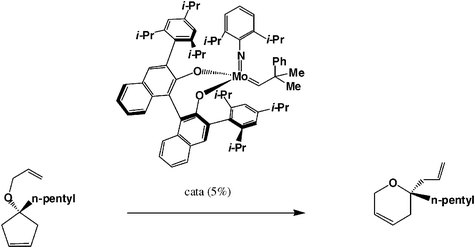 | (10) |
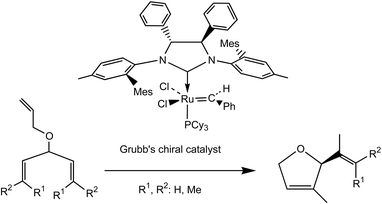 | (11) |
Recent developments and perspectives
σ-Bond metathesis
The metathesis of single C–H and C–C bonds and of double and triple carbon-carbon bonds forms a unified field of catalytic organometallic chemistry that is expanding. Indeed, Basset and his group have disclosed well-defined silica-supported high-oxidation-state alkylidene complexes such as [(SiO)xTaV(CH–t-Bu)(CH2–t-Bu)3−x] inspired from Schrock’s alkylidene complexes. These compounds react with alkanes at 150 °C to give both C–H and C–C cross metathesis products resulting from the reactions between the neopentylidene fragment and the incoming alkanes (Fig. 6). For instance, the metathesis of propane yields ethane and butane.54 In Basset’s σ-bond metathesis chemistry, the silica-supported d0-metal-alkyl complexes are readily hydrogenized by σ-bond metathesis to very active silica-supported d0-metal-hydrides. The latter react with alkanes by σ-bond metathesis to yield dihydrogen and silica-supported d0-metal-alkyls. Silica-supported d0-metal-polyhydride-alkyls generated by hydrogenolysis of silica-supported d0-metal-polyalkyls readily reductively eliminate dihydrogen at such high temperatures to give silica-supported d2-metal-alkyls. The latter can now undergo α-elimination to silica-supported d0-metal-alkylidene-hydrides or β-elimination to silica-supported d0-metal-hydride-alkenes. Thus, silica-supported metal-alkylidenes are generated and, very interestingly, Basset’s group has recently proposed, based on stereochemical experiments, that alkane disproportionation by silica-supported d0-metal-hydrides follows the same mechanism, with metal-alkylidene metallacyclobutane intermediates, as in olefin metathesis. It should be noted that silica-supported d0-metal-alkyls do not directly react with alkanes by σ-bond metathesis of C–C bonds, because electrophilic early transition metals would then have to form metallo-squares in which the β carbon would be pentacoordinated with a fractional negative charge, a highly unfavorable situation.54![Hydrogenolysis and methanolysis by σ-bond metathesis for alkane disproportionation disclosed by Basset’s group with the silica-supported catalyst [(SiO)xTav(CH–t-Bu)(CH2--t-Bu)3 − x].](/image/article/2005/NJ/b412198h/b412198h-f6.gif) | ||
| Fig. 6 Hydrogenolysis and methanolysis by σ-bond metathesis for alkane disproportionation disclosed by Basset’s group with the silica-supported catalyst [(SiO)xTaV(CH–t-Bu)(CH2--t-Bu)3−x]. | ||
Catalyst design for olefin metathesis (see also the supported catalysts below)
Most of the present research focuses on the modification and attempts to improve Grubbs’ ruthenium catalysts. Catalyst design from this starting point43 involves perfluorinated aryloxide ligands to replace the chlorides,43g modifications using Fischer-type carbene ligands,41c the use of chelating bis-phosphines43i and modifications of Hoveyda’s catalyst,14g as well as active on-going studies in the Grubbs group.Alkyne metathesis
High-oxidation-state Schrock carbyne complexes, [M(CR)(OR′)3] (M = Mo or W; OR′ = bulky and electron withdrawing),55 and Fürstner’s analogous tris-amido complexes56 are the only unimolecular catalysts for alkyne metathesis; no unimolecular low-oxidation-state catalyst is known. Thus, it is believed that the Mortreux system, [Mo(CO)6] + a phenol, and its recently improved versions by the Bunz, Grela–Ignatowska and Lavigne–Chauvin groups (with p-ClPhOH, p-F3CPhOH and o-FPhOH),14 involve [Mo(CR)(OAr)3] (where R and Ar are bulky groups) with the alkylidene ligand coming from CO or the alkyne. Fürstner’s very efficient bulky triamido Mo precatalyst [Mo{NR(Ar)}3Cl] is generated by addition of CH2Cl2 to Cummin’s complex [Mo{NR(Ar)}3], but the way it reacts with alkynes is unknown. For instance, the methylidyne complex [Mo{NR(Ar)}3(CH)], also formed along with the precatalyst, does not sustain catalytic turnover. The precatalyst [Mo{NR(Ar)}3Cl] exhibits a remarkable tolerance towards many polar functional groups, unlike Schrock’s prototype [W(CC–t-Bu)(O–t-Bu)3]. Thus, Fürstner has successfully explored synthetic applications of alkyne metathesis in organic chemistry using this catalyst precursor.56Acyclic diyne metathesis (ADIMET), developed by Bunz, has produced polymer materials with various interesting physical properties and Bunz’s recent version of the Mortreux-type catalyst pre-activated with heptyne works at only 30–60 °C higher than the Schrock and Fürstner catalysts.14f Improvement of the Mo(CO)6 catalyst with 2-flurorophenol was reported by Grela and Ignatowska,14c and improvement using [Mo(CO)6] + p-chlorophenol and a polyether (best efficiency: 1,2-diphenylethane) over a bed of molecular sieves, leading to metathesis of phenylpropene at 50 °C, was recently published by the Lavigne–Chauvin group.14h Moore has synthesized highly reactive Mo(VI) alkylidene catalysts by a reductive recycle strategy.14k Then, he very recently reported the use of EtCMo[NAr(t-Bu)]3 for the remarkable synthesis of arylene ethynylene macrocycles by precipitation-driven alkyne metathesis.14m It is likely that discrete alkylidene W and Mo complexes on silica prepared by Basset’s group will also produce useful catalysts that are intermediate between homogeneous and heterogeneous with the advantages of both types. Metathesis of 2-pentyne by the silica-supported alkylidyne-alkylidene complex [(SiO)(Re(C–t-Bu)(CH–t-Bu)(CH2--t-Bu)] has indeed already been reported.57,58
Metathesis in water
ROMP was shown to be faster than slow decomposition of Grubbs’ catalyst in water, which now allows living ROMP and polymerization of heptadiynes.59 The Hoveyda–Grubbs catalyst was attached to a water-soluble, amphiphilic block copolymer based on poly(2-oxazoline)s. Recycling was successful and the micellar conditions accelerated the conversion of the hydrophobic diene, yielding a turnover number of up to 390 for the RCM of diethyldiallylmalonate.60aSupported metathesis catalysts
This area presently concentrates a good deal of the research activity on metathesis. The main issues are the access to highly active catalysts, their separation and recycling, and the removal of catalysts from products. Mo-, Ru- and Re-based metathesis catalysts have been immobilized onto soluble polymers such as poly(ethylene)glycol (PEG) and poly(7-oxanorborn-2-ene), on the supported polymers polystyrene-divinylbenzene and on silica. Immobilization was achieved by halogen, phosphine or alkylidene exchange, or via the N-heterocyclic carbene of the second generation Grubbs catalyst. Most classic supported catalysts (except those involving the silica ligand, vide infra) were reported for ruthenium catalysts, because they are easier to handle in air and water (Scheme 11). The first enantiomerically pure solid-supported catalyst was published in 2002 by the Schrock–Hoveyda groups and gives similar enantioselectivity as the monomeric complexes, although the reactions are slower, presumably due to inefficient diffusion of substrate molecules into the polymer.28e This area of supported metathesis catalysts has recently been reviewed by Buchmeister,7b who also is the author of a review article on ROMP in 2000.7a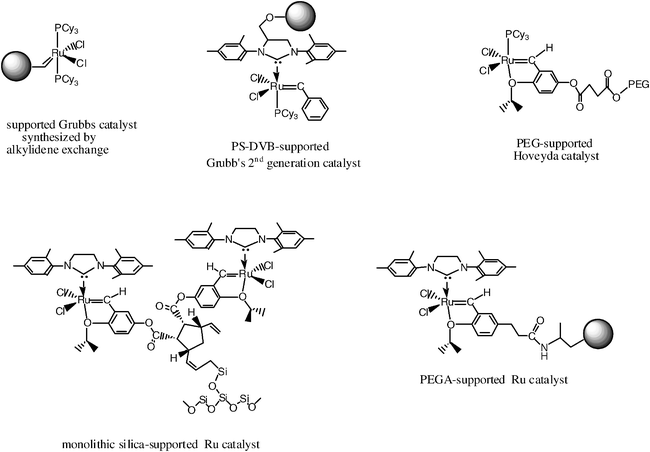 | ||
| Scheme 11 Selected examples of supported ruthenium metathesis catalysts (see text) | ||
Surface organometallic chemistry (SOMC)
SOMC by Basset’s group has provided well-defined heterogeneous catalysts for olefin metathesis. This efficient concept consists in coordinating active metal centers (Mo, W, Re) to silica, with the metal bearing ligands that have already proved useful in homogeneous catalysis and with silica as an additional ligand.61 Recall that Schrock had turned metathesis-inactive alkylidene complexes into active ones by the introduction of alkoxy groups. In Basset’s catalysts, this beneficial role is played by a silyloxy ligand from silica. Thus, the catalysts [(SiO)M(CH–t-Bu)(CH2–t-Bu)2], M = Mo or W,61 and [(SiO)Mo(NH)(CH–t-Bu)(CH2–t-Bu)]58,61 are active at 25 °C, unlike previously reported ill-defined heterogeneous catalysts and the early Mo and W oxides on silica or alumina. The only oxide that had catalyzed olefin metathesis at 25 °C was Re2O7/Al2O3, but it suffers from a low number of active sites, side reactions caused by the acid support and deactivation of the catalyst. On the other hand, Basset and Copéret’s silica-supported rhenium catalyst [(SiO)(Re(C–t-Bu)(CH–t-Bu)(CH2–t-Bu)] catalyzes the metathesis of propene at 25 °C with an initial rate of 0.25 mol per mol Re per s. The formation of 3,3-dimethylbutene and 4,4-dimethylpentene in a 3∶1 ratio results from cross metathesis between propene and the neopentylidene ligand, and the ratio of cross metathesis products matches the relative stability of the metallacyclobutane intermediates. Cross metathesis of propene and isobutene and self-metathesis of methyl oleate can also be achieved, and TON reaches 900 for the latter reaction, which is unprecedented for heterogeneous and most homogeneous catalysts (Fig. 7).62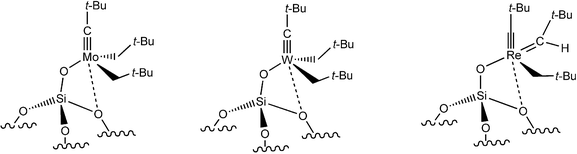 | ||
| Fig. 7 Active, well-defined silica-supported Mo, W and Re alkene metathesis catalysts disclosed by Basset’s group | ||
Applications of metathesis to organic synthesis
It suffices here to refer to the chapters in Volume II of Grubbs’ 2003 Handbook.1a The groups of Schrock and Hoveyda have reported “user-friendly” and practical Mo-based chiral catalysts simply prepared from inexpensive and commercially available, optically pure (R)- or (S)-binaphthol and chiral [Mo(NAr)(CHCMe2Ph)(η2-MeOCH2CH2OMe)(OTf)], also available from Strem (the coordinated dimethoxyethane solvent being replaced by the dinaphthoate). The catalyst generated in this way promotes enantioselective metathesis and its isolation is not required.2 Deithers and Martin also published in 2004 a review article on the synthesis of oxygen and nitrogen-containing heterocycles (ethers, lactones, azacycles) by RCM, some of which were synthesized by tandem metathesis or involved enantioselective cyclizations.9 A study of the tolerance of Mo metathesis catalysts has indicated that Schrock’s Mo catalysts, although they are air- and moisture-sensitive, are effective in the presence of phosphanes, thioethers, nitriles (whereas the Ru catalysts are decomposed by these substrates), sterically protected free alcohols, metal carbonyls and, in many cases, in the presence of amines (even giving unparalleled enantioselectivity with the latter groups).2 Thus, they are fully complementary to the more air-stable Ru catalysts. Perhaps one of the most important developments among the recent ones is the breakthrough in efficient cross metathesis of terminal olefins with olefins bearing an electron-withdrawing group on the double bond.1 The synthesis of polysaccharides by RCM is very active.63,64 Applications of olefin metathesis to combinatorial chemistry were also the subject of several recent reviews.65Applications of metathesis to polymer and material science
Volume III of Grubbs’ Handbook is devoted to this area.1a Block co-polymer nanoparticles, which are important for applications in biosensors, drug delivery, controlled release of hydrophobic drugs and gene therapy, have been synthesized by the assembly of polymers of controlled dimensions using ROMP.66 Polymeric materials (co-polymers, conjugated, functional, bio-active polymers, etc.) designed for their specific physical and biological properties are now available using various types of metathesis reactions, including living ROMP, acyclic diene metathesis polymerization (ADMET), ADIMET (vide supra) and alkyne polymerizations.1Conclusion
The way paved by the discoverers of the heterogeneous metathesis reaction has been exploited by Schrock and Grubbs in an original way, involving the discovery and engineering of metal-alkylidenes and -alkylidynes to develop one of the most productive and useful catalytic reactions in chemistry. Metathesis is indeed presently used every day by the entire organic community, because of its extraordinary potential for the synthesis of molecular targets used as therapeutic agents and by polymer chemists for the synthesis of new materials, including biodegradable ones.By shortening synthetic paths and providing more facile access to therapeutic agents under increasingly more environmentally friendly catalytic conditions, metathesis now is at the forefront of “green chemistry”. The course of the development of the ideas delineated in this review and that led to the progressive discovery and optimization of the now very efficient and selective catalysts involved the careful investigation of molecular mechanisms. The latter is only possible with homogeneous catalysts, which makes the strength of this area of science involving molecular chemistry. Yves Chauvin was the pioneer of the successful mechanistic approach to metathesis and is now universally recognized as such. Moreover, his metallo-square mechanistic schemes are not only important in the olefin metathesis reaction, but for most organometallic reactions involved in catalysis such as the σ-bond and single and double π-bond metathesis and the β-elimination reactions summarized in Scheme 1.
Having learnt Geoffrey Wilkinson’s ideas in organometallic synthesis and John Osborn’s ones in homogeneous catalysis at the beginning of their development, Dick Schrock combined these two essential aspects of inorganic chemistry to bring to the chemical community the first stable metal-methylene, -alkylidene and -alkylidene complexes, then the unambiguous evidence of the validity of the Chauvin mechanism, and finally the first entire family of unimolecular, very efficient catalysts of alkene and alkyne metathesis compatible with a variety of organic functional groups, including the first examples of enantioselective metathesis catalysis. The recent numerous applications of Schrock’s chiral metathesis catalysts to asymmetric synthesis have burst in the organic chemistry.
Likewise, Bob Grubbs used a pragmatic approach, from RuCl3 in water to the sophisticated chiral version of the second generation ruthenium benzylidene catalyst, to provide the organic and polymer communities with what are now the most environmentally friendly metathesis catalysts, because of their stability to air and even to some extent to aqueous media and their high compatibility with most organic functionalities. Schrock-type and Grubbs-type metathesis catalysts are also complementary in terms of efficiency, compatibility of functional groups and stability. Considerable benefits result from the discovery of unimolecular catalysts, in particular by these two groups: tolerance of functional groups for organic synthesis, freedom from side reactions, stereoselectivity, control of polymer molecular weight and rationale for synthetic strategies.
In conclusion, by the breadth and impact of their investigations and their immense success in the discovery of the most efficient and useful catalysts for this key reaction, Schrock and Grubbs have brought a new dimension to both organic chemistry and polymer science.
Acknowledgements
Financial support from the Institut Universitaire de France (IUF), the Centre National de la Recherche Scientifique (CNRS) and the University Bordeaux I is gratefully acknowledged.References
- (a) Handbook of Metathesis, ed. R. H. Grubbs, Wiley-VCH, Weinheim, 2003, vols. 1–3: Search PubMedCatalyst Development (vol. 1); Applications in Organic Synthesis (vol. 2); Applications in Polymer Synthesis (vol. 3); (b) For a major account of Ru metathesis catalysts, see: S. T. Nguyen and T. M. Trnka, in Handbook of Metathesis, ed. R. H. Grubbs, Wiley-VCH, Weinheim, 2003, vol. 1, ch. 1.6 Search PubMed; (c) T. M. Trnka and R. H. Grubbs, Acc. Chem. Res., 2001, 34, 18 CrossRef CAS (this article also contains many references to reviews); (d) For mechanistic aspects of Ru metathesis catalysts, see M. S. Sanford and J. Love, in Handbook of Metathesis, ed. R. H. Grubbs, Wiley-VCH, Weinheim, 2003, vol. 1, ch. 1.9 Search PubMed; (e) R. H. Grubbs, Tetrahedron, 2004, 60, 7117 CrossRef CAS (this review article also contains the references to more than 400 Grubbs’ articles); (f) R. H. Grubbs and S. Chang, Tetrahedron, 1998, 54, 4413 CrossRef CAS.
- (a) R. R. Schrock, J. Mol. Cat. A, 2004, 213, 21 CrossRef CAS; (b) R. R. Schrock and A. H. Hoveyda, Angew. Chem., Int. Ed., 2003, 42, 4592 CrossRef CAS; (c) R. R. Schrock, Chem. Rev., 2002, 102, 145 CrossRef CAS; (d) R. R. Schrock, Top. Organomet. Chem., 1998, 1, 1 Search PubMed; (e) R. R. Schrock, Tetrahedron, 1999, 55, 8141 CrossRef CAS; (f) R. R. Schrock, in Handbook of Metathesis, ed. R. H. Grubbs, Wiley-VCH, Weinheim, 2003, vol. 1, ch. 1.3 and 1.11 Search PubMed.
- (a) K. J. Ivin and J. C. Mol, Olefin Metathesis and Metathesis Polymerization, Academic Press, New York, 1997 Search PubMed; (b) J. C. Mol, J. Mol. Cat. A, 2004, 213, 39 CrossRef CAS; (c) J. C. Mol, Top. Catal., 2004, 27, 95 CrossRef.
- (a) C. Copéret, M. Chabanas, R. Petroff Saint-Arroman and J.-M. Basset, Angew. Chem., Int. Ed., 2003, 42, 156 CrossRef CAS; (b) C. Copéret, F. Lefebvre, J.-M. Basset, Handbook of Metathesis, ed. R. H. Grubbs, Wiley-VCH, Weinheim, 2003, vol. 1, ch. 1.4 and 1.12 Search PubMed; (c) C. Copéret, New J. Chem., 2004, 28, 1 RSC.
- (a) A. Fürstner, Angew. Chem., Int. Ed., 2000, 39, 3012 CrossRef CAS (review); (b) Alkene Metathesis in Organic Chemistry, ed. A. Fürstner, Springer, Berlin, 1998 Search PubMed; (c) A. Fürstner, Top. Organomet. Chem., 1998, 1, 37 Search PubMed; (d) A. Fürstner, Top. Catal., 1997, 36, 2037; (e) A. Fürstner, L’Actualité Chimique, 2003, 4–5, 57 Search PubMed; (f) For a review on alkyne metathesis, see: A. Fürstner, in Handbook of Metathesis, ed. R. H. Grubbs, Wiley-VCH, Weinheim, 2003, vol. 2, ch. 2.12 Search PubMed.
- (a) Reviews on cross metathesis: S. J. Connor and S. Blechert, Angew. Chem., Int. Ed., 2003, 42, 1900 Search PubMed; (b) M. Schuster and S. Blechert, Angew. Chem,. Int. Ed. Engl., 1997, 36, 2036 CrossRef; (c) For a review on tandem ring closing metathesis, see: S. Randl and S. Blechert, in Handbook of Metathesis, ed. R. H. Grubbs, Wiley-VCH, Weinheim, 2003, vol. 2, ch. 2 Search PubMed.
- (a) For reviews on metathesis polymerization, see: M. R. Buchmeiser, Chem. Rev., 2000, 100, 1565 Search PubMed and M. R. Buchmeiser, in Handbook of Metathesis, ed. R. H. Grubbs, Wiley-VCH, Weinheim, 2003, vol. 3, ch. 3.7 CrossRef CAS; (b) For a review on supported metathesis catalysts, see: M. R. Buchmeiser, New J. Chem., 2004, 28, 549 Search PubMed; (c) For reviews on metathesis polymerization, see the Handbook of Metathesis, ed. R. H. Grubbs, Wiley-VCH, Weinheim, 2003, vol. 3, ch. 11 Search PubMed.
- A. H. Hoveyda, D. G. Gillingham, J. J. Van Veldhuizen, O. Kataoka, S. B. Garber, J. S. Kingsbury and J. P. A. Harrity, Org. Biol. Chem., 2004, 2, 8 Search PubMed (review); see also ref. 2b.
- (a) A. Deithers and S. F. Martin, Chem. Rev., 2004, 104, 2199 CrossRef CAS (heterocycle synthesis); (b) For applications of ring-closing metathesis to alkaloid synthesis, see: S. F. Martin in Handbook of Metathesis, ed. R. H. Grubbs, Wiley-VCH, Weinheim, 2003, vol. 2, ch. 2.9. Search PubMed For other applications of metathesis to organic synthesis, see the Handbook of Metathesis, ed. R. H. Grubbs, Wiley-VCH, Weinheim, 2003, vol. 3, ch. 14 Search PubMed.
- (a) D. Evans, J. A. Osborn, F. H. Jardine and G. Wilkinson, Nature (London), 1965, 208, 1203; (b) H. Kagan and T. P. Dang, J. Chem. Soc., Chem. Commun., 1971, 481 RSC; (c) H. Kagan and T. P. Dang, J. Am. Chem. Soc., 1972, 94, 6429 CrossRef CAS.
- A. Loupy, B. Tchoubar and D. Astruc, Chem. Rev., 1992, 92, 1141 CrossRef CAS.
- (a) N. Calderon, Tetrahedron Lett., 1967, 34, 3327 CrossRef; (b) N. Calderon, Acc. Chem. Res., 1972, 5, 127 CrossRef CAS.
- (a) R. L. Banks and G. C. Bailey, Int. Eng. Prod. Dev., 1964, 3, 170 Search PubMed; (b) For a historical account of the early days, see: H. S. Eleuterio, J. Mol. Catal., 1991, 65, 55 Search PubMed (in particular, compare ref. 13c and 13d); (c) H. S. Eleuterio, German Pat. 1072811, 1960 Search PubMed; (d) W. L. Truett, D. R. Johnson, I. M. Robinson and B. P. Montague, J. Am. Chem. Soc., 1960, 82, 2337 CrossRef CAS; (e) M. R. Rouhi, Chem. Eng. News, 2002, 29 (December 23).
- (a) M. Blanchard and A. Mortreux, Bull. Soc. Chim., 1972, 4, 1641 Search PubMed; (b) M. Blanchard and A. Mortreux, J. Mol. Catal., 1975–6, 1, 101 Search PubMed; (c) A. Mortreux, J. C. Delgrange, M. Blanchard and B. Lubochinsky, J. Mol. Catal., 1977, 2, 73 CrossRef CAS; (d) A. Mortreux, F. Petit and M. Blanchard, Tetrahedron Lett., 1978, 49, 4967 CrossRef; (e) A. Mortreux, F. Petit, M. Petit and T. Szymanska-Buzar, J. Mol. Catal. A: Chem., 1995, 96, 95 CrossRef CAS (for recent improvements of the system Mo(CO)6 + p-ClC6H4OH or other activated phenols, see text); (f) U. H. F. Bunz, in Modern Arene Chemistry, ed. D. Astruc, Wiley-VCH, Weinheim, 2002, pp. 217–249 Search PubMed; (g) H. H. F. Bunz, in Handbook of Metathesis, ed. R. H. Grubbs, Wiley-VCH, Weinheim, 2003, vol. 3, ch. 3, 10 Search PubMed; (h) K. Grela and J. Ignatowska, Org. Lett., 2002, 4, 3747 CrossRef CAS; (i) A. Michrowska, R. Bujok, S. Harutyunyan, V. Sashuk, G. Dolgonos and K. Grela, J. Am. Chem. Soc., 2004, 126, 9318 CrossRef CAS; (j) V. Huc, R. Weihofen, I. Martin-Jimenez, P. Oulié, C. Lepetit, G. Lavigne and R. Chauvin, New J. Chem., 2003, 27, 1412 RSC; (k) W. Zhang, S. Kraft and J. S. Moore, Chem. Commun., 2003, 832 RSC; (l) W. Zhang, S. Kraft and J. S. Moore, J. Am. Chem. Soc., 2004, 126, 329 CrossRef CAS; (m) W. Zhang and J. S. Moore, J. Am. Chem. Soc., 2004, 126, 12796 CrossRef CAS.
- E. O. Fischer, Angew. Chem., Int. Ed. Engl., 1964, 580 CrossRef.
- G. Natta, Angew. Chem., Int. Ed. Engl., 1964, 3, 723 CrossRef.
- Y. Chauvin and J.-L. Hérisson, Makromol. Chem., 1971, 141, 161 CrossRef CAS.
- (a) Y. Chauvin, C. R. Seances Acad. Sci., Ser. C, 1973, 276, 169 Search PubMed; (b) Y. Chauvin, D. Commereuc and D. Cruypelinck, Makromol. Chem., 1976, 177, 2637 CrossRef CAS.
- (a) F. N. Tebbe, G. W. Parshall and G. S. Reddy, J. Am. Chem. Soc., 1978, 100, 3611 CrossRef CAS; (b) T. R. Howard, J. B. Lee and R. H. Grubbs, J. Am. Chem. Soc., 1980, 102, 6876 CrossRef CAS; (c) E. V. Anslyn and R. H. Grubbs, J. Am. Chem. Soc., 1987, 109, 4880 CrossRef CAS; (d) J. M. Hawkins and R. H. Grubbs, J. Am. Chem. Soc., 1988, 110, 2821 CrossRef CAS.
- (a) C. P. Casey and T. J. Burkhardt, J. Am. Chem. Soc., 1974, 96, 7808 CrossRef CAS; (b) T. J. Katz and S. J. Lee, J. Am. Chem. Soc., 1980, 102, 422 CrossRef CAS; (c) T. J. Katz, Adv. Organomet. Chem., 1978, 16, 283; (d) This alkyne polymerization mechanism (ref. 20b,c) can be considerably accelerated by coupling with electron-transfer-chain catalysis: M.-H. Desbois and D. Astruc, New J. Chem., 1989, 13, 595 Search PubMed.
- (a) G. Wilkinson, Science, 1974, 185, 109 CAS; (b) G. Wilkinson, Pure Appl. Chem., 1959, 71, 627; (c) R. R. Schrock and G. W. Parshall, Chem. Rev., 1976, 76, 243 CrossRef CAS; (d) M. F. Lappert and R. Pierce, Chem. Rev., 1976, 76, 219 CrossRef CAS.
- (a) R. R. Schrock, J. Am. Chem. Soc., 1974, 96, 6796 CrossRef CAS; (b) R. R. Schrock, Acc. Chem. Res., 1979, 12, 98 CrossRef CAS; (c) R. R. Schrock, Science, 1983, 219, 13 CrossRef CAS; (d) J. Feldman and R. R. Schrock, Prog. Inorg. Chem., 1991, 39, 1; (e) R. R. Schrock, S. M. Rocklage, J. H. Wengrovius, G. Rupprecht and J. Feldmann, J. Mol. Catal., 1980, 8, 73 CrossRef CAS; (f) J. H. Wengrovius, R. R. Schrock, M. R. Churchill, J. R. Missert and W. J. Youngs, J. Am. Chem. Soc., 1980, 102, 4515 CrossRef CAS.
- (a) R. R. Schrock, J. S. Murdzek, G. C. Bazan, J. Robbins, M. DiMare and M. O’Regan, J. Am. Chem. Soc., 1990, 112, 3875 CrossRef CAS; (b) G. C. Bazan, E. Khosravi, R. R. Scrock, W. J. Feast, V. C. Gibson, M. B. O’Regan, J. K. Thomas and W. M. Davis, J. Am. Chem. Soc., 1990, 112, 8378 CrossRef CAS; (c) G. C. Bazan, J. H. Oskam, H. N. Cho, L. Y. Park and R. R. Schrock, J. Am. Chem. Soc., 1991, 113, 6899 CrossRef CAS.
- R. R. Schrock, S. McLain and J. Sancho, J. Am. Chem. Soc., 1979, 101, 4558 CrossRef CAS.
- (a) A. Bre, Y. Chauvin and D. Commereuc, Nouv. J. Chim., 1986, 10, 535 Search PubMed; (b) Y. Chauvin and H. Olivier, in Applied Homogeneous Catalysis, eds. B. Cornils and W. A. Herrmann, VCH, Weinheim, 1996, vol. 1, ch. 2.3.1 and 2.3.6 Search PubMed.
- (a) J. Kress, M. Wesolek and J. A. Osborn, J. Chem. Soc., Chem. Commun., 1982, 514 RSC; (b) J. Kress, M. Wesolek and J. A. Osborn, J. Am. Chem. Soc., 1883, 105, 6346; (c) J. Kress, J. A. Osborn, R. M. E. Green, K. J. Ivin and J. J. Rooney, J. Am. Chem. Soc., 1987, 109, 899 CrossRef CAS; (d) J. Kress, A. Aguero and J. A. Osborn, J. Mol. Catal., 1986, 36, 1 CrossRef CAS.
- (a) F. Quignard, M. Leconte and J.-M. Basset, J. Chem. Soc., Chem. Commun., 1985, 1816 RSC; (b) M. Leconte, J.-M. Basset, F. Quignard and C. Larroche, in Reactions of Coordinated Ligands, ed. P. S. Braterman, Plenum, New York, 1986, vol. 1, pp. 371–420 Search PubMed; (c) F. Lefebvre, M. Lecomte, S. Pagano, A. Mutch and J.-M. Basset, Polyhedron, 1995, 14, 3209 CrossRef CAS.
- (a) J. B. Alexander, D. S. La, D. R. Cefalo, A. H. Hoveyda and R. R. Schrock, J. Am. Chem. Soc., 1998, 120, 4041 CrossRef CAS; (b) S. S. Zu, D. R. Cefalo, D. S. La, J. Y. Jamieson, W. M. Davis, A. H. Hoveyda and R. R. Schrock, J. Am. Chem. Soc., 1999, 121, 8251 CrossRef CAS; (c) S. L. Aeilts, D. R. Cefalo, P. J. Bonitatebus, J. H. Houser, A. H. Hoveyda and R. R. Schrock, Angew. Chem., Int. Ed., 2001, 40, 1452 CrossRef CAS; (d) X. Teng, D. R. Cefalo, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2002, 124, 10779 CrossRef CAS; (e) K. H. Hultzsch, J. A. Jernelius, A. H. Hoveyda and R. R. Schrock, Angew. Chem., Int. Ed., 2002, 114, 609 CrossRef; (f) W. C. P. Tsang, J. A. Jernelius, G. A. Cortez, G. S. Weatherhead, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2003, 125, 2591 CrossRef CAS; (g) W. C. P. Tsang, K. C. Hultzsch, J. B. Alexander, P. J. Bonitatebus and R. R. Schrock, J. Am. Chem. Soc., 2003, 125, 2652 CrossRef CAS.
- D. Astruc, Chimie Organométallique, EDP Sciences, Les Ulis, 2000, ch. 10 Search PubMed on metal-carbene and carbyne complexes (Spanish version: Reverté, Barcelona, 2004).
- (a) P. W. Jolly and R. Pettit, J. Am. Chem. Soc., 1966, 88, 5044 CrossRef CAS; (b) V. Guerchais and D. Astruc, J. Chem. Soc., Chem. Commun., 1985, 835 RSC.
- M. L. H. Green, L. C. Mitchard and M. G. Swanwick, J. Chem. Soc. A, 1971, 794 RSC.
- R. H. Grubbs, J. Am. Chem. Soc., 1972, 94, 2538 CrossRef CAS.
- (a) G. Natta, G. Dall’Asta and L. Porri, Makromol. Chem., 1965, 81, 253 CrossRef CAS; (b) The capture by Grubbs of the renaissance of polymerization chemistry is represented by his review article: R. H. Grubbs and W. J. Tumas, Science, 1989, 243, 907 Search PubMed.
- (a) B. M. Novak and R. H. Grubbs, J. Am. Chem. Soc., 1988, 110, 7542 CrossRef CAS; (b) M. B. France, R. H. Grubbs, V. McGrath and R. A. Paciello, Macromolecules, 1993, 26, 4742 CrossRef CAS; (c) S. T. Nguyen, L. K. Johson, R. H. Grubbs and J. W. Ziller, J. Am. Chem. Soc., 1992, 114, 3974 CrossRef CAS; (d) P. Schwab, M. B. France, J. W. Ziller and R. H. Grubbs, Angew. Chem., Int. Ed. Engl., 1998, 37, 1124 CrossRef CAS; (e) A. Demonceau, A. F. Noels, E. Saive and A. J. Hubert, J. Mol. Catal., 1992, 76, 123 CrossRef CAS; (f) A. W. Stumpf, E. Saive, A. Demonceau and A. F. Noels, J. Chem. Soc., Chem. Commun., 1995, 1127 RSC.
- (a) M. Olivan and K. Caulton, Chem. Commun., 1997, 1733 RSC; (b) J. Wolf, W. Stüer, C. Grünwald, H. Werner, P. Schwab and M. Schulz, Angew. Chem., Int. Ed. Engl., 1998, 37, 1124 CrossRef CAS; (c) H. Werner, J. Wolf, in Handbook of Metathesis, ed. R. H. Grubbs, Wiley-VCH, Weinheim, 2003, vol. 1, ch. 1.8 Search PubMed; (d) M. Gandelman, B. Rybtchinski, N. Ashkenazi, R. M. Gauvin and D. Milstein, J. Am. Chem. Soc., 1997, 119, 3887 CrossRef CAS.
- (a) A. J. Arduengo, Acc. Chem. Res., 1999, 32, 913 CrossRef; (b) W. A. Herrmann and C. Köcher, Angew. Chem., Int. Ed. Engl., 1997, 109, 2256; (c) D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39 CrossRef; (d) T. Wetskamp, W. C. Schattenmann and W. A. Herrmann, Angew. Chem., Int. Ed., 1998, 37, 2490 CrossRef CAS.
- (a) J. Huang, E. D. Stevens, S. P. Nolan and J. L. Petersen, J. Am. Chem. Soc., 1999, 121, 2647 CrossRef; (b) M. Scholl, T. M. Trnka, J. P. Morgan and R. H. Grubbs, Tetrahedron Lett., 1999, 40, 2247 CrossRef CAS; (c) L. Ackermann, A. Fürstner, T. Weskamp, F. J. Kohl and W. A. Herrmann, Tetrahedron Lett., 1999, 40, 4787 CrossRef CAS; (d) M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953 CrossRef CAS; (e) T. M. Trnka, J. P. Morgan, M. S. Sanford, T. E. Wilhelm, M. Scholl, T. L. Choi, S. Ding, M. W. Day and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 2546 CrossRef CAS; (f) J. A. Love, M. S. Sanford, M. W. Day and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 10103 CrossRef CAS; (g) A. K. Chatterjee, T.-L. Choi, D. P. Sanders and R. H. Grubb, J. Am. Chem. Soc., 2003, 125, 11360 CrossRef CAS (this article is particularly useful for the selectivity in cross metathesis); (h) C. Morril and R. H. Grubbs, J. Org. Chem., 2003, 68, 603; (i) F. M. Kilbinger, S. J. Candrill, A. W. Waltman, M. W. Day and R. H. Grubbs, Angew. Chem., Int. Ed., 2003, 42, 3281 CrossRef CAS; (j) J. A. Love, J. P. Morgan, T. M. Trnka and R. H. Grubbs, Angew. Chem., Int. Ed., 2003, 42, 4035; (k) The second-generation Grubbs’ catalyst thermally decomposes at 55 °C in benzene to give, after 72 h, a 46% isolated yield of a hydridic dinuclear carbyne (HRuCRu) complex that catalyzes olefin isomerization: H. Hong, M. W. Day and R. H. Grubbs, J. Am. Chem. Soc., 2004, 126, 7414 Search PubMed.
- (a) J. S. Kingsbury, J. P. A. Harrity, P. J. Bonitatebus and A. H. Hoveyda, J. Am. Chem. Soc., 1999, 121, 791 CrossRef CAS; (b) K. Grela, S. Harutyunyan and A. Michrowska, Angew. Chem., Int. Ed., 2002, 41, 4038 CrossRef CAS; (c) J. O. Krause, M. T. Zarka, U. Anders, R. Weberskirch, O. Nuyken and M. R. Buchmeiser, Angew. Chem., Int. Ed., 2003, 42, 5965 CrossRef CAS; (d) T. Honda, H. Namiki, K. Kaneda and H. Misutani, Org. Lett., 2004, 6, 87 CrossRef CAS.
- (a) S. J. Connon, A. Dunne and S. Blechert, Angew. Chem., Int. Ed., 2002, 41, 3835 CrossRef CAS; (b) H. Wakamatsu and S. Blechert, Angew. Chem., Int. Ed., 2002, 41, 2403 CrossRef CAS (with a biphenyl group, see Fig. 5); (c) H. Wakamatsu and S. Blechert, Angew. Chem.. Int. Ed., 2002, 41, 794 CrossRef CAS (with a binaphtol group); (d) A. M. Dunne, S. Mix and S. Blechert, Tetrahedron Lett., 2003, 44, 2733 CrossRef CAS (RCM and ROCM).
- (a) P. Wijkens, J. T. B. H. Jastrzebski, P. A. van der Schaaf, R. Kolly, A. Hafner and G. van Koten, Org. Lett., 2000, 122, 8168; (b) S. B. Garber, J. S. Kingsbury, B. L. Gray and A. H. Hoveyda, J. Am. Chem. Soc., 2000, 122, 8168 CrossRef CAS.
- (a) S. M. Hansen, M. A. O. Volland, F. Rominger, F. Eisenträger and P. Hofmann, Angew. Chem., Int. Ed., 1999, 38, 1273 CrossRef CAS; (b) S. M. Hansen, F. Rominger, M. Metz and P. Hofmann, Chem.-Eur. J., 1999, 5, 557 CrossRef CAS; (c) M. A. O. Volland, S. M. Ansen, F. Rominger and P. Hofmann, Organometallics, 2004, 23, 800 CrossRef CAS.
- C. Six, K. Beck, A. Wegner and W. Leitner, Organometallics, 2000, 19, 4639 CrossRef CAS.
-
(a) A. Fürstner, C. Picquet, C. Bruneau and P. H. Dixneuf, Chem. Commun., 1998, 1315 RSC;
(b) A. Fürstner, M. Liebl, C. W. Lehmann, M. Picquet, R. Kunz, C. Bruneau, D. Touchard and P. H. Dixneuf, Chem.-Eur. J., 2000, 6, 1847 CrossRef CAS;
(c) A. Fürstner, R. Ackermann, B. Gabor, R. Goddart, W. Lehmann, R. Mynott, F. Stelzer and O. R. Thiel, Angew. Chem., Int. Ed., 2001, 7, 3236 CAS;
(d) A. Fürstner, A. S. Castanet, K. Radkovski and C. W. Lehman, J. Org. Chem., 2003, 68, 1521 CrossRef;
(e) J. C. Conrad, D. Amoroso, P. Czechura, G. P. A. Yap and D. E. Fogg, Organometallics, 2003, 22, 3634 CrossRef CAS;
(f) J. L. Snelgrove, J. C. Conrad, G. P. A. Yap and D. E. Fogg, Inorg. Chim. Acta, 2003, 345, 268 CrossRef CAS;
(g) RuCH(SR) catalysts: H. Katayama, M. Nagao and F. Ozawa, Organometallics, 2003, 22, 586 Search PubMed;
(h) P. A. van der Schaaf, R. Kolly, H.-J. Kirner, F. Rime, A. Mühlebach and A. Hafner, J. Organomet. Chem., 2000, 606, 65 CrossRef;
(i) Supercritical CO2 in metathesis catalysis: A. Fürstner, D. Koch, W. Langemann and C. Leitner, Angew. Chem., Int. Ed. Engl., 1997, 36, 2466 Search PubMed.
- (a) A. Demonceau, A. W Stumpf, E. Saive and A. F. Noels, Macromolecules, 1997, 30, 3127 CrossRef CAS; (b) J. Cossy, F. Bargiggia and S. Bouzbouz, Org. Lett., 2003, 5, 459 CrossRef CAS; (c) S. Bouzbouz, E. De Lemos and J. Cossy, Adv. Synth. Catal., 2002, 344, 627 CrossRef CAS; (d) Y. Ki, M. F. Mayer and S. C. Zimmerman, Angew. Chem., Int. Ed., 2003, 42, 1121 CrossRef CAS; (e) R. V. Anana, S. Baktaraman and V. K. Singh, J. Org. Chem., 2003, 68, 3356 CrossRef; (f) J. H. Cho and B. M. Kim, Org. Lett., 2003, 5, 531 CrossRef CAS; (g) D. F. Taber and K. J. Frankowski, J. Org. Chem., 2003, 68, 6047 CrossRef CAS; (h) B. Alcaide and P. Almendros, Chem.-Eur. J., 2003, 9, 1258 CrossRef CAS.
- (a) S. Gatard, S. Nlate, E. Cloutet, G. Bravic, J.-C. Blais and D. Astruc, Angew. Chem., Int. Ed., 2003, 42, 452 CrossRef CAS; (b) S. Gatard, S. Kahlal, D. Méry, S. Méry, S. Nlate, E. Cloutet, J.-Y. Saillard and D. Astruc, Organometallics, 2004, 23, 1313 CrossRef CAS.
- (a) M. Lynn, B. Mohr and R. H. Grubbs, J. Am. Chem. Soc., 1998, 120, 1627 CrossRef CAS; (b) D. M. Lynn, B. Mohr, R. H. Grubbs, L. M. Henling and M. W. Day, J. Am. Chem. Soc., 2000, 122, 6601 CrossRef CAS.
- (a) N. Audic, H. Clavier, M. Mauduit and J.-C. Guillemin, J. Am. Chem. Soc., 2003, 125, 9248 CrossRef CAS; (b) D. Sémeril, H. Olivier-Bourbigou, C. Bruneau and P. H. Dixneuf, Chem. Commun., 2002, 146 RSC.
- (a) V. Martinez, J.-C. Blais and D. Astruc, Org. Lett., 2002, 4, 651 CrossRef CAS; (b) V. Martinez, J.-C. Blais, G. Bravic and D. Astruc, Organometallics, 2004, 23, 861 CrossRef CAS.
- D. Astruc and F. Chardac, Chem. Rev., 2001, 101, 2991 CrossRef CAS.
- (a) M. Mori, Top. Organomet., 1998, 1, 133 Search PubMed; (b) C. Dietrich-Bucherer, G. Rapenne and J.-P. Sauvage, Chem. Commun., 1997, 2053 RSC; (c) T. O. Schrader and M. L. Snapper, in in Handbook of Metathesis, ed. R. H. Grubbs, Wiley-VCH, Weinheim, 2003, vol. 2, ch. 2.6 (review on ring-opening cross metathesis) Search PubMed.
- (a) V. Martinez, J.-C. Blais and D. Astruc, Angew. Chem., Int. Ed., 2002, 42, 2586; (b) A. M. Martin-Alvarez, F. Hampel, A. M. Arif and J. A. Gladysz, Organometallics, 1999, 18, 955 CrossRef CAS; (c) See also: P. L. Ng and J. N. Lambert, Synlett., 1999, 1749 Search PubMed.
- S. D. Burke, N. Müller and C. M. Beaudry, Org. Lett., 1999, 1, 1827 CrossRef CAS.
- (a) M. Hirama, Science, 2001, 294, 1904 CrossRef CAS; (b) T. Seiders, D. W. Ward and R. H. Grubbs, Org. Lett., 2001, 3, 3225 CrossRef CAS; (c) L. F. Tieze and F. Haunert, in Stimulating Concepts in Chemistry, eds. F. Vögtle, J. F. Stoddart and M. Shibasaki, Wiley-VCH, Weinheim, 2000, pp. 39–64 Search PubMed.
- (a) J. Corker, F. Lefebvre, C. Lecuyer, V. Dufaud, F. Quignard, A. Choplin, J. Evans and J.-M. Basset, Science, 1996, 271, 966 CAS; (b) V. Vidal, A. Théollier, J. Thivolle-Cazat and J.-M. Basset, Science, 1997, 276, 99 CrossRef CAS; (c) O. Maury, L. Lefort, V. Vidal, J. Thivolle-Cazat and J.-M. Basset, Angew. Chem., Int. Ed., 1999, 38, 1952 CrossRef CAS; (d) C. Copéret, O. Maury, J. Thivolle-Cazat and J.-M. Basset, Angew. Chem., Int. Ed., 2001, 40, 2331 CrossRef CAS; (e) F. Rataboul, A. Baudouin, C. Thieuleux, L. Veyre, C. Copéret, J. Thivolle-Cazat, J.-M. Basset, A. Lesage and L. Emsley, J. Am. Chem. Soc., 2004, 126, 12541 CrossRef CAS; (f) E. Le Roux, M. Chabanas, A. Baudouin, A. de Mallmann, C. Copétet, E. A. Quadrelli, J. Thivolle-Cazat, J.-M. Basset, W. Lukens, A. Lesage, L. Emsley and G. Sunley, J. Am. Chem. Soc., 2004, 126, 13391 CrossRef CAS; (g) For recent mechanistic studies of the methanolysis reaction of d0 complexes, see: D. Soulivong, C. Copéret, J. Thivolle-Cazat, J.-M. Basset, B. M. Maunderf, R. Pardi and G. Sunley, Angew. Chem., Int. Ed., 2004, 43, 5366 Search PubMed; (h) For a DFT study of the σ-bond metathesis reaction of H2 and CH4 with d0 complexes, see: L. Maron, L. Perrin and O. Eisenstein, J. Chem. Soc., Dalton Trans., 2002, 534 Search PubMed (H2); (i) L. Maron and O. Eisenstein, J. Am. Chem. Soc., 2001, 123, 1036 CrossRef CAS (CH4).
- R. R. Schrock, D. N. Clark, J. Sancho, J. H. Wengrovius and S. F. Pederson, Organometallics, 1982, 1, 1645 CrossRef CAS.
- (a) A. Fürstner, C. Mathes and C. W. Lehmann, J. Am. Chem. Soc., 1999, 121, 9453 CrossRef; (b) A. Fürstner, C. Mathes and C. W. Lehmann, Chem.-Eur. J., 2001, 7, 5299 CrossRef CAS; (c) A. Fürstner, A.-S. Castanet, K. Radkowski and C. W. Lehmann, J. Org. Chem., 2003, 68, 1521 CrossRef; (d) O. S. Miljanic, K. P. C. Vollhardt and G. D. Whitener, Synlett., 2003, 29.
- R. Petroff Saint-Aroman, M. Chabanas, C. Copéret, J.-M. Basset and L. Emsley, J. Am. Chem. Soc., 2001, 123, 3820 CrossRef CAS.
- D. M. Lynn, B. Mohr and R. H. Grubbs, J. Am. Chem. Soc., 1998, 120, 1627 CrossRef CAS.
- (a) J. O. Krause, M. T. Zarka, U. Anders, R. Weberkirch, O. Nuyken and M. Buchmeiser, Angew. Chem., Int. Ed., 2003, 42, 5965 CrossRef CAS; (b) M. T. Zarka, O. Nuyken and R. Weberskirch, Macromol. Rapid Commun., 2004, 25, 858 CrossRef CAS.
- N. E. Leadbeater and M. Marco, Chem. Rev., 2002, 102, 3217 CrossRef CAS.
- M. Chabanas, A. Baudouin, C. Copéret and J.-M. Basset, J. Am. Chem. Soc., 2003, 125, 492 CrossRef CAS.
- (a) W. A. Herrmann, A. W. Stumpf, T. Priermeier, S. Bogdanovic, V. Dufaud and J.-M. Basset, Angew. Chem., Int. Ed. Engl., 1996, 35, 2803 CrossRef CAS; (b) R. Petroff Saint-Aroman, M. Chabanas, C. Copéret, J.-M. Basset and L. Emsley, J. Am. Chem. Soc., 2001, 123, 3820 CrossRef CAS.
- (a) Synthesis of medium-sized rings by RCM: M. E. Maier, Angew. Chem., Int. Ed., 2000, 39, 2073 Search PubMed; (b) Extensive review on ring synthesis by RCM: K. C. Nicolaou, N. P. King and Y. He, Top. Organomet. Chem., 1998, 1, 73 Search PubMed.
- M. H. D. Postema, J. L. Piper, V. Komanduri and L. Liu, Angew. Chem., Int. Ed., 2004, 43, 2915 CrossRef CAS.
- (a) M. T. Reetz, Comp. Coord. Chem. II, 2004, 9, 509 Search PubMed; (b) A. D. Piscopio and J. E. Robinson, Curr. Opin. Chem. Biol., 2004, 8, 245 CrossRef CAS; (c) B. Schmidt, Eur. J. Org. Chem., 2004, 1865 CrossRef CAS.
- (a) A. Carrillo and R. Cane, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 3352 CrossRef CAS; (b) V. Lapinte, P. de Frémont, V. Montembault and L. Fontaine, Macromol. Chem. Phys., 2004, 205, 1238 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2005 |