N,N′-Bis(2,6-diisopropylphenyl)formamidinate complexes of the heavy alkaline earths
Marcus L.
Cole†
and
Peter C.
Junk
*
School of Chemistry, Monash University, Victoria 3800, Australia. E-mail: peter.junk@sci.monash.edu.au; Fax: +61 (0)3 9905 4597
First published on 14th December 2004
Abstract
The heavy alkaline earth elements calcium, strontium and barium react with two equivalents of N,N′-bis(2,6-diphenyl)formamidine (DippFormH) in the presence of bis(pentafluorophenyl)mercury to afford the bis(formamidinate) species [M(DippForm)2(THF)n] (M = Ca, n = 1 (1), M = Sr (2) or Ba (3), n = 2) in good to moderate yield. Use of one equivalent of DippFormH during the preparation of 1–3 putatively results in the formation of the organoamide heavy alkaline earth Grignard species “[(X)M(DippForm)]” (M = Ca, Sr or Ba, X = F or C6F5). These compounds participate in the nucleophilic substitution of the para-fluorine of pentafluorobenzene, a by-product of prior ligand exchange, to render DippForm(4-HC6F4).
Introduction
The advancement of heavy alkaline earth chemistry (Ca, Sr and Ba) within the discipline of organometallic/organoamide synthesis has largely been defined by developments in cyclopentadienide chemistry.1,2 One motivation for this has been the reliance of contemporary anaerobic syntheses on soluble metal halides (for salt-elimination type syntheses) or dependable sources of metal alkyl (for protolysis type reactions). The anhydrous halides of the alkaline earths are notoriously insoluble in common laboratory solvents and extremely expensive to purchase while, aside from dibutylmagnesium, alkaline earth metal alkyls are not commercially available. One means of circumventing these draw-backs is to employ a metal based synthesis like the redox transmetallation followed by ligand exchange path A (see below, M = alkaline earth metal, L = anionic ligand, R = protolysable donor).3 This obviates preparation of air-sensitive group 1 metallated ligands, use of insoluble halides and the handling of pyrophoric metal alkyls and can be undertaken as a “one-pot synthesis” thereby keeping synthetic steps to a minimum.| HgR2 + M0 + 2LH → “MR2” + Hg0 + 2LH → ML2 + Hg0 + 2RH | (A) |
![Reagents and conditions: (i) 0.5 equiv. M, 0.5 equiv. [Hg(C6F5)2], THF, ambient temperature, 24 h, −0.5 equiv. Hg(l), −2 equiv. C6F5H; 1, M = Ca, n
= 1; 2, M = Sr, n
= 2; 3, M = Ba, n
= 2. (ii) 1.0 equiv. M (M = Ca, Sr or Ba), 1.0 equiv. [Hg(C6F5)2], THF, ambient temperature, 3–5 d, −1.0 equiv. Hg(i), −“MX2”.](/image/article/2005/NJ/b411494a/b411494a-s1.gif) | ||
| Scheme 1 Reagents and conditions: (i) 0.5 equiv. M, 0.5 equiv. [Hg(C6F5)2], THF, ambient temperature, 24 h, −0.5 equiv. Hg(l), −2 equiv. C6F5H; 1, M = Ca, n = 1; 2, M = Sr, n = 2; 3, M = Ba, n = 2. (ii) 1.0 equiv. M (M = Ca, Sr or Ba), 1.0 equiv. [Hg(C6F5)2], THF, ambient temperature, 3–5 d, −1.0 equiv. Hg(I), −“MX2”. | ||
Results and discussion
The heavy alkaline earth metals calcium, strontium and barium react cleanly with two equivalents of DippFormH in tetrahydrofuran in the presence of one equivalent of bis(pentafluorophenyl)mercury to afford [M(DippForm)2(THF)n], where M = Ca, n = 1 (1) and M = Sr or Ba, n = 2 (2 and 3 respectively, see Scheme 1). 19F NMR spectra of the reaction medium after 24 h reveal pentafluorobenzene as the sole fluorine-containing by-product, while FTIR spectra of the filtered vacuum dried reaction medium indicate complete deprotonation of the DippFormH ligand precursor (strong broad C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretch at 1666 cm−1) in that they display strong C–N stretches between 1520 and 1530 cm−1. These are consistent with those of alkali metal complexes of DippForm (e.g.
[Li(DippForm)(THF)2], strong C–N stretch at 1538 cm−1).16 In the absence of the mercurial no reaction takes place, indicating an [M(C6F5)2] or [Hg(DippForm)2] intermediate is generated prior to the formation of 1–3. The latter can be discounted as DippFormH fails to react with bis(pentafluorophenyl)mercury, instead providing the formamadine upon work-up. The intermediacy of an [M(C6F5)2] species is also supported by the isolation of bis(pentafluorophenyl)europium by Forsyth and Deacon21 from the stoichiometric reaction of elemental europium with Hg(C6F5)2.22
N stretch at 1666 cm−1) in that they display strong C–N stretches between 1520 and 1530 cm−1. These are consistent with those of alkali metal complexes of DippForm (e.g.
[Li(DippForm)(THF)2], strong C–N stretch at 1538 cm−1).16 In the absence of the mercurial no reaction takes place, indicating an [M(C6F5)2] or [Hg(DippForm)2] intermediate is generated prior to the formation of 1–3. The latter can be discounted as DippFormH fails to react with bis(pentafluorophenyl)mercury, instead providing the formamadine upon work-up. The intermediacy of an [M(C6F5)2] species is also supported by the isolation of bis(pentafluorophenyl)europium by Forsyth and Deacon21 from the stoichiometric reaction of elemental europium with Hg(C6F5)2.22
Complexes 1–3 are soluble in benzene permitting their composition (in terms of both formamidinate binding and THF ∶ DippForm ratio) to be assessed by 1H and 13C{1H} NMR spectroscopy. Assuming the absence of fluxional processes, the spectra for 1–3 suggest symmetrical binding of the amidinate (well defined isopropyl methyne/methyl and NCN backbone resonances) contrary to related potassium species,10,14 and THF ∶ DippForm ratios of 2 ∶ 1 for 1 and 1 ∶ 1 for 2 and 3. In tandem with the smaller ionic radius of Ca2+ relative to those of Sr2+ and Ba2+ (for six-coordinate species r = 1.00 Å, 1.18 Å and 1.35 Å, respectively),17 this implies that the alkaline earth metal atoms of 1–3 are chelated or bridged by symmetrical DippForm ligands wherein the calcium of 1 is five-coordinate and possesses one less THF donor than the respective metals of 2 and 3, which are both six-coordinate.
Low temperature single crystal X-ray structure determinations of compounds 1 to 3 identify all three species as mononuclear with equivalent N,N′-chelating DippForm ligands. Two cisoid THF donors complement formamidinate coordination for compounds 2 and 3, while for 1 a single THF donor completes calcium coordination. X-Ray collection and refinement data for all compounds described herein have been summarised in the experimental section.
As depicted in Fig. 1 (POV-RAY illustration, 40% thermal ellipsoids, see figure caption for relevant bond lengths and angles), compound 1 possesses one terminal THF-donor coordinated to a calcium that sits 0.545(2) Å above the rms plane of the four nitrogen donors. These belong to two NCN donors that possess an NCN–NCN torsion angle of 51.1(4)°. The Ca–N bond lengths vary in a manner consistent with the magnitude of the O(1)–Ca(1)–N angle. This is exemplified by N(1) (N(1)–Ca(1) 2.361(3) Å, O(1)–Ca(1)–N(1) 110.8(1)°) and N(2) (N(2)–Ca(1) 2.406(3)Å, N(2)–Ca(1)–O(1) 94.9(1)°) which exhibit O–Ca–N angles that decrease markedly with increased Ca–N bond length. In contrast to archival data, the Ca–O (Ca(1)–O(1) 2.323(3) Å) and Ca–N (mean 2.38 Å) bond lengths of 1 are shorter than the mean structurally authenticated Ca–OTHF bond (2.38 Å)23 or those of the compound trans-[Ca{C(Ph){N(SiMe3)}2}2(THF)2] (mean Ca–O, 2.38 Å; mean Ca–N, 2.43 Å)24 which, with the exception of 1, is the only example of a structurally authenticated calcium species bearing an anionic β-dinitrogen ligand. This reflects the lower coordination number of 1 (five versus six), the steric bulk of the formamidinate ligand frustrating further THF donation.
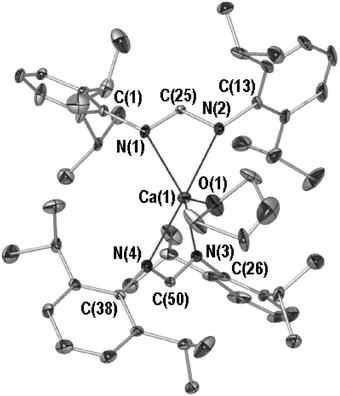 | ||
| Fig. 1 Molecular structure of 1, hydrogen atoms and lesser occupancy disordered atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Ca(1)–N(1) 2.361(3), Ca(1)–N(2) 2.406(3), Ca(1)–N(3) 2.374(3), Ca(1)–N(4) 2.389(3), Ca(1)–O(1) 2.323(3), N(1)–C(25) 1.317(4), N(2)–C(25) 1.316(4), N(3)–C(50) 1.333(4), N(4)–C(50) 1.314(4), N(1)–Ca(1)–N(2) 57.3(1), N(1)–C(25)–N(2) 120.5(3), N(3)–Ca(1)–N(4) 57.8(1), N(3)–C(50)–N(4) 120.7(3), O(1)–Ca(1)–N(1) 110.8(1), O(1)–Ca(1)–N(2) 94.9(1), O(1)–Ca(1)–N(3) 116.3(1), O(1)–Ca(1)–N(4) 92.1(1), N(1)–Ca(1)–N(3) 132.9(1), N(1)–Ca(1)–N(4) 120.7(1). | ||
Compounds 2 and 3 are isostructural and possess six-coordinate metal centres ligated by two cisoid terminal THF donors. This is perhaps surprising given the transoid nature of magnesium bis(formamidinate) species coordinated by two THF donors.9 The molecular structure of 2 is depicted in Fig. 2 (POV-RAY illustration, 40% thermal ellipsoids), with relevant bond lengths and angles for both 2 and 3 (the latter in parentheses) listed in the figure caption. Like 1, compounds 2 and 3 possess a metal centre that sits above the rms plane generated by the four nitrogen donors (0.788(2) Å and 0.908(2) Å, respectively). These donors emanate from two NCN donor sets that exist at an appreciable NCN–NCN angle to one another (33.9(4)° and 31.8(4)°, respectively). However, this angle is significantly smaller than that in 1 (see above) owing to the inclusion of an extra THF donor. This renders the metal centres of 2 and 3 six-coordinate although the actual geometry about the alkaline earth is probably best described as a distorted tetrahedron if one considers the DippForm ligands as point charges located at the carbon of the formamidinate NCN backbone. This provides inter-ligand angles ranging from 103.4(1)° to 108.9(1)° for the C–Sr–O angles of 2, and 104.4(1)° to 108.8(1)° for the analogous angles of 3. The inclusion of the second THF donor, as is consistent with the increased ionic radii of Sr2+ and Ba2+ relative to Ca2+ (increase of 0.18 and 0.35 Å, respectively for six-coordinate M2+),17 leads to Sr–OTHF and Sr–N bond lengths in 2 that are consistent with the mean archival strontium to tetrahydrofuran oxygen bond length (Sr–O 2.55 Å;232: Sr(1)–O(1) 2.542(3) Å, Sr(1)–O(2) 2.583(2) Å) and strontium to nitrogen bond lengths that compare favourably to those of the only other structurally authenticated strontium species bearing amidinate ligands; trans-[Sr{C(Ph){N(SiMe3)}2}2(THF)2] (note: transoid) and [Sr{C(Ph){N(SiMe3)}2}2(diglyme)] (mean Sr–N 2.58 and 2.69 Å respectively;252: mean Sr–N, 2.58 Å), the latter isolated as a diphenylacetylene clathrate.25 By contrast, the increased ionic radius of the barium of 3 (with the same donor ensemble as 2) exhibits Ba–O and Ba–N bond lengths that are considerably shorter than the mean archival Ba–OTHF bond length (2.77 Å;233: Ba(1)–O(1) 2.709(3) Å, Ba(1)–O(2) 2.730(9) Å) or the mean barium to nitrogen contact (2.92 Å—inclusive of amide, imine and amine Ba–N bond lengths;233: mean Ba–N, 2.73 Å), and metallocycle NMN bite angles that are more acute than those of 2 (2: 52.9(1) and 52.9(1)°; 3: 49.9(1) and 49.8(1)°). There is no precedent for Ba–Namidinate bond lengths as 3 represents the first β-dinitrogen compound of barium to be characterised by single crystal X-ray structure determination.26
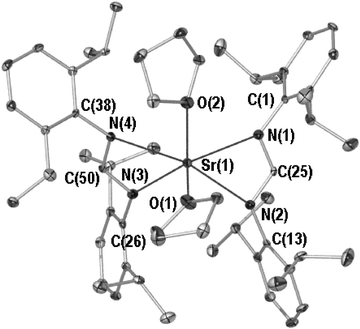 | ||
| Fig. 2 Molecular structure of 2, hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°) for 2 and 3 (latter in parentheses): M(1)–N(1) 2.537(3) (2.685(3)), M(1)–N(2) 2.617(3) (2.763(3)), M(1)–N(3) 2.565(3) (2.693(3)), M(1)–N(4) 2.619(3) (2.769(3)), M(1)–O(1) 2.542(2) (2.709(3)), M(1)–O(2) 2.583(3) (2.730(3)), N(1)–C(25) 1.320(4) (1.313(4)), N(2)–C(25) 1.318(4) (1.315(4)), N(3)–C(50) 1.329(4) (1.318(4)), N(4)–C(50) 1.326(4) (1.320(4)), N(1)–M(1)–N(2) 52.9(1) (49.9(1)), N(1)–C(25)–N(2) 120.9(3) (122.2(3)), N(3)–M(1)–N(4) 52.9(1) (49.8(1)), N(3)–C(50)–N(4) 120.8(3) (121.5(3)), O(1)–M(1)–O(2) 78.4(1) (80.1(1)), O(1)–M(1)–N(1) 112.7(1) (114.5(1)), O(1)–M(1)–N(2) 93.6(1) (100.6(1)), O(1)–M(1)–N(3) 110.6(1) (112.3(1)), O(1)–M(1)–N(4) 107.3(1) (107.1(1)), N(1)–M(1)–N(3) 126.7(1) (126.7(1)), N(1)–M(1)–N(4) 133.6(1) (131.1(4)). | ||
Although rare, the heterolytic cleavage of C–F bonds at highly electropositive metal centres constitutes one of the most common instances of C–F activation.27 This process generally occurs via α-fluoride abstraction with the strength of the metal–fluoride bond acting as a significant driving force. Given the inferred presence of an [M(C6F5)2] intermediate prior to the generation of 1–3 (see above), we thought it plausible that limiting the amount of DippFormH used in the synthesis of the bis(formamidinate) species would result in the formation of either “[M(C6F5)(DippForm)]” species (which may be subject to heterolytic C–F cleavage to provide [(F)M(DippForm)] compounds) or a 50 ∶ 50 mixture of 1–3 and [M(C6F5)2]. As previously mentioned, [Eu(C6F5)2] has been isolated by Forsyth and Deacon.21 This compound, like its relative [Yb(C6F5)2],28 decomposes over a short period (<24 h) furnishing a complex mixture of fluorinated bi- and terphenyls and the metal fluoride, which is consistent with the addition of tetrafluorobenzyne (the putative by-product of heterolytic C–F cleavage) to europium or ytterbium substituted perfluoroaryls. Put simply, this suggests that the “[M(C6F5)(DippForm)]” species, if stable with respect to disproportionation, will yield the heavy alkaline earth organoamide Grignard species [(F)M(DippForm)], by tetrafluorobenzyne elimination.27 Grignard reagents of the heavy alkaline earths are very rare and to our knowledge none have been structurally characterised.1,23 This contrasts with the related chemistry of divalent ytterbium which can be accessed, like that of magnesium, by the direct reaction of ytterbium metal with aryl and alkyl halides.29
In this vein, the preparation of compounds 1–3 was repeated using half the required DippFormH and monitored by 19F NMR for 3–5 days depending on the metal concerned (see Scheme 1). Throughout this period resonances attributable to the typical redox transmetallation ligand exchange by-product; pentafluorobenzene, and a second unknown product (with two fluorine environments, broad multiplet at δ −138.0 and broad singlet at −141.2 ppm, in a 1 ∶ 1 ratio) were observed (in all three instances) with minor associated fluorinated by-products. Beyond this time a 1 ∶ 2 ratio of these compounds prevailed,30 while the reaction mixtures appeared rich in metallic by-product. Filtration followed by removal of volatiles in vacuo gave solely the unidentified by-product 4 (by 19F NMR) as a clean colourless powder that was recrystallised from diethyl ether after washing with cold hexane. Spectroscopic characterisation of 4, in particular 1H and 13C{1H} NMR spectra, indicate at least one rotationally constrained DippForm ligand, as evidenced by the presence of two inequivalent isopropyl methyls attached to the same methyne carbon. This kind of rotational hindrance is common for N-Dipp substituted amidinates with bulkier groups on the NCN backbone carbon31 and, in tandem with the N–C–N asymmetry evident in the DippForm backbone (4: broad strong FTIR C–N stretches at 1657, 1628, 1589 and 1500 cm−1), suggest the presence of further steric bulk at nitrogen beyond that of the Dipp substituent.
To identify the exact nature of this spatial influence, and elucidate the source of the unidentified 19F NMR resonances, a low temperature single crystal X-ray structure determination of 4 was undertaken. Crystal data and refinement parameters are summarised in the experimental section, while the molecular structure of 4 can be seen in Fig. 3 (POV-RAY illustration, 40% thermal ellipsoids, relevant bond lengths and angles given in the figure caption). Compound 4 comprises a DippForm with a 4-HC6F4 substituent bonded to the amine nitrogen, hence the observed rotational hindrance about the proximal Dipp isopropyl groups. Aside from the bond lengths across the NCN backbone, which are consistent with discrete single (C(25)–N(1) 1.381(2) Å) and double (C(25)–N(2) 1.263(2) Å) carbon to nitrogen bonds, the bond lengths and angles of 4 are unremarkable and will not be discussed further, however, the presence of a 4-HC6F4 group at nitrogen is intriguing. We believe this arises from nucleophilic attack of [DippForm]− upon the pentafluorobenzene by-product of A. This has precedent in the organoamide chemistry of platinum(II),32,33 while pentafluorobenzene is known to be susceptible to facile nucleophilic displacement of the para-fluorine.34 It is noteworthy that the abject absence of compound 4 during the preparation of compounds 1 to 3 (as evidenced by 19F NMR) suggests the bis(formamidinate) compounds are incapable of participating in this substitution despite being generated in the presence of pentafluorobenzene. This may arise due to steric congestion about the “MNCN” metallocycles. Thus, we conject that the species responsible for the formation of 4 are indeed the organoamide analogues of heavy Grignard reagents with composition “[(X)M(DippForm)]”, where X = C6F5 or F. We are inclined to suggest the latter of these is more likely due to increased stability21,28 and the limited but disparate fluorine-containing by-products that were observed during the synthesis of 4 (19F NMR, see above). These would emanate from the addition of tetrafluorobenzyne to alkaline earth metal bound perfluoroaryls prior to elimination by C–F cleavage,27 thereby rendering insoluble metal fluoride by-products that would not be visible during reaction due to the dark metallic precipitate that accompanies redox transmetallation syntheses of this type.
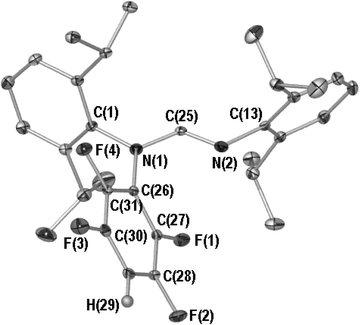 | ||
| Fig. 3 Molecular structure of 4, all hydrogen atoms except H(29) are omitted for clarity. Selected bond lengths (Å) and angles (°): N(1)–C(25) 1.381(2), N(2)–C(25) 1.263(2), N(1)–C(26) 1.422(2), N(1)–C(25)–N(2) 123.9(1), C(1)–N(1)–C(25) 119.2(1), C(1)–N(1)–C(26) 120.2(1), C(25)–N(1)–C(26) 120.6(1), C(13)–N(2)–C(25) 118.3(1). | ||
We are presently applying this facile “one-pot” preparation to the synthesis of divalent and trivalent lanthanoid N,N′-di(aryl)formamidinate species. The outcomes of this study will form the basis of a forthcoming publication.
Experimental section
N,N′-Bis(2,6-diisopropylphenyl)formamidine, DippFormH, was synthesised according to a modified published procedure.35 Alkaline earth metals were purchased from Aldrich, stored under purified nitrogen and freshly filed prior to use. Bis(pentafluorophenyl)mercury was prepared as per a published procedure.36 Tetrahydrofuran (THF), hexane and diethyl ether were dried over sodium, freshly distilled from sodium benzophenone ketyl and freeze–thaw degassed prior to use. Toluene was dried over sodium and freshly distilled from potassium prior to use. All manipulations were performed using conventional Schlenk or glovebox techniques under an atmosphere of high purity dinitrogen in flame-dried glassware. Infrared spectra were recorded as Nujol mulls using sodium chloride plates on a Nicolet Nexus FTIR spectrophotometer. 1H NMR spectra were recorded at 300.13 MHz, 13C{1H} NMR spectra were recorded at 75.46 MHz and 19F NMR spectra were recorded at 282.40 MHz using a Bruker DPX 300 spectrometer, with chemical shifts referenced to the residual 1H or 13C resonances of the deutero-benzene solvent used or an external CCl3F standard (δ = 0). Melting points were determined in sealed glass capillaries under dinitrogen and are uncorrected. All microanalyses were conducted by the Campbell Microanalytical Laboratory, Chemistry Department, University of Otago, P.O. Box 56, Dunedin, New Zealand.General procedure for compounds 1–3: Tetrahydrofuran (20 cm3) was added to a Schlenk flask charged with alkaline earth metal filings (0.03 g, 0.75 mmol Ca (1), 0.07 g, 0.80 mmol Sr (2) or 0.10 g, 0.73 mmol Ba (3)), bis(pentafluorophenyl)mercury (0.37 g, 0.69 mmol) and DippFormH (0.50 g, 1.37 mmol) under purified nitrogen. The resulting slurry was stirred at ambient temperature for 24 h before filtration (to remove elemental mercury and excess alkaline earth metal) and the removal of all volatiles in vacuo. The colourless powdered material obtained was worked-up as follows.
[Ca(DippForm)2(THF)] (1)
Extraction into fresh tetrahydrofuran (10 cm3) followed by moderate concentration (to ca. 8 cm3) gave colourless rectangular blocks of 1 after standing at ambient temperature for several hours (0.29 g, 50% by DippFormH), mp 262 °C. Elemental analyses calculated (%) for C54H78N4O1Ca1: C 77.28, H 9.37, N 6.68; found: C 76.90, H 9.35, N 6.16; IR (Nujol): ν = 1920 w, 1856 w, 1794 w, 1664 m, 1592 m, 1520 s, 1459 s, 1439 s, 1380 m sh, 1360 m sh, 1315 m, 1288 s sh, 1254 m, 1191 m, 1100 m, 1050 m, 1030 m, 938 m, 877 m, 801 m, 758 m cm−1; 1H NMR (300.1 MHz, C6D6, 300 K): δ = 8.16 (s, 1H; NC(H)N), 7.20–7.09 (m, 12H; Ar–H), 3.73 (m, 4H; OCH2, THF), 3.49 (septet, 3J(H,H) = 6.9 Hz, 8H; CH, iPr), 1.33 (m, 4H; CH2, THF), 1.21 (d, 3J(H,H) = 6.9 Hz, 48H; CH3, iPr); 13C{1H} NMR (75.5 MHz, C6D6, 300 K): δ = 169.4 (s; NC(H)N), 147.6 (s; Ar–C), 142.8 (s; Ar–C), 123.9 (s; Ar–CH), 123.7 (s; Ar–CH), 69.6 (s; OCH2, THF), 29.0 (s; CH, iPr), 25.7 (s; CH2, THF), 24.8 (s; CH3, iPr).[Sr(DippForm)2(THF)2] (2)
Extraction into toluene (20 cm3) followed by concentration in vacuo (to ca. 15 cm3) and placement at −5 °C overnight yielded 2 as colourless rhombohedral plates (0.47 g, 72% by DippFormH), mp 242 °C. Elemental analyses calculated (%) for C58H86N4O2Sr1: C 72.64, H 9.04, N 5.84; found: C 72.41, H 9.25, N 5.98; IR (Nujol): ν = 1906 w, 1850 w, 1789 w, 1666 m, 1593 w, 1530 s, 1460 s, 1378 m sh, 1308 m, 1250 m, 1236 m, 1182 m, 1105 w, 1037 m sh, 932 m, 879 m, 800 m, 752 m sh, 722 w cm−1; 1H NMR (300.1 MHz, C6D6, 300 K): δ = 8.12 (s, 1H; NC(H)N), 7.23–7.11 (m, 12H; Ar–H), 3.64 (m, 8H; OCH2, THF), 3.61 (partially obscured septet, 3J(H,H) = 6.9 Hz, 8H; CH, iPr), 1.37 (m, 8H; CH2, THF), 1.26 (d, 3J(H,H) = 6.9 Hz, 48H; CH3, iPr); 13C{1H} NMR (75.5 MHz, C6D6, 300 K): δ = 168.5 (s; NC(H)N), 149.6 (s; Ar–C), 143.0 (s; Ar–C), 123.6 (s; Ar–CH), 123.3 (s; Ar–CH), 68.7 (s; OCH2, THF), 28.6 (s; CH, iPr), 25.8 (s; CH2, THF), 25.1 (s; CH3, iPr).[Ba(DippForm)2(THF)2] (3)
Extraction into toluene (20 cm3) followed by concentration in vacuo (by <1 cm3) induced slight precipitation. Filtration followed by standing (7 days) at ambient temperature rendered 3 as colourless rhombohedral plates (0.54 g, 78% by DippFormH), mp 202 °C (dec.). Elemental analyses calculated (%) for C58H86N4O2Ba1: C 69.07, H 8.59, N 5.55; Found: C 68.92, H 8.67, N 5.81; IR (Nujol): ν = 1909 w, 1851 w, 1789 w, 1666 m, 1593 m, 1528 s, 1461 s, 1434 s, 1378 m sh, 1358 m sh, 1315 s, 1250 m, 1236 m, 1182 m sh, 1107 m, 1040 m, 1012 w, 926 m, 880 m, 799 m, 754 s sh cm−1; 1H NMR (300.1 MHz, C6D6, 300 K): δ = 8.17 (br s, 1H; NC(H)N), 7.22–7.10 (m, 12H; Ar–H), 3.56 (m, 16H; coincident resonances for OCH2, THF and CH, iPr), 1.34 (m, 8H; CH2, THF), 1.28 (d, 3J(H,H) = 6.9 Hz, 48H; CH3, iPr); 13C{1H} NMR (75.5 MHz, C6D6, 300 K): δ = 166.9 (s; NC(H)N), 150.2 (s; Ar–C), 142.6 (s; Ar–C), 123.5 (s; Ar–CH), 123.0 (s; Ar–CH), 68.4 (s; OCH2, THF), 28.8 (s; CH, iPr), 25.8 (s; CH2, THF), 24.9 (s; CH3, iPr).DippForm(4-HC6F4) (4)
Tetrahydrofuran (20 cm3) was added to a Schlenk flask charged with alkaline earth metal filings (0.75 mmol), bis(pentafluorophenyl)mercury (0.37 g, 0.69 mmol) and DippFormH (0.25 g, 0.69 mmol) under purified nitrogen. The resulting slurry was stirred at ambient temperature for 3–5 days before filtration (to remove elemental mercury, excess alkaline earth metal and precipitated by-products) and the removal of all volatiles in vacuo to yield a waxy colourless solid. Washing with cold (0 °C) hexane (2 × 1 cm3) and extraction into diethyl ether (5 cm3) yielded large irregular plates of 4 in three batches after sequential concentration in vacuo and placement at −5 °C over several days (overall mass 0.29 g, 82%), mp 178 °C. Elemental analyses calculated (%) for C31H36N2F4: C 72.63, H 7.08, N 5.46; found: C 72.69, H 6.83, N 5.36; IR (Nujol): ν = 3062 s sh, 1943 w, 1919 w, 1870 w, 1857 w, 1795 w, 1657 s, 1628 s, 1589 s sh, 1500 s, 1464 s, 1406 m, 1384 m sh, 1365 m sh, 1032 s, 1258 m, 1229 m, 1204 m, 1177 s, 1104 m, 1054 m, 1016 m sh, 976 m, 957 m, 932 m, 910 m, 847 m, 800 m, 754 m, 738 w, 718 m, 675 m, 631 m, 609 m cm−1; 1H NMR (300.1 MHz, C6D6, 300 K): δ = 7.68 (br s, 1H; NC(H)N), 7.16–7.03 (m, 6H; Ar–H), 6.07 (tt, 3J(H,F) = 9.6 Hz, 4J(H,F) = 7.2 Hz, 1H; 4-HC6F4), 3.48 (partially obscured septet, 3J(H,H) = 6.9 Hz, 2H; imino Dipp CH, iPr), 3.45 (partially obscured septet, 3J(H,H) = 6.8 Hz, 2H; amino Dipp CH, iPr), 1.22 (d, 3J(H,H) = 6.9 Hz, 12H; imino Dipp CH3, iPr), 1.17 (d, 3J(H,H) = 6.8 Hz, 6H; amino Dipp CH3, iPr), 1.05 (d, 3J(H,H) = 6.8 Hz, 6H; amino Dipp CH3, iPr); 13C{1H} NMR (75.5 MHz, C6D6, 300 K): δ = 150.6 (s; NC(H)N), 148.8 (m; Ar–CF), 148.4 (s; Ar–C), 146.4 (s; Ar–C), 145.5 (m; Ar–CF), 141.9 (m; Ar–C), 140.5 (s; Ar–C), 136.7 (s; Ar–C), 130.0 (s; Ar–CH), 125.4 (s; Ar–CH), 124.7 (s; Ar–CH), 124.0 (s; Ar–CH), 103.3 (t, 2J(C,F) = 22.9 Hz), 29.2 (s; amino Dipp CH, iPr), 28.3 (s; imino Dipp CH, iPr), 25.7 (s, amino Dipp CH3, iPr), 24.7 (s, imino Dipp CH3, iPr), 23.8 (s, amino Dipp CH3, iPr); 19F NMR (282.4 MHz, C6D6, 300 K): δ = −138.0 (m; Ar–CF), −141.2 (br s, width at half peak height = 340 Hz; Ar–CF).X-Ray structure determination
Crystalline samples of compounds 1–4 were mounted on glass fibres in silicone oil at −150(2) °C (123(2) K). Data were collected on an Enraf-Nonius Kappa CCD diffractometer using graphite monochromated Mo Kα X-ray radiation (λ = 0.71073 Å). Data were corrected for absorption by the DENZO-SMN package.37 Lorentz polarisation and absorption corrections were applied. Structural solution and refinement was carried out using the SHELX suite of programs38,39 with the graphical interface X-Seed.40 Crystal data for 1: C54H78N4O1Ca1, M = 839.28, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a
= 11.174(2), b
= 14.010(3), c
= 17.346(4)
Å, α
= 93.49(3), β
= 97.46(3), γ
= 109.51(3)°, V
= 2522.0(9)
Å3, Z
= 2, Dc
= 1.105 g cm−3, F000
= 916, μ
= 0.164 mm−1, 2θmax
= 55.80°, 28677 reflections collected, 11710 unique (Rint
= 0.1398). Final GooF = 0.947, R1 = 0.0820, wR2 = 0.1437, R indices based on 11710 reflections with I > 2σ(I)
(refinement on F2), 648 parameters, 0 restraints. Crystal data for 2: C58H86N4O2Sr1, M
= 958.93, triclinic, P
(no. 2), a
= 12.0689(3), b
= 12.7964(5), c
= 19.6889(7)
Å, α
= 84.466(3), β
= 86.449(3), γ
= 64.2010(10)°, V
= 2724.21(16)
Å3, Z
= 2, Dc
= 1.169 g cm−3, F000
= 1032, μ
= 1.033 mm−1, 2θmax
= 56.14°, 26694 reflections collected, 12922 unique (Rint
= 0.1028). Final GooF = 1.038, R1 = 0.0723, wR2 = 0.1359, R indices based on 12922 reflections with I > 2σ(I)
(refinement on F2), 602 parameters, 0 restraints. Crystal data for 3: C58H86N4O2Ba1, M
= 1008.65, triclinic, P
(no. 2), a
= 11.9811(4), b
= 12.8483(4), c
= 20.3537(9)
Å, α
= 84.4630(10), β
= 86.0010(10), γ
= 63.451(3)°, V
= 2788.47(18)
Å3, Z
= 2, Dc
= 1.201 g cm−3, F000
= 1068 μ
= 0.754 mm−1, 2θmax
= 55.76°, 40603 reflections collected, 13111 unique (Rint
= 0.1199). Final GooF = 0.953, R1 = 0.0533, wR2 = 0.0913, R indices based on 13111 reflections with I > 2σ(I)
(refinement on F2), 635 parameters, 0 restraints. Crystal data for 4: C31H36N2F4, M
= 512.62, triclinic, P
(no. 2), a
= 9.4525(4), b
= 11.4180(5), c
= 14.3724(7)
Å, α
= 110.256(3), β
= 91.904(2), γ
= 106.1040(10)°, V
= 1383.70(11)
Å3, Z
= 2, Dc
= 1.230 g cm−3, F000
= 544, μ
= 0.090 mm−1, 2θmax
= 55.58°, 20737 reflections collected, 6442 unique (Rint
= 0.0762). Final GooF = 0.984, R1 = 0.0490, wR2 = 0.1425, R indices based on 6442 reflections with I > 2σ(I)
(refinement on F2), 342 parameters, 0 restraints.
(no. 2), a
= 11.174(2), b
= 14.010(3), c
= 17.346(4)
Å, α
= 93.49(3), β
= 97.46(3), γ
= 109.51(3)°, V
= 2522.0(9)
Å3, Z
= 2, Dc
= 1.105 g cm−3, F000
= 916, μ
= 0.164 mm−1, 2θmax
= 55.80°, 28677 reflections collected, 11710 unique (Rint
= 0.1398). Final GooF = 0.947, R1 = 0.0820, wR2 = 0.1437, R indices based on 11710 reflections with I > 2σ(I)
(refinement on F2), 648 parameters, 0 restraints. Crystal data for 2: C58H86N4O2Sr1, M
= 958.93, triclinic, P
(no. 2), a
= 12.0689(3), b
= 12.7964(5), c
= 19.6889(7)
Å, α
= 84.466(3), β
= 86.449(3), γ
= 64.2010(10)°, V
= 2724.21(16)
Å3, Z
= 2, Dc
= 1.169 g cm−3, F000
= 1032, μ
= 1.033 mm−1, 2θmax
= 56.14°, 26694 reflections collected, 12922 unique (Rint
= 0.1028). Final GooF = 1.038, R1 = 0.0723, wR2 = 0.1359, R indices based on 12922 reflections with I > 2σ(I)
(refinement on F2), 602 parameters, 0 restraints. Crystal data for 3: C58H86N4O2Ba1, M
= 1008.65, triclinic, P
(no. 2), a
= 11.9811(4), b
= 12.8483(4), c
= 20.3537(9)
Å, α
= 84.4630(10), β
= 86.0010(10), γ
= 63.451(3)°, V
= 2788.47(18)
Å3, Z
= 2, Dc
= 1.201 g cm−3, F000
= 1068 μ
= 0.754 mm−1, 2θmax
= 55.76°, 40603 reflections collected, 13111 unique (Rint
= 0.1199). Final GooF = 0.953, R1 = 0.0533, wR2 = 0.0913, R indices based on 13111 reflections with I > 2σ(I)
(refinement on F2), 635 parameters, 0 restraints. Crystal data for 4: C31H36N2F4, M
= 512.62, triclinic, P
(no. 2), a
= 9.4525(4), b
= 11.4180(5), c
= 14.3724(7)
Å, α
= 110.256(3), β
= 91.904(2), γ
= 106.1040(10)°, V
= 1383.70(11)
Å3, Z
= 2, Dc
= 1.230 g cm−3, F000
= 544, μ
= 0.090 mm−1, 2θmax
= 55.58°, 20737 reflections collected, 6442 unique (Rint
= 0.0762). Final GooF = 0.984, R1 = 0.0490, wR2 = 0.1425, R indices based on 6442 reflections with I > 2σ(I)
(refinement on F2), 342 parameters, 0 restraints.
Variata
For compound 1, three isopropyl groups were disordered over two sites of partial occupancy. For the isopropyls attached to C(18) and C(27) this comprised disorder of the methyl groups (C(23)/C(24) and C(33)/C(34), respectively). These were modelled successfully with 56 ∶ 44 and 67 ∶ 33 occupancies respectively. For the isopropyl group attached to C(2), methyne and methyl protons (C(7), C(8) and C(9)) were disordered. The disorders of these were modelled successfully with occupancies of 54 ∶ 46. Two methylene carbons (C(52) and C(54)) of the coordinated THF molecule also exhibited disorder. This disorder was successfully refined with occupancies of 51 ∶ 49.For compound 3, the methyls of three isopropyl groups attached to C(6), C(18) and C(27) were disordered over two sites of partial occupancy (C(12) and C(23)/C(24) and C(33)/C(34) respectively). These were modelled successfully with occupancies of 68 ∶ 32, 63 ∶ 37 and 52 ∶ 48, respectively.
Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre.‡
Acknowledgements
The authors would like to sincerely thank Prof. Glen B. Deacon for invaluable discussions. They also thank the Australian Research Council (ARC) for continued financial support.References
- J. S. Alexander and K. Ruhlandt-Senge, Eur. J. Inorg. Chem., 2002, 2761 CrossRef CAS.
- T. P. Hanusa, Organometallics, 2002, 21, 2559 CrossRef CAS.
- G. B. Deacon, C. M. Forsyth and P. C. Junk, J. Organomet. Chem., 2000, 607, 112 CrossRef CAS.
- G. B. Deacon, E. E. Delbridge, B. W. Skelton and A. H. White, Eur. J. Inorg. Chem., 1998, 543 CrossRef CAS.
- G. B. Deacon, C. M. Forsyth and S. Nickel, J. Organomet. Chem., 2002, 647, 50 CrossRef CAS.
- G. B. Deacon, C. M. Forsyth, P. C. Junk, B. W. Skelton and A. H. White, Chem. Eur. J., 1999, 5, 1452 CrossRef CAS.
- G. B. Deacon, C. M. Forsyth, A. Gitlits, R. Harika, P. C. Junk, B. W. Skelton and A. H. White, Angew. Chem., Int. Ed., 2002, 41, 3249 CrossRef CAS.
- During the preparation of this manuscript the first heavy alkaline earth metal pyrazolates were reported by Deacon, Ruhlandt-Senge and co-workers using metal-based syntheses: J. Hitzbleck, A. Y. O’Brien, C. M. Forsyth, G. B. Deacon and K. Ruhlandt-Senger, Chem. Eur. J., 2004, 10, 3315 Search PubMed.
- M. L. Cole, D. J. Evans, P. C. Junk and L. M. Louis, New J. Chem., 2002, 1015 RSC.
- J. Baldamus, C. Berghof, M. L. Cole, D. J. Evans, E. Hey-Hawkins and P. C. Junk, J. Chem. Soc., Dalton Trans., 2002, 2802 RSC.
- J. Baldamus, C. Berghof, M. L. Cole, E. Hey-Hawkins, P. C. Junk and L. M. Louis, Eur. J. Inorg. Chem., 2002, 2878 CrossRef CAS.
- M. L. Cole, P. C. Junk and L. M. Louis, J. Chem. Soc., Dalton Trans., 2002, 3906 RSC.
- J. Baldamus, C. Berghof, M. L. Cole, D. J. Evans, E. Hey-Hawkins and P. C. Junk, J. Chem. Soc., Dalton Trans., 2002, 4185 RSC.
- M. L. Cole and P. C. Junk, J. Organomet. Chem., 2003, 666, 55 CrossRef CAS.
- M. L. Cole, D. J. Evans, P. C. Junk and M. K. Smith, Chem. Eur. J., 2003, 9, 415 CrossRef CAS.
- M. L. Cole, A. J. Davies, C. Jones and P. C. Junk, J. Organomet. Chem., 2004, 689, 3093 CrossRef CAS.
- E. Fluck and K. G. Heumann, Periodic Table of the Elements, VCH, Weinheim, Germany, 1991 Search PubMed.
- One divalent lanthanoid complex supported by amidinate ligands has been reported: M. Wedler, M. Noltemeyer, U. Pieper, H. G. Schmidt, D. Stalke and F. T. Edelmann, Angew. Chem., Int. Ed. Eng., 1990, 29, 894 Search PubMed.
- M. N. Bochkarev, Coord. Chem. Rev., 2004, 248, 835 CrossRef CAS.
- By extrapolation from the increase in ionic radii for six-coordinate Eu3+ to Eu2+ (+0.22 Å), the radii of La (La3+ = 1.03 Å) and Ce (Ce3+ = 1.01 Å) in the divalent oxidation state will approach 1.25 and 1.23 Å, respectively.
- C. M. Forsyth and G. B. Deacon, Organometallics, 2000, 19, 1205 CrossRef CAS.
- Attempts to isolate the intermediates [M(C6F5)2(THF)n], where M = Ca, Sr or Ba, provided the insoluble alkaline earth fluorides and a complex mixture of fluorinated aromatic species, the latter discerned by 19F NMR. For a comprehensive description of the decomposition of related divalent lanthanoid species please consult ref. 21 and 28.
- From a survey of the CCDC (CSD Version 5.25, Nov. 2003 plus updates for Januarty, April and July 2004).
- M. Westerhausen and W. Schwarz, Z. Naturforsch., B: Chem. Sci., 1992, 47, 453 CAS.
- M. Westerhausen, H. D. Hausen and W. Schwarz, Z. Anorg. Allg. Chem., 1992, 618, 121 CAS.
- Barium to nitrogen bond lengths for the compound [Ba{N(SiMe3)2}2(THF)2] have been reported, however these are not directly comparable to 3 due to the low-coordination of the metal centre (2.587(6) and 2.596(6) Å): B. A. Vaartstra, J. C. Huffman, W. E. Streib and K. G. Caulton, Inorg. Chem., 1991, 30, 121 Search PubMed.
- J. L. Kiplinger, T. G. Richmond and C. E. Osterberg, Chem. Rev., 1994, 94, 373 CrossRef CAS.
- See: G. B. Deacon and C. M. Forsyth, Organometallics, 2003, 22, 1349 Search PubMed and references therein.
- F. A. Cotton, G. Wilkinson, C. A. Murillo and M. Bochmann, Advanced Inorganic Chemistry, Wiley-Interscience, New York, USA, 6th edn., 1999 Search PubMed.
- Ratio based on the greatest intensity 19F resonance of pentafluorobenzene divided by two.
- R. T. Boeré, M. L. Cole and P. C. Junk, New J. Chem., 2005 10.1039/b409086a (this issue).
- D. P. Buxton, G. B. Deacon, B. M. Gatehouse, I. L. Grayson, R. J. Thomson and D. St. C. Black, Aust. J. Chem., 1986, 39, 2013 CAS.
- D. P. Buxton, G. B. Deacon, B. M. Gatehouse, I. L. Grayson and D. St. C. Black, Aust. J. Chem., 1988, 41, 943 CAS.
- R. D. Chambers, W. K. R. Musgrave, J. S. Waterhouse, D. L. H. Williams, J. Burdon, W. B. Hollyhead and J. C. Tatlow, J. Chem. Soc., Chem. Commun., 1974, 239 RSC.
- R. M. Roberts, J. Org. Chem., 1949, 14, 277 CrossRef CAS.
- G. B. Deacon, J. E. Cosgriff, E. T. Lawrence, C. M. Forsyth and D. L. Wilkinson, in Herrmann–Brauer, Synthetic Methods of Organometallic and Inorganic Chemistry, ed. W. A. Herrmann, Thieme, Stuttgart, Germany, 3rd edn., 1997 Search PubMed.
- Z. Otwinowski and W. Minor, in Macromolecular Crystallography, Part A: Processing of X-ray Diffraction Data Collected in Oscillation Mode, Methods Enzymol., ed. C. W. Cater and R. M. Sweet, Academic Press, New York, USA, 1997, vol. 276, p. 307 Search PubMed.
- G. M. Sheldrick, SHELXL-97, University of Göttingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SHELXS-97, University of Göttingen, Germany, 1997 Search PubMed.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189 Search PubMed.
Footnotes |
| † Present address: Department of Chemistry, University of Adelaide, South Australia 5005, Australia. |
| ‡ CCDC reference numbers 247616–247619. See http://www.rsc.org/suppdata/nj/b4/b411494a/ for crystallographic data in .cif or other electronic format. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2005 |