Functional gene-discovery systems based on libraries of hammerhead and hairpin ribozymes and short hairpin RNAs
Masayuki
Sano
a,
Yoshio
Kato
a and
Kazunari
Taira
*ab
aGene Function Research Center, National Institute of Advanced Industrial Science and Technology (AIST), Central 4, 1-1-1 Higashi, Tsukuba Science City 305-8562, Japan
bDepartment of Chemistry and Biotechnology, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Tokyo 113-8656, Japan. E-mail: taira@chembio.t.u-tokyo.ac.jp; Fax: 81-3-5841-8828 or 81-29-861-3019; Tel: 81-3-5841-8828 or 81-29-861-3015
First published on 13th April 2005
Abstract
Abundant information about the nucleotide sequence of the human genome has become readily available and it is now necessary to develop methods for the identification of genes that are involved in important cellular, developmental and disease-related processes. Identification methods based on the activities of hammerhead and hairpin ribozymes and of short hairpin RNAs (shRNAs), whose target specificities are coupled with loss-of-function phenotypes, have received increasing attention as possible tools for the rapid identification of key genes involved in such processes. We describe here recent advances that have been made with libraries of ribozymes and shRNAs and compare the advantages of the different types of library. The use of such libraries has already revealed new details of several important physiological phenomena.
 Masayuki Sano | Masayuki Sano is a research scientist at the Gene Function Research Center of the National Institute of Advanced Industrial Science and Technology (AIST) in Tsukuba Science City. He received both his MSc (2000) and PhD (2003) from University of Tsukuba, where he conducted research on the ribozyme technology under the guidance of Professor Kazunari Taira. From 2003–2004 he was a postdoctoral fellow in Prof. Taira’s group at AIST. His current research interest is RNAi technology and microRNA function. |
 Yoshio Kato | Yoshio Kato was born in 1976 in Japan. He received his BA in chemistry from the University of Tsukuba in 1999 and his PhD in biotechnology from the University of Tokyo in 2004. At the University of Tokyo, he worked with Prof. Kazunari Taira on the expression system of small functional RNAs, including ribozymes and siRNAs. He is currently a researcher at the National Institute of Advanced Industrial Science and Technology (AIST), Tsukuba, Japan. |
 Kazunari Taira | Kazunari Taira is a Professor at the University of Tokyo. He also holds a Director position at the Gene Function Research Center of the National Institute of Advanced Industrial Science and Technology (AIST) in Tsukuba Science City and he is a founder of iGENE Therapeutics, Inc. Taira obtained his PhD degree (1984) in chemistry from the University of Illinois. After three years of postdoctoral work at Pennsylvania State University, he returned to Japan and joined the faculty of AIST in 1987. In 1994 he was appointed Full Professor at the Institute of Applied Biochemistry, University of Tsukuba. In 1999 he moved from University of Tsukuba to the University of Tokyo. The main focus of his research is the use of the tools of physical organic chemistry and molecular biology to design intracellularly active biomolecules. Around 1990, he became interested in the mechanisms of action of ribozymes and their possible application to medicine. More recently, he has also become interested in functional protein selections and RNAi/microRNA technologies in the medical field. |
Introduction
With the accumulation of vast amounts of data as a result of the sequencing of the human genome, it is now necessary to identify human genes that are involved in various cellular, developmental and disease-related processes and to clarify their functions and potential utility as targets in the treatment of disease.1,2 Before the human genome was sequenced, identification of such genes was a slow and laborious process. For example, it was often necessary to define a cellular phenotype, purify the protein of interest, sequence the peptide, screen a cDNA library with a degenerate probe and finally clone the gene that encoded the protein. However, with the new availability of sequence information and “high-throughput” technologies, such as DNA microarrays and yeast two-hybrid systems, it is now possible to identify functional genes much more rapidly.3,4DNA microarray technology allows the monitoring of changes in gene expression during particular cellular processes and is very useful for the identification of multiple genes simultaneously. However, it does not allow specific discrimination of genes that are directly responsible for a given phenotype from many other genes that are involved only indirectly. Yeast two-hybrid systems allow detection of direct interactions between proteins in vivo and, thus, they allow the identification of genes whose products interact with target proteins derived from a cDNA library. However, in such systems, the resultant interactions do not always reflect roles in the process that is being studied and many false-positive results are generated, most probably because high-level expression of bait proteins induces non-specific interactions. Moreover, although DNA microarrays and yeast two-hybrid systems are sensitive and powerful tools, the involvement of identified genes in the phenotype of interest must often be validated by additional experiments. The isolation of pseudo-positive genes that have to be filtered out in additional experiments makes use of these technologies rather tedious.
A simpler technology for the more definitive identification of functional genes involved in a particular phenotype is clearly required and novel gene-discovery systems based on the activities of hammerhead and hairpin ribozymes and short hairpin RNAs (shRNAs) seem likely to be particularly useful. This review focuses on recent advances that have been made with these novel gene-discovery systems and their respective advantages for the identification of functional genes.
Novel gene-discovery systems using randomized libraries of hammerhead and hairpin ribozymes
Libraries of randomized ribozymes provide a novel tool for the identification of functional genes.5–15 Hammerhead and hairpin ribozymes, which have been extensively employed in such systems, are small RNA molecules with catalytic activity.16–20 They contain conserved catalytic domains, as well as substrate-binding arms that determine target specificity (Fig. 1A). These hammerhead and hairpin ribozymes recognize a target gene via interactions between the binding arms of the ribozyme and the transcript of the gene and they can cleave any RNA substrate that contains an NUH triple, where N is any nucleotide and H is A, C or U in the case of the hammerhead ribozyme, or GUC in the case of the hairpin ribozyme. The mechanism of action of hammerhead ribozymes has been investigated in great detail21–25 and these ribozymes have been used by molecular biologists, biotechnologists and medical researchers.26–30 While the binding arms can be designed to include sequences complementary to the target RNA, randomization of binding arms generates a large pool of ribozymes that are capable of cleaving many mRNA substrates. Such a pool of randomized ribozymes is referred to as a library.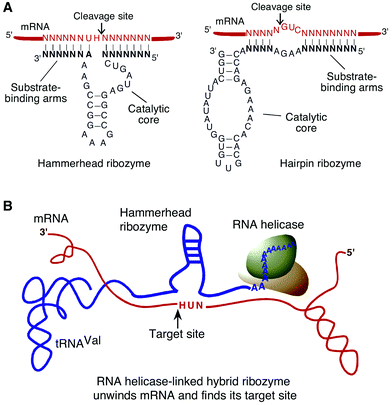 | ||
| Fig. 1 (A) secondary structures of a hammerhead ribozyme (left) and a hairpin ribozyme (right). Bold letters indicate the binding arms of the ribozymes. (B) representation of an RNA helicase-linked hybrid ribozyme. A ribozyme that is coupled to an RNA helicase can cleave a hidden target site after the local secondary structure of the mRNA has been disrupted. | ||
In a ribozyme-based gene-discovery system, a library of randomized ribozymes is introduced into cells, where it causes phenotypic changes as a result of the cleavage of transcripts that are responsible for the particular phenotype. The ribozymes can be recovered from the cells and subsequent sequence analysis, which involves searching for sequences of target site in databases with, for example, the BLAST search program, leads to the identification of the targets of the ribozymes (Fig. 2).6–13,31–35 Alternatively, a partial cDNA fragment that subsequently facilitates the cloning of full-length cDNA can be obtained by 5′- and 3′-RACE (rapid amplification of cDNA ends), which is a PCR-based cloning technique.6–10 Then, limitations related to cleavable triplets, such as GUC, allow the discrimination of real targets from the many false-positive targets. The system is simple and direct, as compared to other “high-throughput” gene-screening methods and no prior sequence information is required. Moreover, because the system allows the identification of direct effectors of a change in phenotype, it reduces the level of background and false-positive results and produces, for the most part, interpretable results.
 | ||
| Fig. 2 Gene discovery using a library of randomized ribozymes. The gene-discovery system includes the introduction of the library into cells, the isolation of cells with an altered phenotype, the recovery of ribozymes from isolated cells, the reintroduction of positive ribozymes into fresh cells and a repeat of the entire assay (if needed). Positive ribozymes are then sequenced and the target genes involved in a particular phenotype are identified. | ||
The design of libraries of randomized ribozymes is clearly important. In the design of libraries of hammerhead ribozymes, our group has had considerable success with the promoter of a gene for human tRNAVal because this promoter ensures the high-level expression of the ribozymes in cells. In our experiments, hammerhead ribozymes transcribed from the tRNAVal promoter were able to efficiently suppress the expression of target genes.36–38 The expression cassette that incorporates the library can be introduced into cells using a viral vector or plasmid vector, depending on the change in phenotype to be examined. Ribozymes that cause a change in the phenotype of interest must be recovered for identification of the complementary target mRNAs that are potential candidates for transcripts of genes of interest. These candidates must be validated by use of other ribozymes that are capable of cleaving a specific target mRNA at other sites. Inactive ribozymes with point mutations within the catalytic site are convenient controls in such analysis. Alternatively, inactivation by small interfering RNAs (siRNAs) can be also used to validate candidate genes.34,35 Once target genes have been identified, they can be characterized individually with respect to the specific involvement of each in the phenotype of interest. The process can then be refined and repeated.
Application of libraries of randomized ribozymes to the analysis of particular phenotypes
There have been several successful attempts to use libraries of randomized ribozymes to identify genes involved in apoptosis,11–13 cell migration and invasion,31,32,34 tumor transformation,7,8 viral replication,6,39 Alzheimer's disease33,40 and the differentiation of normal tissues, such as muscle.35 For example, using libraries of hairpin ribozymes, the laboratories of Wong-Staal and Barber have succeeded in identifying certain tumor-suppressor genes. Non-transformed revertant derivatives of HeLa cells, namely HF cells, were transduced with a library of randomized hairpin ribozymes using a retroviral vector, with the subsequent identification and cloning of a human homolog of the Drosophila gene ppan (the Peter Pan gene) as an important suppressor of the anchorage-independent growth of HF cells.8 A murine telomerase reverse transcriptase (mTERT), the catalytic subunit of telomerase, was also identified as an enzyme that might be involved in the transformation of mouse NIH3T3 cells.7Members of our group have used novel libraries of hybrid hammerhead ribozymes in efforts to improve the access of ribozymes to their targets.11–13,32,35 In general, the secondary and tertiary structures of target mRNAs play very important roles in determining the activity of ribozymes. Extensive double-stranded regions within target mRNAs hinder access by ribozymes to potentially cleavable triplets. Thus, to enhance the accessibility to ribozymes, computer-generated predictions are often used to identify target sites that are most likely to have an open conformation.41 Other groups have exploited oligonucleotide-directed cleavage of target mRNAs by RNase H in cell extracts.42–44 In our laboratory, efforts to solve the problem of target accessibility have included the creation of hybrid ribozymes via attachment of a constitutive transport element (CTE)45 or a naturally occurring RNA motif, the poly(A) sequence,12,13 to the 3′ end of the ribozyme sequence. Thus, for example, since the poly(A) sequence is known to interact with an endogenous RNA helicase, eIF4AI, via interactions with poly(A)-binding protein (PABP) and PABP-interacting protein-1, we are able to combine the cleavage activities of ribozymes with the unwinding activity of the RNA helicase. When RNA helicase is coupled to a ribozyme, the helicase can guide the ribozyme to its target site in a transcript by unwinding any inhibitory secondary structure, with very efficient resultant cleavage of the transcript (Fig. 1B).5,12,13,45
Using a library of poly(A)-attached hybrid randomized ribozymes, our group has been able to identify genes involved in Fas-mediated apoptosis (Fig. 3A).12 We introduced such a library into HeLa-Fas cells using a retroviral vector and then treated the cells with Fas-specific antibodies. We collected surviving cells that harbored hybrid ribozymes targeted to genes involved in Fas-induced apoptosis and sequenced the randomized region of each effective hybrid ribozyme. In this way, we were able to rapidly identify the genes for the Fas-associated death-domain protein (FADD) and for caspases 3, 8 and 9. In addition, we also identified novel genes. In these experiments, use of the library of hybrid ribozymes resulted in only a very small number of false-positive results. Indeed, since the genes for FADD and for caspase 8 were not identified when we used a library of conventional randomized ribozymes without the poly(A) sequence, it was very clear that the hybrid system enhanced the effectiveness of our gene-discovery system.
 | ||
| Fig. 3 Systems for the identification of genes involved in particular processes using libraries of hybrid hammerhead ribozymes. The diagrams show, schematically, the specific use of the libraries to identify genes involved in Fas-mediated apoptosis (A), cell invasion (B) and the differentiation of muscle cells (C). | ||
Our group has also identified genes that are involved in enhancement of the ability of noninvasive cells to invade the extracellular matrix (ECM) by using a library of hybrid ribozymes (Fig. 3B).32 The filter prepared with an ECM gel was used as a barrier to block the migration of noninvasive cells. Cells that had been transduced with the library of hybrid ribozymes were subjected to the assay of invasive activity, as shown schematically in Fig. 3B, and cells that passed through the ECM gel were collected. We were able to identify a gene for Gem GTPase and previously uncharacterized genes that encoded proteins that resemble myosine phosphatase and protein tyrosine phosphatase.
The same technology has been used to identify genes involved in muscle differentiation (Fig. 3C).35 Using a library of hybrid ribozymes, we identified genes for the tumor suppressors p19ARF and p21WAF1, the gene for a member of the sex-determining family of proteins (Fem1), genes for the major muscle-regulatory proteins and genes for six novel proteins. Our study showed clearly that genes for a tumor suppressor, for regulators of the cell cycle (p19ARF and p21WAF1) and a novel gene (Fem1) were all involved in the differentiation of myoblasts. Further investigations of the genes identified in our study should help to characterize the complex pathways of muscle differentiation.
We have also used a ribozyme library for studies of metastasis in vivo by injecting mice with minimally invasive mouse melanoma cells that harbored the library.34 Although a library of hybrid ribozyme yields a larger number of potentially informative clones than a library of conventional ribozymes, poly(A)-containing vectors are not very stable in vivo as A residues are lost at high frequency. Therefore, in this study in vivo we used a library of conventional ribozymes, which allowed us to identify eight genes. The roles of these genes in cell migration and invasion are currently being investigated in efforts to clarify their roles in pathways that promote metastasis.
The randomization of the binding arms of hammerhead and hairpin ribozymes has the potential to generate ribozymes that can target and cleave transcripts of specific genes to cause phenotypic changes of interest. However, randomization also generates many ineffective ribozymes whose binding arms are not complementary to any genome sequence. In practical terms, the small populations of effective ribozymes in a large library might seem to be a disadvantage in attempts at efficient screening. However, the very strong specificity and potential activity of ribozymes against their targets renders this unique technology is extremely valuable for the identification of key genes that are responsible for specific cellular processes. Furthermore, the use of libraries of randomized ribozymes has advantages in terms of the potential for the identification of previously unidentified genes. Moreover, this system has led also to the identification of a novel non-coding double-stranded RNA, namely, small modulatory dsRNA (smRNA), which plays a critical role in the regulation of neuronal differentiation.46 Numerous small non-coding RNAs, including siRNAs and microRNAs (miRNAs), have recently been recognized as crucial regulators of important cellular processes, such as differentiation, cell proliferation, cell death and cell metabolism.47 As structural and sequence information about these non-coding RNAs accumulates, the gene-discovery system that exploits libraries of randomized ribozymes should help to reveal the roles of these RNAs within the cell.
Development of effective vectors for the expression of shRNAs
A second gene-discovery system has been developed that exploits RNA interference (RNAi), which is an evolutionarily conserved phenomenon that involves mediation of the cleavage of a cognate mRNA by double-stranded RNA (dsRNA).48,49 In systems that exploit RNAi, dsRNA molecules are delivered into cells where they are subsequently processed to yield small RNAs of 21–25 nucleotides (nt) in length by an RNase III-like enzyme, known as Dicer.50 The small RNAs (siRNAs) are incorporated into a multicomponent nuclease complex, the RNA-induced silencing complex (RISC).51 The single-stranded RNA derived from the siRNA then acts as a guide sequence, directing the complex to the target mRNA,52,53 where a RISC-associated endoribonuclease cleaves the target mRNA specifically.54–56RNAi has been detected in organisms from protozoans to mammals. However, whereas long dsRNAs can efficiently elicit RNAi in a wide variety of organisms, long dsRNAs cannot be exploited in mammalian cells because they induce the interferon response and activation of a dsRNA-dependent protein kinase (PKR), with the resultant nonspecific inhibition of protein synthesis. However, Elbashir et al. demonstrated that RNA duplexes of approximately 21 nt in length with two-nucleotide overhangs at their 3′-ends (siRNA) do not stimulate the interferon response and can efficiently induce RNAi in mammalian cells.57 As a result, RNAi-based gene-silencing technology has become widely used as a powerful tool for the analysis of the biological functions of genes in mammals, as well as many other organisms.
Both synthetic siRNAs and vector-based siRNAs have been used successfully for the regulation of the expression of specific genes in mammalian cells.58–61 Although the introduction of appropriate synthetic siRNA can strongly suppress the expression of the gene of interest, the use of such siRNA has certain limitations including the short duration of gene silencing and the cost of synthesis of siRNAs. To solve these problems several groups, including our own, established vector-based siRNA-expression systems.62–68 These vector-based systems are designed to produce siRNAs for prolonged periods of time within target cells. Furthermore, even if the efficiency of transfection of particular cells with the plasmid vector is relatively low, a viral vector can be used for the efficient delivery of an expression cassette to specific cells and tissues.69,70 Moreover, one can select transfected cells exclusively by exploiting vector-encoded drug resistance. Retroviral and lentiviral vectors allow the straightforward construction of stable “knockdown” cell lines and “knockdown” animals via integration of the viral vector into the host genome.
We optimized the vector system in which shRNAs are transcribed under the control of the U6 promoter64,71–73 by, for example, introducing C to U (or A to G) point mutations into the sense strand of our constructs (Fig. 4A). When constructing a vector for the expression of short hairpin-type siRNA, we face serious technical problems caused by the hairpin region within the construct. In general, since such a hairpin region consists of a tightly hydrogen-bonded palindromic structure, it is difficult to sequence the constructs. In addition, unfavorable mutations and deletions, due to homologous recombination, appear in approximately 20% to 40% of constructs during amplification and maintenance in Escherichia coli. However, appropriate C to U (or A to G) point mutations in the sense strand have allowed us to solve such problems efficiently and to generate effective constructs. In practice, the resultant G–U base pairings within the transcripts had no negative effects on the silencing activity of the constructs. Furthermore, in an effort to enhance the activities of shRNAs with appropriate mutations, we compared the activities of shRNAs with loop sequences derived from several human miRNAs and found that the shRNA with the loop sequence 5′-GUGUGCUGUCC-3′ had elevated suppressive activity. Thus, the introduction of appropriate point mutations into the sense strand and incorporation of the loop of a natural miRNA have proved useful for the production of active and effective shRNAs.
 | ||
| Fig. 4 (A) schematic representation of a short hairpin RNA with C to U and A to G mutations in the sense strand. (B) a system for the identification of genes involved in apoptosis using a library of shRNAs. Each shRNA-carrying vector directed against a single respective gene is introduced individually into cells. The shRNAs in surviving cells are directed against genes involved in apoptosis and allow the identification of genes of interest. | ||
Although siRNAs have strong suppressive activity and their effects are usually specific, siRNAs can, in some cases, direct the silencing of non-target genes with partial sequence-similarity to the target gene. Indeed, Jackson et al. demonstrated that as few as 11 contiguous nucleotides with sequence similarity can be sufficient to affect the expression of non-target genes.74 Thus, when designing siRNAs a BLAST search must be made for genes with sequences that are strongly homologous. Sequences with strong homology to those of other genes must be excluded as potential targets. In addition, recent work has demonstrated that the internal stability, in particular the base pairing at the 5′-antisense termini, appears to determine the choice of strand that is integrated into RISC and the subsequent activity of the siRNA.75,76 Since both strands of an siRNA duplex have potentially interfering activity, care must be taken in the design of siRNAs to reduce off-target effects. In our vector system it is possible to design constructs such that only the sense strand of the shRNA is rapidly degraded, thereby reducing the off-target effects (unpublished data).
In a recent study we demonstrated that appropriate mutations within long dsRNAs allow us to bypass the interferon response in mammalian cells.77 Although it has been proposed that siRNA does not induce the interferon response, Bridge et al. reported the induction of the interferon response by shRNAs in mammalian cells.78 An shRNA transcribed from the H1 promoter in a lentiviral vector was able to induce the expression of 2′,5′-oligoadenylate synthetase (OAS1), one of the factors involved in the interferon response. Moreover, Sledz et al. demonstrated that synthetic siRNAs were also able to trigger the interferon-mediated activation of the Jak–Stat pathway and the enhanced expression of interferon-stimulated genes.79 These unfavorable side effects, induced by shRNAs and siRNAs via the activation of interferon-related genes, hamper the effective use of siRNAs as basic research tools and therapeutic agents. Careful attention must be paid to interferon responses, which must be avoided when mammalian cells are treated with siRNAs. We have been able to reduce the interferon response by introducing appropriate C to U (or A to G) mutations within the sense strands of long hairpin-type RNAs (>50 bp).77 This strategy is clearly advantageous in efforts to avoid unfavorable side effects when hairpin-type RNAs are used to suppress the expression of specific genes. Moreover, since long hairpin RNAs can direct cleavage at extended target sites that span more than 100 nucleotides, they are probably the RNAs that are most likely to target the viral genomes regardless of the rapid mutation of such genomes.
Our extensive efforts have led to the development of methods for the construction of effective shRNA-expression systems. The appropriate design of the hairpin-type expression vector can stabilize the vector's genome and enhance RNAi activity significantly in addition to reducing non-specific effects. Our shRNA-expression systems are very useful for the application of siRNAs to efforts directed towards an improved understanding of gene function and disruption of the expression of aberrant genes.
Gene discovery using libraries of shRNAs
Genome-wide screening, using libraries of dsRNAs to exploit the phenomenon of RNAi in Caenorhabditis elegans and in Drosophila cells, has proved to be extremely effective for the identification of genes involved in various process.80–84 For the analysis of human cells, several systems have been developed for the establishment of large-scale libraries of siRNAs for application to screening for specific genes. For example, members of Schultz's laboratory constructed an arrayed siRNA-expression cassette library in which a synthetic DNA that encoded an siRNA targeted against a specific gene was inserted between two different opposing pol III promoters.85 Moreover, members of Blau's and Hirose's laboratories developed, independently, new methods for generating shRNA libraries in which cDNA is converted enzymatically into libraries of different shRNA-expression vectors.86,87 Buchholz's group generated a genome-scale library of endoribonuclease-generated siRNAs from long dsRNAs that corresponded to a large number of cDNAs.88Several groups, including our own, have established large-scale shRNA-expression libraries for the identification of genes of unknown function.61,89–92 These libraries are composed of large numbers of sequence-verified shRNA-expression vectors that target different human genes. Each vector targeting a particular gene is introduced individually into cells in a single well of a 96-well plate. The effective shRNAs that cause the phenotypic change of interest reveal potential candidates for genes that might be involved in the chosen phenotype. The use of such libraries has clear advantages for the comprehensive analysis of signaling networks and pathways.
Berns et al. demonstrated that an shRNA library could be used for identification of genes in pathways that are involved in the tumor-suppressive activity of p53.89 They studied BJ-TERT-tsLT cells, which are normally grown at 32 °C. Growth of these cells is arrested when the temperature is raised to 39 °C because of the induced activation of p53 and the subsequent arrest of cell proliferation. Berns et al. introduced their shRNA library into cells using a retroviral vector at 32 °C and then they isolated colonies of cells that continued to proliferate at 39 °C. Sequencing revealed the importance of one known and five novel genes, including genes for ribosomal S6 kinase 4 (RSK4), histone acetyl transferase (TIP60) and histone deacetylase 4, which modulate the p53-dependent arrest of cell proliferation. Suppression of these genes allowed cells to escape from both p53- and p19ARF-dependent growth arrest and it abolished cell cycle arrest at the G1-phase in response to DNA damage.
Paddison et al. constructed an shRNA-expression library directed against a large number of human and mouse genes.90 They developed a unique strategy using a DNA “barcode” to facilitate the screening of cells. An individual 60 nt DNA barcode sequence was linked to each shRNA vector and the barcodes allowed the monitoring of relative frequencies of individual shRNAs in transduced cells using a microarray that consist of DNAs that were complementary to each barcode. The authors focused, in the cited study, on the identification of genes that are responsible for proteasome function. Positive shRNAs that compromised proteasome function were directed against mRNAs that encoded subunits of the 26S proteosomes, which included ATPases and non-ATPases.
Using an shRNA library our group has identified many genes of unknown function that are involved in apoptosis in human cells (Fig. 4B;91,92). To generate a high quality library, we used our optimized shRNA-expression system and our original algorithm for the prediction of suitable target sites in mRNAs. We created our algorithm using siRNA data sets obtained with reporter genes such as genes for luciferase and green fluorescent protein and this algorithm allows us to select appropriate sequences in endogenous genes.71–73 Using our algorithm we found that 70% to 80% of our siRNA vectors directed against endogenous genes were able to silence their target genes very efficiently.91
We transfected HCT116 cells with individual shRNAs that targeted to apoptosis-related genes, which include genes for kinases, caspases, transcription factors and other proteins involved in various apoptotic pathways and then treated these cells with thapsigargin (TG), which elicits the endoplasmic reticulum (ER) stress-induced apoptosis. As a result, we were able to identify three anti-apoptotic genes, NOXA, E2F1 and MAPK1, as well as previously characterized genes in the apoptotic pathway. In addition, our study revealed new details of TG-induced apoptosis. Thus, a large-scale library of shRNAs can provide very precise information about signaling pathways and enhance our understanding of numerous cellular processes. We have already constructed shRNA libraries directed against many human genes. Using such shRNA libraries we recently identified genes for proteins that are essential for RNAi, including a slicer, eIF2C2, and a helicase (unpublished data). It should be possible to make complete libraries of shRNAs that encompass entire genomes. Such libraries should be very valuable for the analysis of the human and other genomes.
Conclusion
In this review we have described two novel tools that exploit the activities of hammerhead and hairpin ribozymes and of shRNAs and can be used for the rapid identification of functional genes.Libraries of randomized hammerhead and hairpin ribozymes and libraries of sequence-verified shRNAs have various advantages and disadvantages as gene-discovery tools (see Table 1). For example, shRNAs participate in the intrinsic activities of cells while hammerhead and hairpin ribozymes are not found naturally in mammalian cells. Thus, shRNAs are often more active and, therefore, more useful than such ribozymes. By contrast, these ribozymes have very strong target specificity and do not induce an interferon response. Thus, specific effects can be detected despite the fact that their activity is lower than that of shRNAs. Furthermore, a major advantage of libraries of randomized ribozymes is that no prior sequence information is required and, thus, they allow the identification of previously unidentified genes46 when large numbers of ribozymes are introduced into cells. It is impossible, however, to completely cover an entire genome with such a library.
| Characteristics | Ribozyme library | shRNA library |
|---|---|---|
| Sequence specificity | Very high | High |
| Inhibitory effects | Moderate | Very high |
| Diversity | High | High and adjustable |
| Cost | Low | High |
| Interferon response | No | Optimization is necessary |
| Off-target effects | No | Optimization is necessary |
| Sequence information of genes | Not required | Required |
It is theoretically possible to create a library of randomized siRNAs using two different opposing pol-III promoters, such as the human and mouse U6 promoters. However, the use of such completely randomized siRNAs in vivo is of questionable value because it is impossible completely to eliminate false-positive results. It is also impossible to avoid false-positive results with ribozyme libraries. However, cleavage of target mRNAs by ribozymes requires a cleavable triplet, such as GUC, and, thus, any sequenced potential targets that lack a cleavable triplet are easily recognized as false-positives and can be discarded. Similar discrimination of false-positives from true-positives is impossible when a library of completely randomized siRNAs is used. Moreover, the presence of two potentially active strands of siRNA makes it more difficult to identify true targets because it is not easy to identify the strand that has acted as the antisense strand. Therefore, with respect to completely randomized libraries, libraries of ribozymes rather than of siRNAs appear to be more useful. Theoretically, it is possible to mix individually constructed shRNA vectors to generate mixed populations, but these mixed shRNAs might saturate and, thus, limit the activity of the target-cleaving RISC.
The use of shRNAs is seriously compromised by the non-specific suppressive activity of such RNAs, such as off-target effects and induction of the interferon response. However, we have developed reliable algorithms for the prediction of suitable target sites, as well as effective shRNA-expression vectors that minimize both the interferon response and off-target effects. Our efforts have enhanced the potential utility of shRNAs in vivo and allowed us to construct high-quality libraries of shRNAs. The use of such libraries has provided precise information about various cellular processes.
In conclusion, the high specificity of hammerhead and hairpin ribozymes and the strong activities of shRNAs, as well as improvements of various expression vectors, have yielded tools that are proving extremely helpful in efforts to identify novel functional genes in the human genome.
References
- E. S. Lander, L. M. Linton, B. Birren, C. Nusbaum, M. C. Zody, J. Baldwin, K. Devon, K. Dewar, M. Doyle, W. FitzHugh, R. Funke, D. Gage, K. Harris, A. Heaford, J. Howland et al., Nature, 2001, 409, 860––921 Search PubMed.
- J. C. Venter, M. D. Adams, E. W. Myers, P. W. Li, R. J. Mural, G. G. Sutton, H. O. Smith, M. Yandell, C. A. Evans, R. A. Holt, J. D. Gocayne, P. Amanatides, R. M. Ballew, D. H. Huson, J. R. Wortman et al., Science, 2001, 291, 1304––1351 Search PubMed.
- I. G. Serebriiskii, V. Khazak and E. A. Golemis, BioTechniques, 2001, 30, 634–636 CAS.
- N. L. van Berkum and F. C. Holstege, Curr. Opin. Biotechnol., 2001, 12, 48–52 CrossRef CAS.
- K. Taira, M. Warashina, T. Kuwabara and H. Kawasaki, Japanese Patent Application H11-316133, 1999.
- M. Kruger, C. Beger, Q. X. Li, P. J. Welch, R. Tritz, M. Leavitt, J. R. Barber and F. Wong-Staal, Proc. Natl. Acad. Sci. USA, 2000, 97, 8566–8571 CrossRef CAS.
- Q. X. Li, J. M. Robbins, P. J. Welch, F. Wong-Staal and J. R. Barber, Nucleic Acids Res., 2000, 28, 2605–2612 CrossRef CAS.
- P. J. Welch, E. G. Marcusson, Q. X. Li, C. Beger, M. Kruger, C. Zhou, M. Leavitt, F. Wong-Staal and J. R. Barber, Genomics, 2000, 66, 274–283 CrossRef CAS.
- C. Beger, L. N. Pierce, M. Kruger, E. G. Marcusson, J. M. Robbins, P. Welcsh, P. J. Welch, K. Welte, M. C. King, J. R. Barber and F. Wong-Staal, Proc. Natl. Acad. Sci. USA, 2001, 98, 130–135 CrossRef CAS.
- M. Kruger, C. Beger, P. J. Welch, J. R. Barber, M. P. Manns and F. Wong-Staal, Mol. Cell Biol., 2001, 21, 8357–8364 CrossRef CAS.
- H. Kawasaki and K. Taira, Nucleic Acids Res., 2002, 30, 3609–3614 CrossRef CAS.
- H. Kawasaki and K. Taira, EMBO Rep., 2002, 3, 443–450 Search PubMed.
- H. Kawasaki, R. Onuki, E. Suyama and K. Taira, Nat. Biotechnol., 2002, 20, 376–380 CrossRef CAS.
- J. E. Chatterton, X. Hu and F. Wong-Staal, Targets, 2004, 3, 10–17 Search PubMed.
- D. L. Nelson, E. Suyama, H. Kawasaki and K. Taira, Target, 2003, 2, 191–200 Search PubMed.
- N. Sarver, E. M. Cantin, P. S. Chang, J. A. Zaia, P. A. Ladne, D. A. Stephens and J. J. Rossi, Science, 1990, 247, 1222–1225 CAS.
- N. K. Vaish, A. R. Kore and F. Eckstein, Nucleic Acids Res., 1998, 26, 5237–5242 CrossRef CAS.
- R. Shippy, R. Lockner, M. Farnsworth and A. Hampel, Mol. Biotechnol., 1999, 12, 117–129 Search PubMed.
- M. J. Fedor, J. Mol. Biol., 2000, 297, 269–291 CrossRef CAS.
- Y. Takagi, M. Warashina, W. J. Stec, K. Yoshinari and K. Taira, Nucleic Acids Res., 2001, 29, 1815–1834 CrossRef CAS.
- D. M. Zhou and K. Taira, Chem. Rev., 1998, 98, 991–1026 CrossRef.
- K. Suzumura, Y. Takagi, M. Orita and K. Taira, J. Am. Chem. Soc., 2004, 126, 15504–15511 CrossRef CAS.
- Y. Takagi, A. Inoue and K. Taira, J. Am. Chem. Soc., 2004, 126, 12856–12864 CrossRef CAS.
- Y. Tanaka, Y. Kasai, S. Mochizuki, A. Wakisaka, E. H. Morita, C. Kojima, A. Toyozawa, Y. Kondo, M. Taki, Y. Takagi, A. Inoue, K. Yamasaki and K. Taira, J. Am. Chem. Soc., 2004, 126, 744–752 CrossRef CAS.
- M. Warashina, T. Kuwabara, Y. Nakamatsu, Y. Takagi, Y. Kato and K. Taira, J. Am. Chem. Soc., 2004, 126, 12291–12297 CrossRef CAS.
- Nucleic Acids and Molecular Biology, eds F. Eckstein and D. M. J. Lilley, Springer-Verlag, Berlin, 1996, vol. 10 Search PubMed.
- Ribozyme, Biochemistry and Biotechnology, eds G. Krupp and R. K. Gaur, Eaton Publishing, MA, 2000 Search PubMed.
- N. Usman and L. M. Blatt, J. Clin. Invest., 2000, 106, 1197–1202 CrossRef CAS.
- B. A. Sullenger and E. Gilboa, Nature, 2002, 418, 252–258 CrossRef CAS.
- Methods in Molecular Biology, ed. M. Sioud, Humana Press, Totowa NJ, 2004, vol. 252 Search PubMed.
- E. Suyama, H. Kawasaki, T. Kasaoka and K. Taira, Cancer Res., 2003, 63, 119–124 CAS.
- E. Suyama, H. Kawasaki, M. Nakajima and K. Taira, Proc. Natl. Acad. Sci. USA, 2003, 100, 5616–5621 CrossRef CAS.
- R. Onuki, Y. Bando, E. Suyama, T. Katayama, H. Kawasaki, T. Baba, M. Tohyama and K. Taira, EMBO J., 2004, 23, 959–968 CrossRef CAS.
- E. Suyama, R. Wadhwa, K. Kaur, M. Miyagishi, S. C. Kaul, H. Kawasaki and K. Taira, J. Biol. Chem., 2004, 279, 38083–38086 CrossRef CAS.
- R. Wadhwa, T. Yaguchi, K. Kaur, E. Suyama, H. Kawasaki, K. Taira and S. C. Kaul, J. Biol. Chem., 2004, 279, 51622–51629 CrossRef CAS.
- H. Kawasaki, R. Eckner, T. P. Yao, K. Taira, R. Chiu, D. M. Livingston and K. K. Yokoyama, Nature, 1998, 393, 284–289 CrossRef CAS.
- T. Kuwabara, M. Warashina, T. Tanabe, K. Tani, S. Asano and K. Taira, Mol. Cell, 1998, 2, 617–627 CrossRef CAS.
- S. Koseki, T. Tanabe, K. Tani, S. Asano, T. Shioda, Y. Nagai, T. Shimada, J. Ohkawa and K. Taira, J. Virol., 1999, 73, 1868–1877 CAS.
- S. Waninger, K. Kuhen, X. Hu, J. E. Chatterton, F. Wong-Staal and H. Tang, J. Virol., 2004, 78, 12829–12837 CrossRef CAS.
- R. Onuki, A. Nagasaki, H. Kawasaki, T. Baba, T. Q. Uyeda and K. Taira, Proc. Natl. Acad. Sci. USA, 2002, 99, 14716–14721 CrossRef CAS.
- M. Zuker, Methods Enzymol., 1989, 180, 262–88 CrossRef CAS.
- N. Milner, K. U. Mir and E. M. Southern, Nat. Biotechnol., 1997, 15, 537–541 CrossRef CAS.
- M. Scherr and J. J. Rossi, Nucleic Acids Res., 1998, 26, 5079–5085 CrossRef CAS.
- W. H. Pan, P. Xin, V. Bui and G. A. Clawson, Mol. Ther., 2003, 7, 129–139 CrossRef CAS.
- M. Warashina, T. Kuwabara, Y. Kato, M. Sano and K. Taira, Proc. Natl. Acad. Sci. USA, 2001, 98, 5572–5577 CrossRef CAS.
- T. Kuwabara, J. Hsieh, K. Nakashima, K. Taira and F. H. Gage, Cell, 2004, 116, 779–793 CrossRef CAS.
- D. P. Bartel, Cell, 2004, 116, 281–297 CrossRef CAS.
- A. Fire, S. Xu, M. K. Montgomery, S. A. Kostas, S. E. Driver and C. C. Mello, Nature, 1998, 391, 806–811 CrossRef CAS.
- M. K. Montgomery, S. Xu and A. Fire, Proc. Natl. Acad. Sci. U S A, 1998, 95, 15502–15507 CrossRef CAS.
- E. Bernstein, A. A. Caudy, S. M. Hammond and G. J. Hannon, Nature, 2001, 409, 363–366 CrossRef CAS.
- S. M. Hammond, E. Bernstein, D. Beach and G. J. Hannon, Nature, 2000, 404, 293–296 CrossRef CAS.
- A. Nykanen, B. Haley and P. D. Zamore, Cell, 2001, 107, 309–321 CrossRef CAS.
- J. Martinez, A. Patkaniowska, H. Urlaub, R. Luhrmann and T. Tuschl, Cell, 2002, 110, 563–574 CrossRef CAS.
- J. Liu, M. A. Carmell, F. V. Rivas, C. G. Marsden, J. M. Thomson, J. J. Song, S. M. Hammond, L. Joshua-Tor and G. J. Hannon, Science, 2004, 305, 1437–1441 CrossRef CAS.
- J. Martinez and T. Tuschl, Genes Dev., 2004, 18, 975–80 CrossRef CAS.
- J. J. Song, S. K. Smith, G. J. Hannon and L. Joshua-Tor, Science, 2004, 305, 1434–1437 CrossRef CAS.
- S. M. Elbashir, J. Harborth, W. Lendeckel, A. Yalcin, K. Weber and T. Tuschl, Nature, 2001, 411, 494–498 CrossRef CAS.
- S. M. Hammond, A. A. Caudy and G. J. Hannon, Nat. Rev. Genet., 2001, 2, 110–119 CrossRef CAS.
- M. T. McManus and P. A. Sharp, Nat. Rev. Genet., 2002, 3, 737–747 CrossRef CAS.
- D. M. Dykxhoorn, C. D. Novina and P. A. Sharp, Nat. Rev. Mol. Cell. Biol., 2003, 4, 457–467 CrossRef CAS.
- H. Akashi, S. Matsumoto and K. Taira, Nat. Rev. Mol. Cell Biol., Search PubMed in press.
- T. R. Brummelkamp, R. Bernards and R. Agami, Science, 2002, 296, 550–553 CrossRef CAS.
- N. S. Lee, T. Dohjima, G. Bauer, H. Li, M. J. Li, A. Ehsani, P. Salvaterra and J. Rossi, Nat. Biotechnol., 2002, 20, 500–505 CAS.
- M. Miyagishi and K. Taira, Nat. Biotechnol., 2002, 20, 497–500 CrossRef CAS.
- P. J. Paddison, A. A. Caudy, E. Bernstein, G. J. Hannon and D. S. Conklin, Genes Dev., 2002, 16, 948–958 CrossRef CAS.
- C. P. Paul, P. D. Good, I. Winer and D. R. Engelke, Nat. Biotechnol., 2002, 20, 505–508 CrossRef CAS.
- G. Sui, C. Soohoo, B. Affar el, F. Gay, Y. Shi and W. C. Forrester, Proc. Natl. Acad. Sci. USA, 2002, 99, 5515–5520 CrossRef CAS.
- J. Y. Yu, S. L. DeRuiter and D. L. Turner, Proc. Natl. Acad. Sci. USA, 2002, 99, 6047–6052 CrossRef CAS.
- G. M. Barton and R. Medzhitov, Proc. Natl. Acad. Sci. USA, 2002, 99, 14943–14945 CrossRef CAS.
- X. F. Qin, D. S. An, I. S. Chen and D. Baltimore, Proc. Natl. Acad. Sci. USA, 2003, 100, 183–188 CrossRef CAS.
- M. Miyagishi and K. Taira, Oligonucleotides, 2003, 13, 325–333 Search PubMed.
- M. Miyagishi, S. Matsumoto and K. Taira, Virus Res., 2004, 102, 117–124 CrossRef CAS.
- M. Miyagishi, H. Sunimoto, H. Miyoshi, Y. Kawakami and K. Taira, J. Gene Med., 2004, 6, 715–23 CrossRef CAS.
- A. L. Jackson, S. R. Bartz, J. Schelter, S. V. Kobayashi, J. Burchard, M. Mao, B. Li, G. Cavet and P. S. Linsley, Nat. Biotechnol., 2003, 21, 635–637 CrossRef CAS.
- A. Khvorova, A. Reynolds and S. D. Jayasena, Cell, 2003, 115, 209–216 CrossRef CAS.
- D. S. Schwarz, G. Hutvagner, T. Du, Z. Xu, N. Aronin and P. D. Zamore, Cell, 2003, 115, 199–208 CrossRef CAS.
- H. Akashi, M. Miyagishi and K. Taira, submitted for publication.
- A. J. Bridge, S. Pebernard, A. Ducraux, A. L. Nicoulaz and R. Iggo, Nat. Genet., 2003, 34, 263–264 CrossRef CAS.
- C. A. Sledz, M. Holko, M. J. de Veer, R. H. Silverman and B. R. Williams, Nat. Cell Biol., 2003, 5, 834–839 CrossRef CAS.
- A. G. Fraser, R. S. Kamath, P. Zipperlen, M. Martinez-Campos, M. Sohrmann and J. Ahringer, Nature, 2000, 408, 325–330 CrossRef CAS.
- P. Gonczy, C. Echeverri, K. Oegema, A. Coulson, S. J. Jones, R. R. Copley, J. Duperon, J. Oegema, M. Brehm, E. Cassin, E. Hannak, M. Kirkham, S. Pichler, K. Flohrs, A. Goessen, S. Leider, A. M. Alleaume, C. Martin, N. Ozlu, P. Bork and A. A. Hyman, Nature, 2000, 408, 331–336 CrossRef CAS.
- S. S. Lee, R. Y. Lee, A. G. Fraser, R. S. Kamath, J. Ahringer and G. Ruvkun, Nat. Genet., 2003, 33, 40–48 CrossRef CAS.
- L. Lum, S. Yao, B. Mozer, A. Rovescalli, D. Von Kessler, M. Nirenberg and P. A. Beachy, Science, 2003, 299, 2039–2045 CrossRef CAS.
- M. Boutros, A. A. Kiger, S. Armknecht, K. Kerr, M. Hild, B. Koch, S. A. Haas, H. F. Consortium, R. Paro and N. Perrimon, Science, 2004, 303, 832–835 CrossRef CAS.
- L. Zheng, J. Liu, S. Batalov, D. Zhou, A. Orth, S. Ding and P. G. Schultz, Proc. Natl. Acad. Sci. USA, 2003, 101, 135–140 CrossRef.
- G. Sen, T. S. Wehrman, J. W. Myers and H. M. Blau, Nat. Genet., 2004, 36, 183–189 CrossRef CAS.
- D. Shirane, K. Sugao, S. Namiki, M. Tanabe, M. Iino and K. Hirose, Nat. Genet., 2004, 36, 190–196 CrossRef CAS.
- R. Kittler, G. Putz, L. Pelletier, I. Poser, A. K. Heninger, D. Drechsel, S. Fischer, I. Konstantinova, B. Habermann, H. Grabner, M. L. Yaspa, H. Himmelbaur, B. Korn, K. Neugebauer, M. T. Pisabarro and F. Buchholz, Nature, 2004, 432, 1036–1040 CrossRef CAS.
- K. Berns, E. M. Hijmans, J. Mullenders, T. R. Brummelkamp, A. Velds, M. Heimerikx, R. M. Kerkhoven, M. Madiredjo, W. Nijkamp, B. Weigelt, R. Agami, W. Ge, G. Cavet, P. S. Linsley, R. L. Beijersbergen and R. Bernards, Nature, 2004, 428, 431–437 CrossRef CAS.
- P. J. Paddison, J. M. Silva, D. S. Conklin, M. Schlabach, M. Li, S. Aruleba, V. Balija, A. O'Shaughnessy, L. Gnoj, K. Scobie, K. Chang, T. Westbrook, M. Cleary, R. Sachidanandam, W. R. McCombie, S. J. Elledge and G. J. Hannon, Nature, 2004, 428, 427–431 CrossRef CAS.
- T. Futami, M. Miyagishi and K. Taira, J. Biol. Chem., 2005, 280, 826–831 CAS.
- S. Matsumoto, M. Miyagishi and K. Taira, J. Biol. Chem., 2005 Search PubMed , in press.
| This journal is © The Royal Society of Chemistry 2005 |