Design of compound libraries based on natural product scaffolds and protein structure similarity clustering (PSSC)
Rengarajan
Balamurugan
ab,
Frank J.
Dekker
ab and
Herbert
Waldmann
*ab
aDepartment of Chemical Biology, Max-Planck Institute of Molecular Physiology, Otto-Hahn Str. 11, D-44227 Dortmund, Germany. E-mail: herbert.waldmann@mpi-dortmund.mpg.de; Fax: +49-231-133-2499; Tel: +49-231-133-2400
bFachbereich 3, Organic Chemistry, University of Dortmund, Otto-Hahn Str. 6, D-44227 Dortmund, Germany. E-mail: herbert.waldmann@mpi-dortmund.mpg.de; Fax: +49-231-133-2499; Tel: +49-231-133-2400
First published on 19th April 2005
Abstract
Recent advances in structural biology, bioinformatics and combinatorial chemistry have significantly impacted the discovery of small molecules that modulate protein functions. Natural products which have evolved to bind to proteins may serve as biologically validated starting points for the design of focused libraries that might provide protein ligands with enhanced quality and probability. The combined application of natural product derived scaffolds with a new approach that clusters proteins according to structural similarity of their ligand sensing cores provides a new principle for the design and synthesis of such libraries. This article discusses recent advances in the synthesis of natural product inspired compound collections and the application of protein structure similarity clustering for the development of such libraries.
 Rengarajan Balamurugan | Rengarajan Balamurugan was born in Tiruvarur, India in 1974. He studied chemistry at Bharathidasan University in Tiruchirapalli, where he received his MSc in 1996. He earned his PhD in 2002 under the guidance of Professor Veejendra K. Yadav in the Indian Institute of Technology, Kanpur. His doctoral work involved the exploration of synthetic utilities of bulky silyl-substituted cyclopropylmethylsilanes and π-facial selectivity. After his PhD he took up a postdoctoral assignment to work on the synthesis of sphingolipid derivatives with Professor Richard R. Schmidt at the University of Konstanz, Germany. Currently he is working in the group of Professor Herbert Waldmann at the Max Planck Institute for Molecular Physiology, Dortmund as a Humboldt fellow. His research interests include the synthesis of biologically relevant compounds and the development of synthetic strategies to address regio- and stereocontrol in organic synthesis. |
 Frank J. Dekker | Frank Dekker was born in Middelburg (The Netherlands) in 1977. From 1995 until 2000 he studied pharmacy at the Utrecht University (The Netherlands). He obtained his PhD in 2004 in the department of Medicinal Chemistry, in the faculty of Pharmacy at the Utrecht University, under the supervision of Professor Rob M. J. Liskamp. During the PhD project, research was performed on modulation of protein–protein interactions in signal transduction. The project involved a combination of techniques including organic synthesis, molecular modelling and biophysical characterization. After obtaining his PhD he moved to the Chemical Biology department of Professor Herbert Waldmann in the Max-Planck Institute of Molecular Physiology in Dortmund. His current research interests include solution and solid-phase synthesis of natural product inspired compound libraries, activity of small molecules on protein targets and the integration of information on protein structure and compound library design |
 Herbert Waldmann | Professor Dr Waldmann was born on 11 June 1957 in Neuwied. He studied chemistry at the University of Mainz and received his PhD in organic chemistry in 1985 under the guidance of Professor Kunz. After a postdoctoral appointment with Professor George Whitesides at Harvard University, Dr Waldmann completed his habilitation at the University of Mainz in 1991. In 1991 he was appointed as Professor of Organic Chemistry at the University of Bonn, then in 1993 was appointed to full Professor of Organic Chemistry at the University of Karlsruhe. In 1999 he was appointed as Director of the Max-Planck Institute of Molecular Physiology Dortmund and Professor of Organic Chemistry at the University of Dortmund. |
1. Introduction
Large-scale global efforts along with advancements in analytical techniques have provided a wealth of information about biological systems, particularly, DNA sequence (genomics) and protein structure (structural genomics). This gain in knowledge needs to be translocated into a better understanding of cellular processes at the molecular level, in particular the interplay of proteins and their interactions with other biomolecules. The information gleaned from this knowledge will lead to new modulators of protein function which ultimately may be utilized in the development of new therapies and drugs. In recent years, combinatorial chemistry has emerged as a powerful tool to access diverse small molecules.1 Although initial expectations about hit and lead finding were not matched, these efforts made clear that the underlying structures of compound libraries need to be relevant in a biological context and that quality and not quantity of the library members determines the hit rates. In this process diversity,2 drug-likeness,3 and biological relevance4 have emerged as the most important parameters in the choice of library scaffolds from the number of thinkable small molecule modulators of protein function. In this context, natural product guided compound library development deserves particular attention.2. Natural products as biologically validated ligands to modulate protein functions
The precondition for biological relevance is fulfilled by natural products as they have evolved to interact with multiple proteins and can be regarded as embodying privileged structures. ‘Privileged structures’ are compound classes that can bind to various proteinaceous receptors.5 A typical example of a privileged structure is the benzodiazepine. Natural products often embody privileged structures since they are synthesized and modified by more than one protein and since they often display multiple biological activities mediated by interactions with different proteins.It is to be expected that privileged structures like for example the peptidomimetic benzodiazepine might be present among evolutionarily selected ligands that have been selected by Nature. In this context, the corresponding scaffolds can be privileged in a chemical sense i.e. scaffolds with a necessary balance of flexibility and rigidity combined with the ability to present functional groups in a favourable spatial arrangement. Additionally, they may be privileged in a biological sense since they may equip the corresponding compound class with the ability to bind to different proteins.
This analysis is further supported by the observation that hit rates for natural product collections in biochemical and biological screens often are significantly higher than those found for libraries obtained exclusively on the basis of chemical feasibility.4b Notably, nearly half of the new drugs introduced into the market in the last two decades are natural products or derived therefrom and nearly 60% of the anti-cancer drugs and 75% of the infectious disease drugs are natural products in origin.6
3. Natural products as inspiration source for compound library design
For certain domains of evolutionary or structurally related proteins, when ligand types or frameworks of ligands are already known, the scaffolds of the ligands may be advantageously employed for library development. This reasoning applies in particular to natural products.The libraries designed and synthesised around the core structures of natural products are expected to yield biologically pre-validated modulators of protein functions in relatively high frequency at considerably reduced library size. Fundamentally, this natural product structure-based strategy does not neglect the issue of chemical diversity; rather it builds on the diversity created by Nature itself. Hence this approach offers an opportunity to identify ‘privileged structures’ from Nature and explore their possible application in Chemical Biology and the drug discovery processes.
4. Solid-phase synthesis of natural product derived compound collections
Solid-phase synthesis is exceedingly useful in compound library synthesis.7 Generation of libraries of natural product analogues using solid-phase chemistry has received increasing attention in recent years and the synthesis of new libraries is in steady progress. In designing a multi-step synthesis of a natural product based library on a solid support versatile and high yielding reactions are most desirable that employ a wide range of reactants with different electronic and steric properties. Solution phase combinatorial synthesis is an alternative, however it poses the difficulty of isolating and purifying the library members. Methodologies for solution phase synthesis of compound libraries were recently reviewed by Boger et al.8 In an intermediate strategy natural product cores are synthesised in solution phase and attached to a solid support to proceed further with the synthetic sequences to introduce diversity. Hall et al. have reviewed a number of such libraries.9 A variety of natural product derived libraries such as libraries of carbohydrates, steroids, fatty acid derivatives, polyketides, peptides, terpenoids, flavonoids, and alkaloids has been designed and synthesised in recent years (for representative examples, see Fig. 1).10,11a Strategies for the exploitation of natural products in compound library synthesis on solid supports have been reviewed repeatedly.11 Therefore we focus in this article on several of the most recent developments in the area. | ||
| Fig. 1 Representative examples of natural product derived compound libraries.10,11a | ||
4.1. Natural product libraries based on 6,6′-spiroketal-, indoline-, indole- and tetrahydroquinoline frameworks
6,6′-Spiroketals are structural motifs found in many natural products such as spongistatins, integramycin, tautomycin and okadaic acid. Interestingly they may retain the biological activity of some of the parent natural products and hence constitute a biologically validated starting point for compound library development. Recently asymmetric synthesis of 6,6′-spiroketals has been carried out on a solid support (Scheme 1).12 The solid-phase synthesis essentially involves up to 12 linear steps including two asymmetric boron-mediated aldol reactions which proceed with high enantio- and diastereoselectivity. The salient feature of the synthesis is the use of polymer-bound and soluble chiral boron enolates for the asymmetric induction. The synthesis is also attractive in the sense that the cleavage from the polymeric support (Merrifield resin with Wang linker) is accompanied by PMB (p-methoxybenzyl) deprotection to result in the formation of the spiroketal in an overall yield comparable with that of a corresponding solution phase synthesis. The developed synthetic sequence was employed successfully for the generation of a small compound collection. | ||
| Scheme 1 Asymmetric solid-phase synthesis of the 6,6′-spiroketal skeleton. | ||
Several heterocyclic natural products possess indole and indoline scaffolds in their molecular architecture and show diverse biological activities. Recently several hydroxy indoline based libraries have been synthesized using the IRORI split-and-mix approach by Arya and co-workers.13 A hydroxy indolinol scaffold anchored to a (bromomethyl)phenoxymethyl polystyrene support was used to synthesize tricyclic compound libraries using a Mitsunobu reaction as the key step. Diversity was introduced at two sites; one by the choice of the amino acid and the other during an amide bond formation. A 100-membered compound library was made following the reaction sequence shown in the Scheme 2.
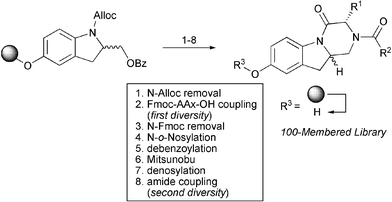 | ||
| Scheme 2 Synthesis of an indoline based tricyclic library. | ||
In making further use of indoline scaffolds in combinatorial library synthesis, the same group has used functionalized aminoindoline scaffolds for the synthesis of a 90-membered indoline-alkaloid-like tricyclic library using the IRORI split-and-pool technique (Scheme 3).14 In this case a stereoselective in situ conjugate aza-Michael addition has been used for cyclization. The stereoselectivity depended on the choice of the amino acid. In the resulting indoline-alkaloid-like tricyclic derivatives, diversification has been accomplished at four sites. Here, (4-methoxyphenyl)diisopropylsilylpropyl polystyrene with a three-carbon spacer between the polymer and the aromatic moiety was employed for better yields.
 | ||
| Scheme 3 Synthesis of a highly functionalized indolinol based library. | ||
The indole derivative indomethacin is a non-steroidal anti-inflammatory drug (NSAID), which is widely used in the treatment of pain, arthritis, cardiovascular diseases, Alzheimer's disease and in the treatment and prevention of cancer. Indomethacin induces G1 arrest and apoptosis of human colorectal cancer cells by induction of the Wnt signal pathway, down regulation of the transcriptional activity of peroxisome proliferation-activated receptor δ (PPARδ), and inhibition of the formation of new blood vessels from pre-existing ones (angiogenesis). A library of 197 indomethacin analogues has been synthesized employing a “resin–capture–release” strategy in overall yields ranging from 4% to quantitative (Scheme 4).15 The synthesis is compatible with a variety of functional groups in each building block, i.e. ketones, acid chlorides, and hydrazines. The overall yields are highest when activating electron-donating substituents were present in the hydrazines.
 | ||
| Scheme 4 Synthesis of a 197-membered library based on the indole scaffold. | ||
Biological investigations of the library members have been performed to evaluate their ability to inhibit angiogenesis-related tyrosine kinase receptors.15a Vascular development depends on endothelium-specific receptor tyrosine kinases, in particular vascular endothelial growth factor receptor 1–3 (VEGFR1–3) and the Tie-2 receptor. All the above receptors have been implicated in tumor angiogenesis. From the total library, 134 representative compounds were assayed as possible inhibitors for VEGFR-2, VEGFR-3, Tie-2, FGFR-1 and also insulin-like growth factor 1 receptor (IGF1R). From the 134 investigated compounds, 6 inhibit the kinases with IC50 values in the low micromolar range, while inodmethacin itself inhibits in a concentration of 100 µM. In addition, some members of the indomethacin analogues were found to modulate the activity of multidrug resistance resistance protein-1 (MRP-1), which is found in normal human and tumor tissues.15b Among 60 of the tested indomethacin analogues, nine were found to potentiate the toxicity of the chemotherapeutic agent doxorubicin in a model system of MRP-1 expressing human glioblastoma cell line T98G.
Derivatisation of functional groups in the core structure of a natural-product scaffold is also practiced to increase diversity of the library. An example for this is a medium-sized (27-compound) library based on the tetrahydroquinoline scaffold.16 A chiral tetrahydroquinoline scaffold was synthesized from 5-hydroxy-2-nitro-benzaldehyde in solution involving a key asymmetric aminohydroxylation step. This building block was anchored to a solid support with a bromo-Wang linker and diversity was introduced by selective deprotection and derivatization of the protected hydroxyl and amino substituents (Scheme 5).
 | ||
| Scheme 5 Synthesis of a tetrahydroquinoline scaffold-containing library. | ||
5. Application of protein structure similarity clustering (PSSC) to select natural products as starting points for library design
The crucial question to be answered in library synthesis is; where can a biologically validated starting point be found in vast structural space? Answers to the question might be provided by Nature itself. Recent results in bioinformatics indicate that folds of different protein domains often show a high structural similarity. We proposed that protein domain cores with similar three dimensional structures can be clustered in so-called protein structure similarity clusters (PSSC) and that the knowledge about known natural product ligands for members of such a cluster can be employed to guide compound library development for other members of the cluster.17 This concept has performed well at its exploratory level and a good generality is expected in the selection of natural product derived starting points.4c,e,176. Proteins—an infinite universe made of limited families
Proteins are modular biomolecules built from different domains, which are formed by folding of regions of a polypeptide chain into distinct, stable and compact secondary structures. Many proteins are multidomain entities.18 Although the number of possible protein domains is huge, Nature has restricted the structural complexity of protein domains to a limited number of folds.19 Many protein domains adopt similar folds either due to functional or biophysical constraints on secondary structure elements or as a result of divergent evolution to a stage where the sequence relationship is not recognizable anymore.20 The sequences of proteins often indicate a possibility of having evolved from different combinations of pre-existing domains.21 Possibly, many protein domains have descended from relatively few ancestral types.22 Therefore these domains can be regarded as evolutionarily mobile units whose coding sequences can be duplicated, diverged and/or recombined.23 While the latest release of SCOP (Structural Classification of Proteins)24 predicts 800 folds corresponding to 20619 entries in the protein data bank (PDB), Orengo and co-workers have predicted 8000 distinct protein folds using the CATH structure database.25 Depending on the models and approximations used, the number of folds ranges from 1000 to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000.19b–e Another feature of the conservatism in protein domain folding is that the distribution of folds is highly non-homogeneous with some folds occurring abundantly and some rarely. It seems certain that a great majority of protein domains can be attributed to about 1000 most commonly observed folds.19b,c,e
000.19b–e Another feature of the conservatism in protein domain folding is that the distribution of folds is highly non-homogeneous with some folds occurring abundantly and some rarely. It seems certain that a great majority of protein domains can be attributed to about 1000 most commonly observed folds.19b,c,e
The ligand binding or catalytic sites are the most relevant parts from the point of view of small molecule ligand development. There is evidence that the topological location of functional sites is also often conserved. However, the correlation patterns of protein fold and protein function remain a matter of debate.26 A vast number of specific functions are carried out by this limited number of protein domain folds due to the presence of diverse functional sites.27 This diversity in functional sites arises from the high amino-acid diversity of proteins that show similar folds. Nevertheless, in some cases a remarkable conservatism in functional sites can be observed like for example conserved catalytic residues in enzyme active sites. This is true for the example described later in the review on similarity of Cdc25A phosphatase, acetylcholinesterase and 11β-hydroxysteroid dehydrogenases.
These findings led to the development of a novel strategy that exploits Nature's structural conservatism concerning protein architecture for the identification of small molecule modulators of protein function (Fig. 2). We introduced Protein Structure Similarity Clustering (PSSC) as a guiding principle for the selection of biologically pre-validated starting points for compound library synthesis.4c,d,17 In this concept the structures of natural or non-natural products that bind to one member protein of a PSSC are taken as guiding structures for compound library synthesis. Proteins that share structural similarity despite low sequence identity are the most interesting cases for PSSC, because classical clustering strategies may not detect and consider these cases. In such a PSSC, significant diversity in the functional sites of the clustered proteins can be expected. Thus, compound libraries addressing a PSSC should display sufficient chemical diversity in order to match the biological diversity occurring in the functional sites of the clustered proteins (Fig. 2).
 | ||
| Fig. 2 From structural conservatism to library design—PSSC guided focused library design. | ||
7. PSSC based reanalysis of the development of nuclear hormone receptor ligands
Analysis of some literature examples provided support for the viability of the PSSC concept and has been described already.4c,d One example is the development of ligands for the farnesoid X receptor. The farnesoid X receptor is as transcriptional sensor for bile acids, the primary products of cholesterol metabolism and plays an important role in lipid homeostasis.28 The farnesoid X receptor was until recently an example of a receptor for which no specific ligands existed. Selective ligands for this receptor have been found in natural product derived libraries described by Nicolaou et al.29 The group of Nicolaou developed solid phase synthesis methods to make combinatorial libraries based on a benzopyran core structure.30 A 10000-membered combinatorial library based on the benzopyran core structure was synthesized31 and screened for activity on the farnesoid X receptor. The first specific ligands for the farnesoid X receptor were found in this library. These ligands were used in a chemical genetic analysis to unravel the farnesoid X receptor functions in lipid metabolism.28a
The farnesoid X receptor is a member of the class of nuclear hormone receptors, which have key roles in homeostasis, as well as in many diseases like obesity, diabetes and cancer.32 The farnesoid X receptor shows structural similarity to the estrogen receptor β (ERβ), which mediates a broad spectrum of physiological functions ranging from regulation of the menstrual cycle and reproduction to modulation of bone density, and cholesterol transport.33 The farnesoid X receptor shows also similarity to the peroxisome proliferation-activated receptor γ (PPARγ), which is involved in fat metabolism, inflammatory and immune responses.34 The estrogen receptor β (ERβ), the peroxisome proliferation-activated receptor γ (PPARγ) and the farnesoid X receptor (FXR) can be clustered in a protein structure similarity cluster (PSSC). These receptors display a similar fold as shown in Fig. 3, however the sequence similarities are less than 20%. According to the PSSC concept, we expected that there is a high chance that proteins in a PSSC recognize derivatives showing the same natural product core structure. The natural product genistein (Fig. 4) is active on both the ERβ and PPARγ proteins and the synthetic compound troglitazone (Fig. 4) is active on the PPARγ protein. The core structures of these natural products show remarkable similarity to the benzopyran core structure. Application of PSSC to find ligands for the nuclear hormone receptors would have indicated the use of the benzopyran core structure as a guiding principle for library synthesis. This example provides support for the applicability of the PSSC concept for de novo development of inhibitors for proteins of a similarity cluster.
 | ||
| Fig. 3 Superimposition of the X-ray structures of the catalytic domains of ERβ, PPARγ, and FXR, each with bound ligand. ERβ with genistein (blue), PPARγ, with rosiglitazone (red), and FXR with ligand E (Fig. 4) (yellow). | ||
 | ||
| Fig. 4 Natural, non-natural and synthetic ligands for ERβ, PPARγ, and FXR receptors. | ||
8. The PSSC of Cdc25A phosphatase–acetylcholinesterase–11β-hydroxysteroid dehydrogenases
The concept of using PSSC for actual de novo compound library design was first applied to the development of inhibitors of Cdc25A phosphatase, acetylcholinesterase (AChE) and 11β-hydroxysteroid dehydrogenase type 1 and type 2 (11βHSD1 and 11βHSD2).17 PSSC was performed as outlined in Fig. 5. Initially, searches in the Dali/FSSP35 and CE36 databases were performed with the 3D coordinates of the query protein Cdc25A (PDB codes). Hits of these searches were listed with decreasing similarity level (3D and sequence similarity). Pharmaceutically relevant proteins displaying low sequence identity (<20%), yet a certain structural similarity (RMSD < 4-5 Å) were selected and analyzed in detail.17 Proteins exhibiting sufficient similarities in their 3D structures as well as in the topological location of their functional sites are assigned to a PSSC. | ||
| Fig. 5 Systematic procedure for the searching of databases to identify protein structure similarity clusters (PSSCs). | ||
The enzymes Cdc25A phosphatase, AChE, 11βHSD1 and 11βHSD2 were clustered using the PSSC approach. The ligand sensing cores of these enzymes show remarkable structural resemblance despite their low sequence similarity (5–8%) as shown in Fig. 6. Moreover, the central catalytic residues of Cdc25A (Cys430) and AChE (Ser200) occupy similar spatial locations. Also the catalytic residues of both 11βHSD isoenzymes occupy similar positions in space with respect to the catalytically important functional groups (sulfur in Cys430 and a phenolic hydroxy group in Tyr183).
 | ||
| Fig. 6 Superposition of the catalytic cores of Cdc25A (red), 11βHSD1 (green), and AChE (blue). The important catalytic residues Cys430 (Cdc25A), Tyr183 (11βHSD1) and Ser200 (AChE), represented in CPK notion, are located identically. | ||
These enzymes represent viable or known targets for the treatment of various diseases. Cdc25A regulates cell cycle progression at the G1→S checkpoint by dephosphorylating the Cdk2/cyclin complex. Thus, it may be an interesting target for antiproliferative drug design.37,38 AChE hydrolyzes the neurotransmitter acetylcholine and thereby terminates impulse transmission at cholinergic synapses. Therefore, it is a classical target in the treatment of myasthenia gravis, glaucoma and Alzheimer's disease.39 The enzyme 11βHSD1 catalyzes the oxoreduction of cortisone to cortisol and is therefore essential for the local and tissue-specific activation of glucocorticoid receptors. Currently, application of 11βHSD1 inhibitors in the treatment of various diseases such as obesity, the metabolic syndrome, diabetes type 2 and cognitive dysfunction is being discussed.40 The 11βHSD2 isoenzyme catalyzes exclusively the oxidation of cortisol and inhibition of 11βHSD2 causes sodium retention resulting in hypertension and therefore, isoenzyme-specificity is a major prerequisite for the clinical use of 11βHSD1 inhibitors.41
According to the proposed concept, a natural product that binds to one of the PSSC member proteins was selected as ‘leitmotiv’ for the generation of a focused compound library. A naturally occurring inhibitor of Cdc25A is the sesterterpene dysidiolide (compound A, Fig. 7).42 This compound was selected as a starting point for library synthesis. Based on earlier investigations43 and literature reports on the phosphate-inhibiting activity of related natural products,37a it was hypothesized that the γ-hydroxybutenolide group incorporated in the natural product is a major determinant of its phosphatase inhibiting activity. Consequently, a 147-membered compound collection of γ-hydroxybutenolides and closely related α,β-unsaturated five-membered lactones was synthesized and screened for inhibition of Cdc25A, AChE, 11βHSD1 and 11βHSD2.17 Compounds that displayed IC50 values of ≤10 µM were considered as hits (Fig. 7). According to these guidelines, 42 out of 147 compounds were qualified as hits in the Cdc25A assay. The most potent compound had an IC50 value of 350 nM, which is significantly lower than the reported value for dysidiolide (9.4 µM42). Moreover, the compound library contained also three AChE inhibitors with IC50 values of 1.3–4.5 µM, three 11βHSD1 inhibitors with IC50 values of 7.8–10 µM and four 11βHSD2 inhibitors with IC50 values of 2.4–6.7 µM. These examples show that a hit rate of approximately 2–3% can be obtained for enzymes that were identified as similar by PSSC. Moreover, a pronounced degree of selectivity was observed for individual enzymes and also for the isoenzymes 11βHSD1 and 11βHSD2, as shown in Fig. 7 (compare compounds B, C and D).
 | ||
| Fig. 7 Active dysidiolide analogs; (A is the naturally occurring Cdc25A inhibitor, B, C, and D are synthetic analogs of dysidiolide which were found to be active against Cdc25A, AChE and 11βHSD1/2). | ||
This example convincingly demonstrates that application of target clustering based on protein structure similarity in conjunction with natural product inspired compound library synthesis provides increased hit rates at comparably small library size. It should be noted that shape complementarity or appropriate orientation of functional groups in the binding site is beyond the scope of the PSSC concept that we proposed here. Application of PSSC is not limited to existing crystal structures of proteins. In addition it could be applied to structures derived from homology model as well. A homology model, as applied for the 11βHSD type 1 and type 2 enzymes performed well in this first de novo application of PSSC for compound library design. The PSSC concept will be useful in the early stage of drug development as a first abstracting rationale to select natural products as biologically validated starting points for library design. After initial natural product selection other library design methods, like for example ligand docking in the binding site, may further improve the quality of library.
9. Conclusions
Natural products are biologically pre-validated and evolutionary selected starting points for focused compound library synthesis and find useful applications in drug discovery and chemical genomics approaches. During the last decade solid-phase chemistry has been developed to a level that multi-step synthesis of natural products and their analogues in library format has become feasible. The strong interest and progress in this approach is illustrated in this review with some of the recent reports on the solid-phase synthesis of libraries of 6,6′-spiroketal-, indole-, indoline-, and tetrahydroquinoline scaffolds. Although these examples highlight the state of the art they demonstrate that novel, versatile, and selective reactions that can be applied in the solid-phase synthesis of structurally more complex molecules are in increasing demand.Protein structure similarity clustering (PSSC), a structure based approach for clustering of proteins, serves as a new guiding principle to select natural products to target structurally similar proteins and to develop compound libraries around natural product scaffolds to address the structural diversity found in the binding sites of PSSC member proteins.
The PSSC concept offers new opportunities for the design and use of small-molecule libraries in the emerging field of ‘chemical genomics’, which basically deals with understanding the functions of members of a gene family by making use of small molecule lead compounds identified for other members of the gene family. We expect that natural product derived compound collections will play an important role in this field of research.
Acknowledgements
We thank Dr. Ingrid Vetter (Max Planck Institute of Molecular Physiology, Dortmund) for continuing inspiring discussions and Mr. Marcus A. Koch (Max Planck Institute of Molecular Physiology, Dortmund) for providing generous assistance and suggestions during the preparation of this article. This work was financially supported by the Max-Planck Gesellschaft and the Deutsche Forschungsgemeinschaft. R. B and F. J. D. thank, respectively, the Alexander von Humboldt (AvH) foundation and the Netherlands Organization for Scientific Research (NWO) for financial support.References
- R. E. Dolle, J. Comb. Chem., 2004, 6, 623 CrossRef CAS
.
-
(a) M. D. Burke and S. L. Schreiber, Angew. Chem. Int. Ed., 2004, 43, 46 CrossRef
-
(a) W. P. Walters and M. A. Murcko, Adv. Drug Deliv. Rev., 2002, 54, 255 CrossRef CAS
-
(a) F. J. Dekker, M. A. Koch and H. Waldmann, Curr. Opin. Chem. Biol., 2005 Search PubMed
- B. E. Evans, K. E. Rittle, M. G. Bock, R. M. DiPrado, R. M. Freidinger, W. L. Whitter, G. F. Lundell, D. F. Veber, P. S. Anderson, R. S. L. Chang, V. J. Lotti, D. J. Cerino, T. B. Chen, P. J. Kling, K. A. Kunkel, J. P. Springer and J. Hirshfield, J. Med. Chem., 1988, 31, 2235 CrossRef CAS
-
(a) D. J. Newman, G. M. Cragg and K. M. Snader, J. Nat. Prod., 2003, 66, 1022 CrossRef CAS
-
(a) R. E. Sammelson and M. J. Kurth, Chem. Rev., 2001, 101, 137 CrossRef CAS
- D. L. Boger, J. Desharnais and K. Capps, Angew. Chem. Int. Ed., 2003, 42, 4138 CrossRef CAS
- D. G. Hall, S. Manku and F. Wang, J. Comb. Chem., 2001, 3, 125 CrossRef CAS
- P. M. Abreu and P. S. Branco, J. Braz. Chem. Soc., 2003, 14, 675 CAS
-
(a) A. M. Boldi, Curr. Opin. Chem. Biol., 2004, 8, 281 CrossRef CAS
- O. Barun, S. Sommer and H. Waldmann, Angew. Chem. Int. Ed., 2004, 43, 3195 CrossRef CAS
- P. Arya, C.-Q. Wei, M. L. Barnes and M. Daroszewska, J. Comb. Chem., 2004, 6, 65 CrossRef CAS
- Z. Gan, P. T. Reddy, S. Quevillon, S. Couve-Bonnaire and P. Arya, Angew. Chem. Int. Ed., 2005, 44, 2
-
(a) C. Rosenbaum, P. Baumhof, R. Mazitschek, O. Müller, A. Giannis and H. Waldmann, Angew. Chem. Int. Ed., 2004, 43, 224 CrossRef CAS
- S. Couve-Bonnaire, D. T. H. Chou, Z. Gan and P. Arya, J. Comb. Chem., 2004, 6, 73 CrossRef CAS
- M. A. Koch, L. Wittenberg, S. Basu, D. A. Jeyaraj, E. Gourzoulidou, K. Reinecke, A. Odermatt and H. Waldmann, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16721 CrossRef CAS
- W. A. Lim, Curr. Opin. Struct. Biol., 2002, 12, 61 CrossRef CAS
-
(a) N. V. Grishin, J. Struct. Biol., 2001, 134, 167 CrossRef CAS
-
(a) A. Lupas, C. P. Ponting and R. B. Russell, J. Struct. Biol., 2001, 134, 191 CrossRef CAS
-
C. P. Ponting, J. Schultz, R. R. Copley, M. A. Andrade and P. Bork, in Advances in Protein Chemistry, ed. P. Bork, Academic Press, 2000, vol. 54, p. 185 Search PubMed
- A. E. Todd, C. A. Orengo and J. M. Thornton, Curr. Opin. Chem. Biol., 1999, 3, 548 CrossRef CAS
- C. Chothia, J. Gough, C. Vogel and S. A. Teichmann, Science, 2003, 300, 1701 CrossRef CAS
-
(a) A. G. Murzin, S. E. Brenner, T. Hubbard and C. Chothia, J. Mol. Biol., 1995, 247, 536 CrossRef CAS
-
(a) C. A. Orengo, A. D. Michie, S. Jones, D. T. Y. Jones, M. B. Swindells and J. M. Thornton, Structure, 1997, 5, 1093 CrossRef CAS
- R. B. Russell, P. D. Sasieni and M. J. E. Sternberg, J. Mol. Biol., 1998, 282, 903 CrossRef CAS
-
(a) V. Anantharaman, L. Aravind and E. V. Koonin, Curr. Opin. Chem. Biol., 2003, 7, 12 CrossRef CAS
-
(a) M. Downes, M. A. Verdecia, A. J. Roecker, R. Hughes, J. B. Hogenesch, H. R. Kat-Woelbern, M. E. Bowman, J.-L. Ferrer, A. M. Anisfeld, P. A. Edwards, J. M. Rosenfeld, J. G. A. Alvarez, J. P. Noel, K. C. Nicolaou and R. M. Evans, Mol. Cell, 2003, 11, 1079 CrossRef CAS
- K. C. Nicolaou, R. M. Evans, A. J. Roecker, R. Hughes, M. Downes and J. A. Pfefferkorn, Org. Biomol. Chem., 2003, 1, 908 RSC
- K. C. Nicolaou, J. A. Pfefferkorn, A. J. Roecker, G.-Q. Cao, S. Barluenga and H. J. Mitchell, J. Am. Chem. Soc., 2000, 122, 9939 CrossRef CAS
- K. C. Nicolaou, J. A. Pfefferkorn, H. J. Mitchell, A. J. Roecker, S. Barluenga, G.-Q. Cao, R. L. Affleck and J. E. Lillig, J. Am. Chem. Soc., 2000, 122, 9954 CrossRef
-
(a) M. Robinson-Rechavi, H. E. Garcia and V. Laudet, J. Cell Sci., 2003, 116, 585 Search PubMed
- V. C. Jordan, Cancer Cell, 2004, 5, 207 Search PubMed
- C. A.d. la Lastra, S. Sanchez-Fidalgo, I. Villegas and V. Motilva, Curr. Pharm. Des., 2004, 10, 3505 Search PubMed
- L. Holm and C. Sander, Science, 1996, 273, 595 CAS
-
(a) I. N. Shindyalov and P. E. Bourne, Protein Eng., 1998, 11, 739 CrossRef CAS
-
(a) M. A. Lyon, A. P. Ducruet, P. Wipf and J. S. Lazo, Nat. Rev. Drug Discov., 2002, 1, 961 CrossRef CAS
- L. Bialy and H. Waldmann, Angew. Chem. Int. Ed., 2005 Search PubMed
- M. Racchi, M. Mazzucchelli, E. Porrello, C. Lanni and S. Govoni, Pharmacol. Res., 2004, 50, 441 CrossRef CAS
-
(a) B. R. Walker and J. R. Seckl, Expert Opin. Ther. Targets, 2003, 7, 771 Search PubMed
- M. I. New and R. C. Wilson, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12790 CrossRef CAS
- S. P. Gunasekera, P. J. McCarthy and M. Kelly-Borges, J. Am. Chem. Soc., 1996, 118, 8759 CrossRef CAS
-
(a) D. Brohm, S. Metzger, A. Bhargava, O. Müller, F. Lieb and H. Waldmann, Angew. Chem. Int. Ed., 2002, 41, 307 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2005 |