Genomics and the second golden era of cancer drug development
Paul Workman
Cancer Research UK Centre for Cancer Therapeutics, The Institute of Cancer Research, Haddow Laboratories, 15 Cotswold Road, Sutton, Surrey, UK SM2 5NG. E-mail: paul.workman@icr.ac.uk; Fax: +44 (0) 20 8722 4324; Tel: +44 (0) 20 8722 4301
First published on 30th March 2005
Abstract
The first golden era of cancer drug development was initiated in the 1940s and gave rise to the cytotoxic agents that dominate current cancer medicine. The second golden era is now underway in which cancer genomics will direct drug development.
 Paul Workman | Professor Paul Workman is the Director of the Cancer Research UK Centre for Cancer Therapeutics and Harrap Professor of Pharmacology and Therapeutics at The Institute of Cancer Research, Sutton, UK. He is also Visiting Professor at Leeds and Manchester Universities. He was previously (1993–1997) Cancer Research Bioscience Section Head at AstraZeneca Pharmaceuticals where he led the biology team on the gefitinib (Iressa) discovery project. Prior to this he was Professor and Director of Laboratory Research in the Department of Medical Oncology, Beatson Laboratories, University of Glasgow (1990–1993), UICC Visiting Fellow at Stanford University (1989) and scientific staff member of the MRC Clinical Oncology Unit, MRC Centre, Cambridge University (1976–1990). Honours include European School of Oncology Award for Excellence in Oncology Research (1985), Cancer Research Campaign (now Cancer Research UK) Life Fellow (1991) and Fellow of the Academy of Medical Sciences (2002). He is a Scientific Founder of Chroma Therapeutics and PIramed Limited. |
Introduction
In October 2004, a cover feature article entitled “Beating cancer” was published in the influential Economist magazine that contained a quote from this author which claimed, “this is the second golden era of cancer research”. The article went on, “while no one expects a cure for cancer in the next decade, many think it could be demoted to the status of a chronic disease that people can live with—in other words, something more like diabetes”.1 Earlier in the same year, the equally influential Fortune magazine ran a piece with the controversial title “Why we’re losing the war on cancer and how to win it”.2 Both articles looked back at progress since the former US president Richard Nixon initiated the War on Cancer in 1971 and questioned how much has been achieved. There have in fact been real advances on many fronts but the treatment of solid cancers that have spread (metastasized) to distant sites remains a considerable challenge. Major cancer organisations remain upbeat about their aspirational targets for cancer treatment. The US National Cancer Institute has announced the objective of eliminating suffering and death due to cancer by 2015.3 Cancer Research UK aims to conquer cancer in two generations.4To achieve these objectives, much faith is being placed in cancer genomics. The elucidation of the molecular basis of cancer has certainly preceded and outpaced the therapeutic advances, but recent progress and future prospects are encouraging. In this article, I will examine the actual and potential therapeutic impact of genomics on cancer drug development and expand on my view that we are now in the second golden era. For reasons that will become apparent, emphasis will be on drugs acting on oncogenic kinases but the emerging principles will be relevant more broadly.
Lessons from the first golden era
The original golden era of cancer drug development began in the 1940s with the discovery of the therapeutic activity of the first DNA alkylating agent, nitrogen mustard, in a patient with non-Hodgkin's lymphoma, together with the demonstration of the effectiveness of the first antimetabolite, the antifolate aminopterin, in children with acute lymphoblastic leukaemia.5 These ground breaking clinical studies have led to the development since that time of a wide range of cytotoxic cancer chemotherapeutic agents that remain to this day the mainstay of drug treatment for cancer.6 Significant improvements over the original drugs were certainly achieved. The aminopterin analogue methotrexate was the first drug to cure a solid tumour, known as choriocarcinoma—a rare tumour usually derived from the placenta—and when given in high doses with leucovorin after surgery resulted in the cure of the bone tumour osteosarcoma.5 Other antimetabolites followed, such as 5-fluorouracil with activity in colorectal cancer. Nitrogen mustard was optimised into the more manageable cyclophosphamide and other DNA reactive agents gained regulatory approval, notably the platinum compounds cisplatin (which cured testicular cancer) and carboplatin with its reduced kidney toxicity and broader range of activity.5 Several natural products have proved effective, including anthracyclines, epipodophyllotoxins and camptothecins that act on DNA topoisomerases and the taxane paclitaxel which stabilises microtubule polymerisation and showed striking activity in ovarian cancer.5Steady progress was made, but without further dramatic breakthroughs in the treatment of solid cancers, particularly in their advanced, metastatic forms. Although undoubtedly useful, cytotoxic agents act on proliferating cells, usually by damaging DNA, inhibiting DNA synthesis or interfering with the mechanics of cell replication. Thus, normal dividing cells are also killed, side-effects are often severe and the therapeutic window between anticancer activity and toxicity is inevitably narrow. Options for further improvement of cytotoxics were therefore very limited. New ideas were needed and these eventually came from the molecular revolution.
Important lessons were learned from the first golden era. The principle of combining drugs, commonly in groups of two to four agents with non-overlapping toxicity, was firmly established. Drug resistance, both intrinsic and acquired, was shown to be a universal and enduring cause of treatment failure. Resistance was demonstrated frequently to occur via mutation or altered expression of the target. Animal models, initially rodent leukaemias and then human tumour xenografts, were shown to be useful but flawed. All of these issues still have resonance for us today.
Into the second golden era
The second golden era of cancer drug discovery is based on the principle that understanding the molecular basis of malignancy will provide the platform for the design of rational, mechanism-based drugs that will act selectively on malignant cells and have fewer serious toxicities compared to cytotoxics.7,8 We now understand that cancer is caused and driven by mutations in DNA that hijack the signal transduction pathways that normally operate to regulate life and death in healthy cells.9 Though still far from complete, our comprehension of the molecular pathways responsible for the malignant characteristics of human cancer, including proliferation, inappropriate survival, immortalisation, invasion, angiogenesis and metastasis, is increasingly sophisticated.10Fig. 1A illustrates schematically how the process of malignant progression is driven by the activation of oncogenes (stimulating the ‘accelerators’ of malignancy) and the inactivation of tumour suppressor genes (disabling the ‘brakes’ on cancer). Also shown is how this process is enhanced by the mutation and loss of DNA repair genes and supported by genes such as those encoding histone deacetylases and the molecular chaperone Hsp90. Many of the players in these oncogenic pathways are known and represent potential targets for therapeutic intervention. Thus, the selection of novel targets for cancer drug discovery has now become heavily dependent on the identification of new cancer genes and the pathways that they exploit.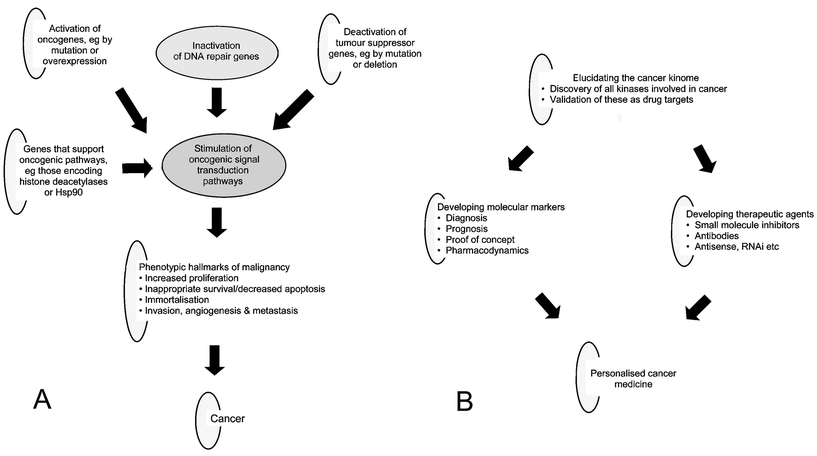 | ||
| Fig. 1 Genomics and contemporary cancer drug development. A; schematic illustration of how the development of cancer is initiated and then driven by the activation of oncogenes (including many kinases such as BRAF) and the deactivation of tumour suppressor genes (including the phosphatase PTEN). The process is enhanced by the mutation and loss of DNA repair genes and is supported by genes such as histone deacetylases and the molecular chaperone Hsp90. Changes in the structure and expression of cancer-causing genes leads to the hijacking of oncogenic signal transduction pathways. This in turn results in the characteristic features of malignant cancers. For more details see references 9–11. Many players in cancer-causing pathways are established or candidate targets for drug development. B; schematic illustration of the exploitation of the cancer genome for the development of targeted molecular therapeutics and personalised cancer treatment. The process is exemplified here by the drugging of the cancer kinome, but the same conceptual and practical approach also applies to other cancer genes. As shown, the discovery and validation of malignancy-inducing kinase genes (e.g.BRC–ABL, EGFR, BRAF and PIK3CA) triggers both the development of targeted drugs and the simultaneous development of molecular biomarkers. These biomarkers are used to identify the patients that will benefit from the drug, i.e. diagnostic and prognostic markers, as well as to demonstrate proof of concept for the molecular mechanism and to measure pharmacodynamic responses that allow rational selection of the optimal dose and schedule. Drugs are targeted to oncogenic kinases at the protein level by the design of small molecular inhibitors of catalytic activity or by the engineering of therapeutic antibodies in the case of cell membrane-associated kinases. Alternatively, and less well validated and exemplified, approaches may be targeted at the messenger RNA level, e.g. using antisense oligonucleotides, the emerging approach of RNA interference or theoretically at the cognate gene level (not shown). The archetypal small molecule kinase inhibitor is imatinib mesylate (Gleevec) which blocks the cancer-inducing drive of the mutant BRC–ABL and KIT oncogenes in chronic myeloid leukaemia and gastrointestinal stromal tumours, respectively, while the best example of a kinase-directed therapeutic antibody is trastuzumab (Herceptin) in ERBB2 overexpressing breast cancers. For more details see references 5,7 and 8. | ||
Discovering cancer genes and drug targets
There is now no doubt whatsoever that cancer is caused by the mutation and aberrant expression of critical genes.11 A recently published census identified 291 cancer-causing genes,12 representing around 1% of the latest prediction of the 20–25![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 genes that are encoded in the human genome.13 Twenty seven (or 6%) of these cancer genes encode protein kinase domains, as compared to the six genes (or 2%) predicted by random selection. In contrast to this over-representation of kinases, G-protein coupled receptors are significantly under-represented with only one known example.12 Kinases are now accepted to be readily druggable as exemplified by the efficacy and regulatory approval of the small molecule catalytic inhibitors imatinib mesylate (Gleevec), gefitinib (Iressa) and erlotinib (Tarceva), as well as therapeutic antibodies to the ERBB2† receptor (trastuzumab, Herceptin), the epidermal growth factor receptor (EGFR) (cetuximab, Erbitux) and the angiogenic vascular endothelial growth factor isoform VEGF-A ligand (bevacizumab, Avastin). See Table 1 for details and Fig. 2 for chemical structures of kinase inhibitors.
000 genes that are encoded in the human genome.13 Twenty seven (or 6%) of these cancer genes encode protein kinase domains, as compared to the six genes (or 2%) predicted by random selection. In contrast to this over-representation of kinases, G-protein coupled receptors are significantly under-represented with only one known example.12 Kinases are now accepted to be readily druggable as exemplified by the efficacy and regulatory approval of the small molecule catalytic inhibitors imatinib mesylate (Gleevec), gefitinib (Iressa) and erlotinib (Tarceva), as well as therapeutic antibodies to the ERBB2† receptor (trastuzumab, Herceptin), the epidermal growth factor receptor (EGFR) (cetuximab, Erbitux) and the angiogenic vascular endothelial growth factor isoform VEGF-A ligand (bevacizumab, Avastin). See Table 1 for details and Fig. 2 for chemical structures of kinase inhibitors. | ||
| Fig. 2 Chemical structures of small molecule kinase inhibitors. | ||
| Drug | Type | Molecular target | Major indication a | Main biological effect | Comments | Example reference |
|---|---|---|---|---|---|---|
| a Abbreviations: CML, chronic myeloid leukaemia; NSCLC, non-small cell lung cancer; GISTs, gastrointestinal stromal tumours. | ||||||
| Imatinib (Gleevec) | Small molecule | BCR–ABL, KIT, PDGFRβ | CML, GIST | Antiproliferative, pro-apoptotic | Preferential activity in leukaemias with the BCR–ABL translocation and GISTs with certain KIT mutations. Resistance develops by kinase mutation | Branford et al.,74Heinreich et al.75 |
| Gefitinib (Iressa) | Small molecule | EGF receptor | NSCLC | Antiproliferative, pro-apoptotic | Lynch et al.,35 Paez et al.36 | |
| Preferential activity in patients with mutant EGFR | ||||||
| Erlotinib (Tarceva) | Small molecule | EGF receptor | NSCLC | Antiproliferative, pro-apoptotic | Pao et al.37 | |
| Trastuzumab (Herceptin) | Humanized monoclonal antibody | ERBB2 | Breast | Antiproliferative, pro-apoptotic | Preferential activity in breast cancers overexpressing ERBB2 | Slamon et al.76 |
| Bevacizumab (Avastin) | Humanized monoclonal antibody | VEGF-A | Colorectal | Antiangiogenic | No prognostic markers yet available | Hurwitz et al.77 |
| Cetuximab (Erbitux) | Humanized monoclonal antibody | EGF receptor | Colorectal | Antiproliferative, pro-apoptotic | Activity not dependent on the degree of EGF receptor expression but patients were selected to be EGF receptor positive | Cunningham et al.78 |
The impact of genomics: drugging the cancer kinome
Given the high proportion of kinases involved in cancer, their proven druggability (Table 1) and the clinical and commercial successes emerging from various kinase-directed drugs, this class of gene products has become the most frequently targeted in cancer drug discovery.14,15 It is not surprising therefore that in the recent application of high throughput genomics together with bioinformatic methods to cancer gene discovery, the initial focus has been on protein kinases and related enzymes. Since the total collection of 518 putative human kinases encoded within the genome is known as the kinome, this approach can conveniently be labelled as drugging the cancer kinome. The availability of the complete human genome sequence,13 together with the use of rapid mutation detection and DNA sequencing technologies, has massively enhanced our capability to discover mutations in selected gene families and the strategy has been spectacularly successful with kinases.16 Several remarkable discoveries have been made using this approach, beginning with two landmark papers in 2002 and 2003, followed by a series of important publications in 2004 (Table 2). Many, if not all, of these discoveries are likely to have a major therapeutic impact and it is therefore timely to review them together from that standpoint.| Observation | Comments and therapeutic significance | Reference |
|---|---|---|
| Activating mutations in BRAF in melanoma, colorectal, thyroid and other cancers | Mutant BRAF is validated as a bona fide oncogene and cancer target | Davies et al.17 |
| Mutations in several tyrosine kinases and related enzymes, defining a tyrosine kinome in colorectal cancer | Further research is required to determine functional and potential therapeutic significance. Could represent possible new drug targets | Bardelli et al.21 |
| Mutations in tyrosine phosphatases defining tyrosine phosphatome in colorectal and other cancers | Again, further research is needed. The corresponding kinases would need to be identified for drug discovery | Wang et al.22 |
| Mutations in PIK3CA which encodes the P110α PI3 kinase were identified in glioblastoma, colorectal, gastric and breast cancer | These may be activating mutations and the significance for the development of PI3 kinase inhibitors is still to be determined | Samuels et al.,26 Bachman et al.27 |
| Activating EGFR mutations that correlated with sensitivity of non-small lung cancer patients to gefitinib and erlotinib | Suggests that drug treatment can be based on the demonstration of mutation as a predictor of molecular dependence | Lynch et al.,35 Paez et al.,36 Pao et al.37 |
| Equivalent mutations to those in EGFR identified in ERBB2 also in non-small cell lung cancer | The functional and therapeutic significance is still to be defined. Could affect sensitivity to trastuzumab or small molecule inhibitors | Stephens et al.42 |
| Point mutations in BCR–ABL that result in resistance to imatinib | This supports the development of alternative inhibitors such as BMS-354825 that retain activity against resistant mutants. Combination regimens containing inhibitors with different binding modes may reduce the incidence of mutations arising | Gorre et al.,43 Shah et al.45 |
| Point mutations in EGFR causing resistance to gefitinib and erlotinib | Demonstrates that sequential kinase mutations can result in sensitivity and then resistance to kinase inhibitors | Pao et al.,47 Kobayashi et al.48 |
The first breakthrough17 came from the use of denaturing capillary gel electrophoresis to detect activating mutations in the serine/threonine kinase BRAF gene which are now known to occur in the dangerous skin cancer malignant melanoma (27–70%), papillary thyroid cancer (5–22%), serous ovarian cancer (∼30%) and 1–2% of other malignancies.17,18 The most common mutation, accounting for 86% of all abnormalities, was originally known as V599E but was recently redesignated as V600E. This converts the valine at amino acid position 600 in the protein to a glutamate.18 Along with most other mutations, this causes catalytic activation, stimulates downstream signalling to the MAP kinase ERK1/2, induces proliferation and oncogenic transformation and causes tumour formation in animals. A detailed molecular explanation was provided by X-ray crystallographic studies19 with wild type and mutant BRAF complexed with the biaryl urea inhibitor BAY 43-9006 (sorofenib), a drug that is now in clinical trials. This work revealed that the mutation promotes the active kinase conformation by disruption of the hydrophobic interaction between the regulatory activation segment (a flexible region located between the conserved DFG and APE motifs) and the glycine-rich region kown as the P-loop (a region which normally serves to clamp ATP into the catalytic cleft). Most of the BRAF mutations cluster around the P-loop and activation segments, destabilising the inactive conformation and stimulating BRAF kinase activity. Together with cell biology studies, including the use of RNA interference to cause knockdown of RAF family members, these thorough structural studies clearly show mutant BRAF to be a bona fide oncogene and validated drug target.18 Though active on BRAF and CRAF, sorefenib is now known to inhibit a broad range of kinases including VEGF receptor tyrosine kinases.20 Inhibition of VEGF receptor kinases probably explains its activity in the highly angiogenic (blood supply-inducing) renal cancers which are driven by VHL tumour suppressor gene mutations that activate VEGF signalling. Thus, more specific BRAF inhibitors are now urgently sought.
In the subsequent studies, exon resequencing (i.e. reanalysis of the coding regions of genes) was used to look for somatic mutations in protein kinases, lipid kinases and phosphatases. In many cases multiple genes were profiled, as in the study which systematically explored the tyrosine kinome in colorectal cancers.21 All exons containing predicted kinase domains were sequenced in 90 tyrosine kinase (TK) genes, 43 tyrosine kinase-like (TKL) genes and five receptor guanylate cyclase (RGC) genes. The results indicated somatic mutations in five TK genes (NTKR3, FES, KDR, EPHA3 and MLK4), one TKL gene (MLK4) and one RGC gene (GUCY2F). Mutations were prevalent in the kinase domains. However, no functional studies were carried out and so the biological, oncological and therapeutic significance of the mutations remains to be determined.
A comparable study of tyrosine phosphatases, or the phosphatome, was also conducted in colorectal cancer.22 Coding exons of all 87 members of the tyrosine phosphatase superfamily were resequenced. Mutations were identified in six genes (PTPRF, PTPRG, PTPRT, PTPN3, PTPN13 and PTPN14). The nature of these mutations suggested the likely inactivation of phosphatase function. This was demonstrated for mutants of PTPRT which also lacked growth inhibiting activity in cells. Thus, PTPRT (and perhaps the others) could be a tumour suppressor gene, but further validation is required. However, even if appropriately validated there is no immediate drug development opportunity. This is because whereas it is technically feasible to inhibit many kinases, activation of phosphatases with drugs is not currently possible. Hence therapeutic exploitation of tumour suppressor phosphatases would require identification of the corresponding kinases which could then be inhibited.
There is already a precedent for this type of thinking. The lipid phosphatase PTEN is well established as the second most common tumour suppressor gene after P53, and is especially implicated in the brain cancer glioblastoma and also in prostate and endometrial cancer.23PTEN reverses the reactions that generate phosphatidylinositol 3,4-bisphosphate and phosphatidylinositol 3,4,5-trisphosphate that are catalysed by the class I members of the phosphatidyl inositol-3 kinase (PI3 kinase) family.24 Thus, the loss of PTEN stimulates the formation of these phosphatidyl inositol lipids leading to activation of the PI3 kinase pathway through their binding to pleckstrin homology domains of downstream signalling proteins. Especially given that there was already evidence that at least one PI3 kinase catalytic subunit may be oncogenic, namely the p110α subunit encoded by the PIK3CA gene which is amplified and overexpressed in ovarian and cervical cancer,25 a systematic search was carried out for mutations in the predicted kinase coding exons of eight PI3 kinase and eight PI3 kinase-like genes.26 Mutations were identified only in PIK3CA and these clustered in regions that could activate the kinase, an effect confirmed biochemically in one example. The mutations are found especially in colorectal (32%), glioblastoma (27%), gastric (25%) and breast cancers (8%).26,27 Functional studies are now emerging which confirm that PIK3CA mutation results in kinase activation and enhanced oncogenic properties.28 These findings add weight to the case for developing inhibitors of the PI3 kinase pathway.29–33 Potent and selective inhibitors of PI3 kinase and the downstream kinase AKT/PKB are in preclinical development but none have yet entered clinical trials.31–33 It will be important to determine whether such inhibitors exert preferential effects against cancers that show molecular dependence on, or so called ‘addiction’ to,34 the PI3 kinase pathway, as occurs to some extent with the rapamycin class of drugs that inhibit mTOR, another kinase downstream of PI3 kinase.29 Evaluation should now include assessment of the impact of PIK3CA mutations.
In another pioneering discovery, precedent has already been established recently for a greater therapeutic sensitivity to kinase inhibition for cancers which harbour activating mutations in the kinase target.35–37 Exemplification was initially provided by two important studies published in May of 2004, which demonstrated that the sensitivity of patients with non-small cell lung cancer to the EGF receptor tyrosine kinase inhibitor gefitinib correlated with EGFR mutation.35,36 This relationship was then extended to erlotinib.37 Two of the studies specifically screened for EGFR mutations35,37 while the other systematically sequenced the activation loops of 47 receptor tyrosine kinases.36 The EGFR mutations cluster in the ATP-binding site and are in positions that are likely to result in activation of the kinase activity (as noted with BRAF, see earlier). Furthermore, EGFR mutation has been shown to enhance EGFR signalling in cells and to cause much greater inhibitory sensitivity.35,36 In addition, gefinitinib-sensitising EGFR mutations selectively activate survival pathways that signal through the downstream proteins AKT/PKB and STAT3/5.38 Furthermore, cancer cells harbouring mutations become highly sensitive to molecular and pharmacological inhibition of these anti-apoptotic pathways, consistent with oncogene dependence. Multiple groups have followed up the original observations in large numbers of lung cancer patients and have confirmed that EGFR mutations are generally seen in those responsive to gefitinib and erlotinib, representing around 5–10% of US patients and 20–30% in Asian populations.39–41 There are nevertheless sensitive patients who do not harbour EGFR mutations and the mechanisms operating in these cases remain to be defined. It will also be important to reassess the disappointingly negative non-small lung cell cancer studies involving gefitinib combined with cytotoxic chemotherapy in the light of EGFR mutations. Will the same relationship hold true in the combination setting?
Interestingly, mutations analogous to those in EGFR have been identified in the related ERBB2 gene in 4% of non-small cell lung cancers overall and in 10% of the lung adenocarcinoma subclass.42EGFR and ERBB2 mutations did not occur in the same cancer, possibly indicative of functional redundancy, as seen with BRAF and the upstream cancer-causing G-protein gene RAS.17 Further work is required to determine the effects of these ERBB2 mutations on enzyme activity and drug sensitivity. In particular it will be important to assess the consequences for sensitivity to trastuzumab and also to the emerging group of small molecule ERBB2 kinase inhibitors.
The EGFR mutation story will have a major impact on future drug development strategies. Not only will it be necessary to monitor the expression of the molecular target in relation to drug sensitivity (as with ERBB2 expression and sensitivity to trastuzumab) but the effect of sequence will also need to be considered for the molecular target itself and potentially for other relevant signalling proteins. Furthermore, it will also be important to look for mutations in the kinase target that might result in resistance to kinase inhibitors.
The precedent for this was established for the first time in the context of resistance to imatinib in chronic myeloid leukaemia. Here the cancer is driven by the activation of ABL kinase following chromosomal translocation to form the BCR–ABL hybrid protein. Once again, kinase sequencing studies have been pivotal. It is clear from these analyses that resistance occurs by selection for cancer cells that have one of at least 17 point mutations in the BCR–ABL kinase domain that interfere with imatinib binding and can also, but to a much lower extent, be due to amplification of the genomic locus.43 Imatinib binding to the ABL kinase requires the activation loop to be closed and causes stabilisation of the closed or inactive conformation, similar to the binding of sorofenib to BRAF (see earlier and ref 19); in addition, it also induces a conformational change in the kinase.44 These effects explain imatinib's fairly high level of selectivity and also its potential to induce resistance. Most of the point mutations appear to cause resistance by preventing the kinase from adopting the closed form to which imatinib binds. However, it would be predicted that most of the mutant forms would retain sensitivity to structurally different kinase inhibitors that have less stringent binding requirements and can dock with either the active or the inactive conformation, but which are also less selective for any given kinase as a result. Consistent with this hypothesis, the new drug BMS-354825, which belongs to a compound class that exhibits these properties, retains activity against all but the most common mutant.45 This latter mutant involves a residue that acts as a so-called ‘gatekeeper’ for kinase inhibitors. BMS-354825 shows promise in early clinical trials.46 Based on this work it can be envisaged that a good strategy to prevent resistance arising would involve giving a combination of a highly selective kinase inhibitor like imatinib that targets the closed inactive confirmation together with an inhibitor like BMS-354825 which is less selective but binds to both active and inactive conformations. This could be a useful generic approach to kinase inhibition and has parallels with AIDS/HIV therapy. However, alternative strategies will be required to deal with the gatekeeper mutation which accounts for 15–20% of the resistant variants.
Supporting the view that kinase mutation may be a more generalised mechanism of resistance, two very recent papers have shown that in patients with EGFR mutations which result in sensitivity to gefitinib/erlotinib (see earlier), second mutations can occur that are analogous to those in BRC–ABL and that confer equivalent resistance to the inhibitors.47,48
Another promising strategy to deal with kinase resistance is to use HSP90 molecular chaperone inhibitors such as 17AAG49,50 that effectively deplete the cell of mutant BCR–ABL.51 Again this may be applicable to other kinases.
Lessons learned so far from drugging the cancer kinome
So what have we learned from our several years of experience with drugging the cancer kinome? A number of lessons in fact, many or all of which may well be applicable to other target classes.Cancer kinome inhibitors are effective but their development remains challenging
It is easily forgotten that as little as ten years ago the approach of small molecule inhibitors of kinases was unprecedented and seen as very high risk, with concerns about the problem of competing with millimolar ATP, the difficulty of achieving kinase selectivity and the likelihood of toxicity due to inhibiting kinases in normal tissues. Antibodies to kinases were also unproven. It is now obvious, a decade on, that kinases are clearly druggable technically by either small molecules or antibodies. Furthermore, the approach is validated clinically and commercially, as well as scientifically, by the regulatory approval of the superstars described in Table 1. Many more drugs targeted to cancer kinases are in preclinical studies and development. For example, inhibitors of MEK1/2, the kinase that connects RAF kinases to ERK1/2, show considerable promise, as exemplified by CI-1040 and a more potent analogue that has now replaced it in clinical trials.52 But it is not all plain sailing. Drugs like the cyclin-dependent kinase (CDK) inhibitor flavopiridol and the CDK/CHK1 inhibitor 17-hydroxystaurosporine (UCN01) have run into difficulties with issues of efficacy, toxicity or physicochemical and pharmacokinetic properties. Second generation inhibitors, such as the CDK inhibitor CYC202 (seliciclib) and others, may address limitations in the initial agents.53 This author's experience as part of the gefitinib discovery team provided a hard lesson in how challenging it can be to optimise pharmacokinetic behaviour, even when a potent and selective inhibitor has been identified. The imatinib development path, though successful, was at times very challenging.54 There were for example commercial concerns about the relatively small leukaemia market, although sales for imatinib now exceed $1 billion. The approval of gefitinib was controversial because of the modest response rate in populations that were not enriched for EGFR mutations.55 Even though the principle for kinase inhibition in cancer is now well established, each new target project will still have many hurdles to jump. Despite the implementation of modern technologies to accelerate drug discovery (for example, high throughput biochemical, in silico and fragment-based screening; combinatorial chemistry; X-ray crystallography and other rational design approaches; see reference 56), the creation of a drug, as distinct from a specific chemical probe, is still a difficult and risky enterprise.57 Given the various pitfalls listed above, particularly pharmacokinetics, selectivity and toxicity, success will not be achieved with every kinase target.Target validation and selection
Here we must balance the need for speed against the need to get it right. This applies throughout the drug development process.58 The time elapsed between the discovery of the BCR–ABL translocation in chronic myeloid leukaemia and the regulatory approval of imatinib was about 40 years. Clearly we need to be faster than this. However, if we move too quickly we may select targets that are not sufficiently validated and which may turn out not to play a key role in human cancer progression. As mentioned earlier, BRAF is now well validated as a target whereas some of the other kinases in which mutations have been identified require their credentials to be polished up further (see examples in Table 2). There is a balance to be struck between prioritising the targets most likely to result in clinical benefit while not being so overly demanding in terms of validation criteria that the result is analysis paralysis. Pharmaceutical companies will be cautious because of the massive costs of industrial drug development. Though controversial, these are calculated at $800 million when the opportunity costs associated with drug failures are taken into account. The failure rate remains high, with only 5% of drugs in cancer and other therapeutic areas surviving the attrition between entry into the clinic and regulatory approval.59 More academically orientated ‘chemical biology’ projects can afford to take greater risk, since the production of small molecule research tools can still be viewed as a success, even if disease modification is not achieved.60 Furthermore, the strategies can differ where the primary goal is to develop tool inhibitors that are used to understand biological systems rather than to develop potential drugs which have much more demanding requirements, particularly in terms of pharmacokinetics, metabolism and toxicity properties.57 This is an area where the academic and commercial sectors can be highly synergistic.In some cancers the number of patients exhibiting molecular dependence on a particular abnormality may be quite low. As mentioned earlier with respect to the early days of imatinib development, this has major implications for the potential market for pharmaceutical companies. Again there is a role here for academic organisations to share the risk and hence to help fulfil unmet medical need in potentially small groups of patients. A noteworthy example of this is the group of paediatric cancers that are not only relatively uncommon but also exhibit quite distinct and unusual genetic changes. Looking at this more positively, the same molecular abnormalities may be seen in small percentages of any one type of cancer but may occur across several cancer types, resulting a total aggregate number of cases that is much more substantial. In this way, cancer treatment seems likely to evolve to target drugs towards particular molecular pathologies rather than towards cancers that are classified by anatomical location, stage and grade. On the other hand, the biological context of a given molecular abnormality (e.g. as dictated by the cell of origin) will almost certainly play a significant role in ways that we do not yet understand at the molecular level.
Selecting patients based on molecular dependence
The experience with imatinib and gefitinib exemplifies the importance of selecting patients for drug treatment that exhibit the molecular characteristics that result in dependence on, or addiction to, the drug target. Thus, patients who are unlikely to respond can be spared unnecessary treatment. Clinical trials can be focussed on the most likely responsive subpopulations, leading to more rapid regulatory approval based on fewer patients. Approved drugs can then be targeted to responsive subpopulations, leading to an overall pharmacoeconomic benefit.Developing biomarkers
In addition to diagnostic and prognostic biomarkers of molecular dependence which are essential, it is also necessary to develop pharmacodynamic markers that can be used to demonstrate proof of concept for target modulation as well as to select the most optimal drug dose and administration schedule (see Fig. 1B and references 61–64). A recent survey shows that the implementation of biomarkers for patient selection and pharmacodynamic analysis is disappointingly uncommon in early clinical trials of molecular cancer therapeutics.65Resistance develops through mutation
It is clear that drug exposure leads to the selection of resistant mutants. Demonstration of this mechanism with both imatinib and gefitinib suggest that it will be a common problem. The rational design of second generation inhibitors that retain activity against the mutant forms is therefore encouraged. Furthermore, the use upfront of combinations of kinase inhibitors that target the inactive and active conformations, respectively, may be particularly effective in preventing resistance arising by target mutation.Combinational treatments for cancers with multi-gene pathologies
Unlike in chronic myeloid leukaemia where in the early phase BCR–ABL appears to be the single causal oncogene, the majority of cancers are likely to be driven by a combination of several different pathological abnormalities. Thus, treatment targeted at any one of these will be necessary but insufficient for an optimal therapeutic outcome. Rather, it will be important to use appropriate combinational approaches involving drug cocktails, multi-targeted kinase inhibitors (i.e. drugs that act on several key oncogenic kinases) or the use of drugs with pleiotropic downstream biological effects.66,67 Such drugs include inhibitors of the proteasome,68 chromatin modifying enzymes such as histone deacetylases69 and the HSP90 molecule chaperone.70 Although these targets may be considered as performing housekeeping functions that would be required for normal as well as cancer cells, the promising therapeutic results obtained with inhibitors in preclinical models and in the clinic suggests that such targets may be especially important in supporting multiple oncogene addiction.Prioritising drug combinations is difficult
Considering both the clinical experience of chemotherapy from the first golden era and the molecular information from the second golden era, it is likely that drug combinations are going to be essential for a meaningful and durable long term therapeutic benefit.66,67 Combinations could involve cocktails of targeted molecular therapeutics or regimens involving mixtures of drugs from the two eras. Either way, the choice and development of such combinations is difficult.71 Animal models are not necessarily predictive. In the clinic, there is evidence for synergy between antibodies such as rituximab (Rituxan), trastuzumab, bevacizumab and cetuximab with conventional cytotoxic chemotherapy,5 but results with small molecule signalling inhibitors such as gefitinib plus cytotoxics have been disappointing.39 Given the overwhelming number of possible combinations and the uncertainty in predicting the ones that are most likely to be effective, it seems appropriate in this new post-genomic era to prioritise combinations based on molecular profiling.5,71 Carefully planned preclinical and clinical studies are required in this area, incorporating extensive molecular characterisation. These investigations are going to be particularly important in the setting in which individual members of the combination may have minimal activity as single agents. Once again, biomarkers will be essential. In practical terms such combination studies are also challenging because of the involvement of more than one pharmaceutical company.Concluding remarks: towards personalised cancer medicine
This article opened by drawing a distinction between the first and second golden eras of cancer drug development. The main differentiating feature is that the second era is based firmly on targeted therapies that are designed specifically to tackle the molecular abnormalities that we now know are responsible for the causation and progression of human cancers. This is a practical as well as intellectual distinction. It influences the choice of drug target, the way the drugs are developed and the selection of patients, all of which can be based rationally on molecular insights and measurements. The examples cited here have been drawn heavily from the experience with kinase targets and drugs since it is in drugging the cancer kinome that we have accrued the most experience. It seems likely, however, that the issues and learning points will be similar for other classes of molecular targets and therapeutics.Differences between the two eras of drug development have been emphasised here in order to ensure that a real appreciation is gained of the advances made and the differences in scientific approach. It is also true that there are transferable lessons from the first golden era that need to be taken on board in the second, in particular the persistent problem of drug resistance and the importance of drug combinations. It should also be remembered that the anti-hormonal agents that have been employed for a long time in breast and prostate cancer were in many ways the first molecular therapeutics targeted to the biology of specific cancers and lessons can also be learned from their use.
It is the opinion of this author that when we look back in twenty years time we will see enormous benefits from the second golden era of cancer drug development, equivalent at least to those achieved in the much more mature cytotoxic era. We are entering a new period in which cancer drug treatment will be based increasingly on the genomic and molecular profile of the individual patient and their cancer. Personalised treatments will be challenging to deliver, but will be of major benefit to cancer patients. Molecular profiling and in particular the identification of patients with molecular dependency on the drug target is likely to be critical.72,73 In terms of the immediate challenges ahead, the focus for the pharmacologist and oncologist should be on the development of appropriate molecular profiling tools (including mutation sequencing, gene expression patterns, proteomic and metabolomic analyses and functional molecular imaging). New approaches and models that can be used to assemble the most effective, rationally based combinations are urgently needed. Meanwhile, the basic research community will continue to uncover the remaining oncogenic abnormalities so that we have a complete molecular portrait of all human cancers. This will in turn uncover more targets for drug discovery. One thing is certainly true: there is much work to do and it is an exciting privilege for all of us that are involved in the endeavour.
Acknowledgements
I thank my many colleagues for collaboration and discussion. I also thank Dr Vasillios Bavetsias for chemical structures and Professor Keith Jones for helpful suggestions. The work of the author's laboratory (http://www.icr.ac.uk/cctherap) is funded primarily by Cancer Research UK [CUK] Programme Grant C309/A2187 and he is a Cancer Research UK Life Fellow.References
- Anonymous, Economist, 2004, October 16, 99––101 Search PubMed.
- C. Leaf, Fortune, 2004, 149(6), 76–82 Search PubMed.
- A. C. Von Eschenbach, Nat. Rev. Cancer, 2001, 4, 820–828.
- http://www.cancerresearchuk.org.
- B. A. Chabner and T. G. Roberts, Nat. Rev. Cancer, 2005, 5, 65–72 CrossRef CAS.
- Cancer Principles and Practice of Oncology, ed. V. T. DeVita Jnr., S. Hellman and S. A. Rosenburg, 2004, Lipincott, Philadelphia, 7th edn Search PubMed.
- Principles of Molecular Oncology, ed. M. H. Bronchud, M. A. Foote, G. Giaccone, O. Olopade and P. Workman, 2004, Humana Press, Totowa, 2nd edn Search PubMed.
- P. Workman, Curr. Opin. Pharmacol., 2001, 1, 342–352 CrossRef CAS.
- G. I. Evan and K. H. Vousden, Nature, 2001, 411, 343–348.
- D. Hanahan and R. A. Weinberg, Cell, 2004, 100, 57–70.
- A. Balmain, J. Gray and B. Ponder, Nat. Genet., 2003, 33(suppl), 238–244 CrossRef CAS.
- A. Futreal, L. Coin, M. Marshall, T. Down, T. Hubbard, R. Wooster, N. Rahman and M. R. Stratton, Nat. Rev. Cancer, 2004, 4, 177–183 CrossRef . For updates see: http://www.sanger.ac.uk/genetics/CGP/Census.
- International Human Genome Sequencing Consortium. Finishing the enchromatic sequence of the human genome, Science, 2004, 431, 931–946 Search PubMed.
- P. Cohen, Nat. Rev. Drug Discovery, 2002, 1, 309–315 CrossRef CAS.
- A. L. Hopkins and C. R. Groom, Nat. Rev. Drug Discovery, 2002, 1, 727–730 CrossRef CAS.
- B. Weir, X. Zhao and M. Meyerson, Cancer Cell, 2004, 6, 433–438 Search PubMed.
- H. Davies, G. R. Bignell, C. Cox, P. Stephens, S. Edkins, S. Clegg, J. Teague, H. Woofendin, M. G. Garnett, W. Bottomley, N. Davis, E. Dicks, R. Ewing, T. Floyd, K. Gray, S. Hall, R. Hawes, J. Hughes, V. Kosmidou, A. Menzies, C. Mould, A. Parker, C. Stevens, S. Watt, S. Hooper, R. Wilson, H. Jayatilake, B. A. Gusterson, C. Cooper, J. Shipley, D. Hargrave, K. Pritchard-Jones, N. Maitland, G. Chenevix-Trench, G. J. Riggins, D. D. Bigner, G. Palmieri, A. Cossu, A. Flanagan, A. Nicholson, J. W. C. Ho, S. Y. Leung, S. T. Yuen, B. L. Weber, H. F. Seigler, T. L. Darrow, H. Paterson, R. Marais, C. J. Marshall, R. Wooster, M. R. Stratton and P. A. Fureal, Nature, 2002, 417, 949–954 CrossRef CAS.
- M. J. Garnett and R. Marais, Cancer Cell, 2004, 6, 313–319 Search PubMed.
- T. C. Wan, M. J. Garnett, S. M. Roe, S. Lee, D. Niculescu-Duvaz, V. M. Good, C. M. Jones, C. J. Marshall, C. J. Springer, D. Barford and R. Marais, Cell, 2004, 116, 855–867 CrossRef.
- S. M. Wilhelm, C. Carter, L. Tang, D. Wilkie, A. McNabola, H. Rong, C. Chen, X. Zhang, P. Vincent, M. McHugh, Y. Cao, J. Shujath, S. Gawlak, D. Eveleigh, B. Rowley, L. Liu, L. Adnane, M. Lynch, D. Auclair, I. Taylor, R. Gedrich, A. Voznesensky, B. Riedl, L. E. Post, G. Bollag and P. A. Trail, Cancer Res., 2004, 64, 7099–7109 CAS.
- A. Bardelli, D. W. Parsons, N. Silliman, J. Ptak, S. Szabo, S. Saha, S. Markowitz, J. K. Willson, K. Parmiagiani, L. W. Kinzler, B. Vogelstein and V. E. Velculescu, Science, 2003, 300, 949 CrossRef CAS.
- Z. Wang, D. Shen, D. W. Parsons, A. Bardelli, J. Sager, S. Szabo, J. Ptak, N. Silliman, B. A. Peters, M. S. van der Hejden, G. Parmigiani, H. Yan, T. L. Wang, G. Riggins, S. M. Powell, J. K. Willison, S. Markowitz, K. W. Kinzler, B. Vogelstein and V. E. Velculescu, Science, 2004, 304, 1164–1166 CrossRef CAS.
- P. L. Dahia, Endocr. Relat. Cancer, 2000, 7, 115–129 Search PubMed.
- B. Vanhaesebroeck, S. J. Leevers, K. Ahmadi, J. Timms, R. Katso, P. C. Driscoll, R. Woscholski, P. J. Parker and M. D. Waterfield, Annu. Rev. Biochem., 2001, 70, 535–602 CrossRef CAS.
- L. Shayesteh, Y. Lu, W. L. Kuo, R. Baldocchi, T. Godfrey, C. Collins, D. Pinkel, B. Powell, G. B. Mills and J. W. Gray, Nat. Genet., 1999, 21, 99–102 CrossRef CAS.
- Y. Samuels, Z. Wang, A. Bardelli, N. Silliman, J. Ptak, S. Szabo, H. Yan, A. Gazdar, S. M. Powell, G. J. Riggins, J. K. Willison, S. Markowitz, K. W. Kinzler, B. Vogelstein and V. E. Velculescu, Science, 2004, 23, 304.
- K. E. Bachman, P. Argani, Y. Samuels, N. Silliman, J. Ptak, S. Szabo, H. Konishi, B. Karakas, B. G. Blair, C. Lin, B. A. Peters, V. E. Velculescu and B. H. Park, Cancer Biol. Ther., 2004, 8, 772–775 Search PubMed.
- S. Kang, A. G. Bader and P. K. Vogt, Proc. Natl. Acad. Sci. U. S. A., 2005, 18(102), 802–807 CrossRef.
- I. Vivanco and C. L. Sawyers, Nat. Rev. Cancer, 2002, 2, 489–500 CrossRef CAS.
- R. Stein and M. D. Waterfield, Mol. Med. Today, 2000, 6, 347–357 CrossRef CAS.
- P. Workman, Biochem. Soc. Trans., 2004, 32(Pt2), 393–396 CrossRef CAS.
- B. E. Drees, G. B. Mills, R. Rommel and G. D. Prestwich, Expert Opin. Ther. Pat., 2004, 14, 703–732 Search PubMed.
- S. G. Ward, Y. Sotsios, J. Dowden, I. Bruce and P. Finan, Chem. Biol., 2003, 10, 207–213 CrossRef CAS.
- I. B. Weinstein, Science, 2002, 297, 63–64 CrossRef CAS.
- T. J. Lynch, D. W. Bell, R. Sordella, S. Gurubhagavatula, R. A. Okimoto, B. W. Brannigan, P. L. Harris, S. M. Haserlet, J. G. Supko, F. G. Haluska, D. N. Louis, D. C. Christiani, J. Settleman and D. A. Haber, New Engl. J. Med., 2004, 350, 2129–2139 CrossRef CAS.
- J. G. Paez, P. A. Janne, J. C. Lee, S. Tracy, H. Greulich, S. Gabriel, P. Herman, F. J. Kaye, N. Lindeman, T. J. Boggon, K. Naoki, H. Sasaki, Y. Fujii, M. J. Eck, W. R. Sellers, B. E. Johnson and M. Meyerson, Science, 2004, 304, 1497–1500 CrossRef CAS.
- W. Pao, V. Miller, M. Zarkowski, J. Doherty, K. Politti, I. Sarkaria, B. Singh, R. Heelan, V. Rusch, L. Fulton, E. Mardis, D. Kupfer, R. Wilson, M. Kris and H. Varmus, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13306–13311 CrossRef CAS.
- R. Sordella, D. W. Bell, D. A. Haber and J. Settleman, Science, 2004, 305, 1163–1167 CrossRef CAS.
- J. E. Dowell and J. D. Minna, Nature Clinical Practice Oncology, 2004, 1, 2–3 Search PubMed.
- H. Shigematsu, L. Lin, T. Takahashi, M. Nomura, M. Suzuki, I. I. Wistuba, K. M. Fong, H. Lee, S. Toyooka, N. Shimizu, T. Fujisawa, Z. Feng, J. A. Roth, J. Herz, J. D. Minna and A. F. Gazdar, J. Natl. Cancer Inst., 2005, 2(97), 339–346 CrossRef.
- W. R. Sellers and M. Meyerson, J. Natl. Cancer Inst., 2005, 2(97), 326–328 CrossRef.
- P. Stephens, C. Hunter, G. Bignell, S. Edkins, H. Davis, J. Teague, C. Stevens, S. O'Meara, R. Smith, A. Parker, A. Barthorpe, M. Blow, L. Brackenbury, A. Butler, O. Clarke, J. Cole, E. Dicks, A. Dyke, A. Drozd, K. Edwards, S. Forbes, R. Foster, K. Gray, C. Greenman, K. Halliday, K. Hills, V. Kosmidou, R. Lugg, A. Menzies, J. Perry, R. Petty, K. Raine, L. Ratford, R. Shepherd, A. Small, Y. Stephens, C. Tofts, J. Varian, S. West, S. Widaa, A. Yates, F. Brasseur, C. S. Cooper, A. M. Flanagen, M. Knowles, S. Y. Leung, D. N. Louis, L. H. J. Looijenga, B. Malkowic, M. A. Pierotti, B. The, G. Chevenvix-Trench, B. L. Weber, S. T. Yuen, G. Harris, P. Goldshaw, A. G. Nicholson, P. A. Futreal, R. Wooster and M. R. Stratton, Nature, 2004, 431, 525–526 CrossRef CAS.
- M. E. Gorre, M. Mohammed, K. Ellwood, N. Hsu, R. Paquette, P. N. Rao and C. L. Sawyers, Science, 2001, 293, 876–880 CrossRef CAS.
- T. Schindler, W. Bornmann, P. Pellicena, W. T. Miller, B. Clarkson and J. Kuriyan, Science, 2000, 289, 1938–1942 CrossRef CAS.
- N. P. Shah, C. Tan, F. Y. Lee, P. Chen, D. Norris and C. L. Sawyers, Science, 2004, 305, 399–401 CrossRef CAS.
- C. L. Sawyers, N. P. Shah, H. M. Kantarjian, N. Donato, J. Nicoll, S. A. Bai, F. Huang, E. Clark, A. P. DeCillis and M. Talpaz, Blood, 2004, 104, abstract 1.
- W. Pao, V. A. Miller, K. A. Politi, G. J. Riely, R. Somwar, M. F. Zakowski, M. G. Kris and H. Varmus, PLoS Med., 2005, 2, 1–11 Search PubMed.
- S. Kobayashi, T. J. Boggon, T. Dayaram, P. A. Jänne, O. Kocher, M. Meyerson, B. E. Johnson, M. J. Eck, D. G. Tenen and B. Halmos, New Engl. J. Med., 2005, 352, 786–92 CrossRef CAS.
- P. Workman, Trends Mol. Med., 2004, 10, 47–51 Search PubMed.
- P. Workman, Curr. Cancer Drug Targets, 2003, 3, 297–300 Search PubMed.
- M. E. Gorre, K. Ellwood-Yen, G. Chiosis, N. Rosen and C. L. Sawyers, Blood, 2002, 100, 3041–3044 CrossRef CAS.
- J. S. Sebolt-Leopold, D. T. Dudley, R. Herrera, K. Van Becelaere, A. Wiland, R. C. Gowan, H. Tecle, S. D. Barrett, A. Bridges, S. Przybranowski, W. R. Leopold and A. R. Saltiel, Nat. Med., 1999, 5, 810–816 CrossRef CAS.
- S. R. Whittaker, M. I. Walton, M. D. Garrett and P. Workman, Cancer Res., 2004, 64, 262–272 CAS.
- R. Capdeville, E. Buchdunger, J. Zimmerman and A. Matter, Nat. Rev. Drug Discovery, 2002, 7, 493–501 CrossRef.
- T. G. Roberts and B. A. Chabner, New Engl. J. Med., 2004, 251, 501–505 CrossRef CAS.
- P. Workman, Cancer Chemother. Pharmacol., 2003, 52 (Suppl 1), S45–S46 CAS.
- C. Lipinsky and A. Hopkins, Nature, 2004, 432, 855–861 CrossRef.
- J. Becker, Nat. Biotechnol., 2004, 22, 15–18 CrossRef CAS.
- I. Kola and J. Landis, Nat. Rev. Drug Discovery, 2004, 3, 711–715 CrossRef CAS.
- C. P. Austin, L. S. Brady, T. R. Inselad and F. S. Collins, Science, 2004, 306, 1138–1139 CrossRef CAS.
- P. Workman, Eur. J. Cancer, 2002, 18, 2189–2193 CrossRef.
- R. Frank and R. Hargreaves, Nat. Rev. Drug Discovery, 2003, 2, 566–580 CrossRef CAS.
- P. Workman, Curr. Pharm. Des., 2003, 9, 891–902 Search PubMed.
- K. A. Gelmon, E. A. Eisenhauer, A. L. Harris, M. J. Ratain and P. Workman, J. Natl. Cancer Inst., 1999, 91, 1281–1287 CrossRef CAS.
- W. R. Paruleker and E. A. Eisenhauer, J. Natl. Cancer Inst., 2004, 96, 990–997 CrossRef.
- P. Workman, Curr. Opin. Invest. Drugs, 2003, 4, 1410–1415 Search PubMed.
- M. V. Blagosklonny, Cell Cycle, 2005, 4, 89–98 Search PubMed.
- J. Adams, Trends Mol. Med., 2002, 8(4 Supp), S49–54 Search PubMed.
- R. Kristeleit, L. Stimson, P. Workman and W. Aherne, Expert Opin. Emerg. Drugs, 2004, 9, 135–154 Search PubMed.
- B. W. Dymock, M. J. Drysdale, E. McDonald and P. Workman, Expert Opin. Ther. Pat., 2004, 14, 837–847 Search PubMed.
- A. Jackman, P. Workman and S. Kaye, Drug Discovery Today: Therapeutic Strategies, 2004, 1, 1–10 Search PubMed.
- C. L. Sawyers, Genes Dev., 2003, 17, 2998–3010 CrossRef CAS.
- T. G. Roberts and B. A. Chabner, New Engl. J. Med., 2004, 351, 501–505 CrossRef CAS.
- S. Branford, Z. Rudzki, S. Walsh, I. Parkinson, A. Grigg, J. Szer, K. Taylor, R. Herrmann, J. F. Seymour, C. Arthur, D. Joske, K. Lynch and T. Hughes, Blood, 2003, 102, 276–283 CrossRef CAS.
- M. C. Heinreich, C. L. Corless, G. D. Demetri, C. D. Blanke, M. von Mehren, H. Joensuu, L. S. McGreevey, C. J. Chen, A. D. van den Abbeele, B. J. Druker, B. Kiese, B. Eisenberg, P. J. Roberts, S. Singer, C. D. Fletcher, S. Siberman, S. Dimitrijevic and J. A. Fletcher, J. Clin. Oncol., 2003, 21, 4324–4349.
- D. J. Slamon, B. Leyland-Jones, S. Shak, H. Fuchs, V. Paton, A. Bajamonde, T. Fleming, W. Eiermann, J. Wolter, M. Pegram, B. J. Baselga and L. Norton, New Engl. J. Med., 2001, 15(344), 783–792 CrossRef.
- H. Hurwitz, L. Fehrenbacher, W. Novotny, T. Cartwright, J. Hainsworth, W. Heim, J. Berlin, A. Baron, S. Griffing, E. Holmgren, N. Ferrara, G. Fyfe, B. Rogers, R. Ross and F. Kabbinavar, New Engl. J. Med., 2004, 350, 2335–2342 CrossRef CAS.
- D. Cunningham, Y. Humblet, S. Siena, D. Khayat, H. Bleiberg, A. Santoro, D. Bets, M. Mueser, A. Harstrick, C. Verslype, I. Chau and E. Van Cutsem, New Engl. J. Med., 2004, 351, 337–345 CrossRef CAS.
Footnote |
| † The convention used in the article is that gene names are given in upper case italic font (e.g.ERBB2) whereas the cognate proteins are shown in upper case regular font (e.g. ERBB2). ERBB2, EGFR and VEGF receptors are membrane receptor tyrosine kinases. |
| This journal is © The Royal Society of Chemistry 2005 |