NMR studies on Cu(II)–peptide complexes: exchange kinetics and determination of structures in solution
Elena
Gaggelli
a,
Henryk
Kozlowski
b,
Daniela
Valensin
a and
Gianni
Valensin
*a
aDepartment of Chemistry, University of Siena, Via Aldo Moro, Siena 53100, Italy. E-mail: valensin@unisi.it; Fax: ++30-0577-234254; Tel: ++39-0577-234231
bFaculty of Chemistry, University of Wroclaw, Joliot-Curie 14, 50-383 Wroclaw, Poland
First published on 8th March 2005
Abstract
The interaction of copper(II) with histidine containing peptides has recently acquired renewed interest following the established link between abnormal protein behaviour in neurodegenerative processes and unpaired copper homeostasis. Five peptide sequences taken from the amyloid precursor protein and the prion protein were considered. Addition of paramagnetic Cu(II) ions to solutions of such peptides was not found to severely affect the appearance of NMR spectra, thus limiting the usual approach for structural determination. Exchange kinetics was shown to play a major role in determining the observed paramagnetic spin-lattice relaxation rates. Two independent methods were suggested for evaluating the exchange rates of His-containing peptides from the copper-coordination sphere and to calculate copper–proton distances. In such a way NMR was demonstrated to have the potential of providing detailed structures of the Cu(II)–peptide complexes in solution.
1. Introduction
Addition of paramagnetic ions to solutions of any NMR detectable ligand has been widely exploited in investigations aimed at delineating structures of metal complexes. In fact, nuclear relaxation rates and chemical shifts are dramatically affected in a way that is strictly related to metal location in the molecular frame. The desired structural information is thus gained by applying developed theories of paramagnetic NMR.1 However, if compared to such general trends, what is commonly observed with histidine-containing peptides strongly binding Cu(II) is that NMR spectra are practically unaffected even at very high metal : ligand ratios;2 only the imidazole protons of the considered peptides are somehow broadened, which ratifies the common view of histidine providing the first and most relevant anchoring point for Cu(II) at pH values consistent with imidazole first deprotonation.3–9This being the case, the use of NMR has generally been limited to qualitatively demonstrate the involvement of His (or other eventual residues) in metal binding without taking any advantage of the potential of the technique in obtaining structural details. Other methods, such as EPR, CD, UV-VIS, potentiometric titration or, whenever possible, X-ray spectroscopy have been given prominence in gaining structural, kinetic and thermodynamic information on systems involving Cu(II), especially on copper-binding domains of proteins playing relevant roles in neurodegenerative processes, where direct links with unpaired copper homeostasis have been demonstrated.10–13
The aim of the present work is to ascertain whether the potential of NMR can be exploited also in the case of peptides strongly interacting with Cu(II) such that delineation of structures of copper(II) complexes is made possible by obtaining nucleus–metal distances from NMR and using them as restraints in molecular mechanics and dynamics calculations. The NMR approach will be exemplified by experiments carried out on His-containing synthetic peptides taken from the sequences of the human prion protein and the amyloid precursor protein. The sequence of the investigated peptides are shown in Table 1.
| PrP_Monomer | Ac-ProHisGlyGlyGlyTrpGly-NH2 |
| PrP_Dimer | Ac-(ProHisGlyGlyGlyTrpGlyGln)2-NH2 |
| APP(145–155) | Ac-GluThrHisLeuHisTrpHisThrValAlaLys-NH2 |
| PrP106–126 | LysThrAsnMetLysHisMetAlaGlyAlaAlaAlaAlaGlyAlaValValGlyGlyLeuGly |
| PrP106–113 | LysThrAsnMetLysHisMetAla-NH2 |
2. Materials and methods
Synthesis of peptides
Peptides were synthesized by the solid-phase method using Fmoc chemistry. The TentaGel S RAM (solutions of Fmoc froups 0.25 meq g−1). (RAPP Polymere, Germany) was used as a support. The syntheses were carried out manually. Deprotections were performed with 50% morpholine in DMF–NMP, (1 : 1, v/v) with addition 1% Triton X-100. Couplings were achieved using 1 mM HOBt–1 mM DIPCDI (1 : 1, v/v) in the mixture DNF–NMP (1 : 1, v/v) with addition 1% Tritron X-100 for 60 min. When required, after completion of the syntheses, the N-terminal amino group was acetylated with 0.2 M 1-acetylimidazole in DMF–DCM (1 : 1, v/v) for 40 min.14 The peptides were removed from the resin together with the side chain protections in one step procedure using Reagent-B + TFA–phenol–triisopropylsilane–H2O (88 : 5 : 2 : 5, v/v) for 90 min.15 The crude peptide was purified on a semipreparative C8 HPLC column (Kromasil-100 10*250 mm, 15 µM, Knauer) with a linear gradient 20–80% of B (A: 0.1% TFA; B: 80% acetonitrile in A) over 30 min. Elution profiles were monitored at 278 nm. The purity of the peptide was checked on an analytical C8 HPLC column (Kromasil-100 4.6*250 mm, Knauer) with a linear gradient 20–80% of B (A: 0.1% TFA; B: 80% acetonitrile in A) over 30 min monitored at 276 nm, and for both peptides was greater than 96%. The synthesized peptides have shown a correct molecular mass measured by mass spectrometry using the ESI-MS technique.NMR experiments
Peptides were dissolved in deionised water containing D2O 10% or in deuterium oxide. The pH was adjusted at desired values with either DCl or NaOD. In some cases, the aqueous suspension was freeze-dried and the lyophilised sample was dissolved in DMSO-d6. The solutions were carefully deoxygenated through a freezing–vacuum pumping–sealing–thawing procedure. The desired concentration of copper ions was achieved by using a stock solution of copper nitrate (Sigma Chemical Co.) either in deionised water or in DMSO-d6. TSP-d4, 3-(trimethylsilyl)-[2,2,3,3-d4] propansulfonate, the sodium salt, was used as an internal reference standard.All the NMR experiments were carried out on a Bruker Avance DRX 600 spectrometer at controlled temperature (±0.2 K). A 5 mm triple broadband inverse probe was used for most of the measurements, a cryoprobe was used for acquisition of 13C and 15N HSQC experiments at natural abundance. Suppression of the residual water signal was achieved by excitation sculpting16 using a selective square pulse on water 2 ms long. A typical NMR spectrum required 16 transients acquired with a 7200 Hz spectral width, and 2.0 s recycling delay. Proton resonance assignments for the ligand were obtained using COSY, TOCSY, NOESY and ROESY experiments. TOCSY experiments were acquired with a total spin-locking time of 75 ms using a MLEV-17 mixing sequence. NOESY and ROESY spectra were obtained at different values of mixing time to optimise the best one. Spectra processing was performed on a Silicon Graphics O2 workstation using XWINNMR 2.6 software.†
The proton spin lattice relaxation rates were measured with an inversion recovery pulse sequence; the same sequence was also used to measure the single- or double-selective relaxation rates by means of suitably shaped π-pulses instead of the usual non-selective π-pulse. All rates were calculated by regression analysis of the initial recovery curves of longitudinal magnetization components leading to errors not larger than ±3%.
Since the simple inversion recovery experiment is suitable only for the well isolated peaks, the IR–TOCSY sequence was used to calculate the relaxation rates of the overlapping 1H NMR signals.17 The obtained results were compared with those obtained from the normal IR sequence. The agreement was found within the error limit of both experiments.
Structure calculations
The proton spin–lattice relaxation rates of the peptide in the metal complex (R1b) were converted into distance constraints. Simulated annealing in the torsional angle space was performed with 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps using 300 random starting positions of the investigated peptides generated with the program DYANA.18
000 steps using 300 random starting positions of the investigated peptides generated with the program DYANA.18
DYANA (DYnamics Algorithm for NMR Applications) is a widely used program for NMR structure calculations. The program uses simulated annealing combined with molecular dynamics in torsion angle space. The target function takes the role of the potential energy, and the system is coupled with a temperature bath which is cooled down slowly from an initial high temperature thereby allowing the system to cross the barriers between the local minima of the target function. In this procedure the potential energy is a function of the difference between the distance constraints provided by the user and the corresponding distances found in a given conformer (target function). No other potential energy terms are present except the Van der Waals repulsion. At the beginning of the calculation an arbitrary number of different conformers is generated by randomly varying torsional dihedral angles. Then, the potential energy is minimised by a simulated annealing procedure in the torsion angle space in which the system is brought to high temperature, to allow all possible high energy starting conformations, and subsequently, cooled down in order to stabilise it in the potential energy minima which better satisfy the imposed constraints.
Molecular structures were generated by the HYPERCHEM software package19 by using the ZINDO-1 semi-empirical method for charge calculations and the MM+ force field for molecular mechanics and dynamics calculations.
3. Results and discussion
In support of the general statement that NMR parameters of His-containing peptides are barely affected by added Cu(II) ions, Fig. 1 shows the effect of the reported addition of Cu(II) ions on the appearance of the 1H-NMR spectra of two peptides taken from the sequence of the amyloid precursor protein (APP) and the prion protein (PrP). If it were not for some broadening experienced by the arrowed aromatic protons of histidine, one might conclude that copper(II) does not change the appearance of the spectra, as if it were not binding the peptides. In contrast with such observation, all other experimental techniques support strong interaction of copper. This apparent paradox can be explained by taking contributions of the exchange rate from the metal coordination sphere into account. In solutions of ligands containing paramagnetic ions as co-solute, the measured spin-lattice relaxation rate is averaged over the values in the two (or more) environments: free (f) and metal-bound (b):1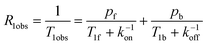 | (1) |
 | (2) |
 | ||
| Fig. 1 1H-NMR spectra of a) APP(145–155) 2 mM in H2O–D2O at pH 7.3; b) APP(145–155) 2 mM in H2O–D2O at pH 7.3 after addition of 0.02 equivalents of Cu(II); c) PrP106–113 2.6 mM in H2O–D2O at pH 5.6; d) PrP106–113 2.6 mM in H2O–D2O at pH 5.6 after addition of 0.1 equivalents of Cu(II). The arrows help locate the aromatic protons of histidine in the NMR spectra. | ||
The paramagnetic contribution is easily measured and has the potential of providing R1b = 1/T1b which is the structure-sensitive term, as accounted for by the Solomon equation for systems with S = ½:20
 | (3) |
When metal–nucleus distances are sought, R1b must be calculated from eqn (2) and, therefore, the contribution of exchange, if not negligible, must be evaluated in independent ways. The same equation suggests that the exchange rate may become a limiting factor and level off the paramagnetic contribution to spin-lattice relaxation rates and, in the same way, to the line-width. This may effectively be the case for copper(II) strongly associated to His-containing peptides, where the inverse of the off-rate kinetic constant (k−1off) may become so long that the dipolar contribution to nuclear relaxation rates is washed out. This being the case, the exclusive line-broadening of the imidazole protons might arise from a faster exchange rate rather than from its unique involvement in binding.
The relevance of the exchange contribution is ratified by comparing the output of different NMR experiments performed on the same sample. Fig. 2A reports the 1H-NMR spectrum of the PrP106–126 peptide in the presence of Cu(II) at 1 : 5 metal : ligand ratio. It is evident that only the Hδ and Hβ of His-111 are broadened, while all other resonances are poorly affected. However, Fig. 2B shows the α-region of the 1H–13C HSQC spectra of the same peptide before and after the addition of 0.2 equivalents of Cu(II) ions. The circled Lys-106, Thr-107, Lys-110, His-111 and Gly-126 Hα–Cα connectivities are somehow affected by the presence of paramagnetic ions. Such different broadening of Hα in the two spectra is accounted for by Hα and Cα experiencing different paramagnetic contributions (due to different distances from the metal and magnetogyric ratios) such that k−1off does not limit, in the case of Cα, the detection of the dipolar interaction.
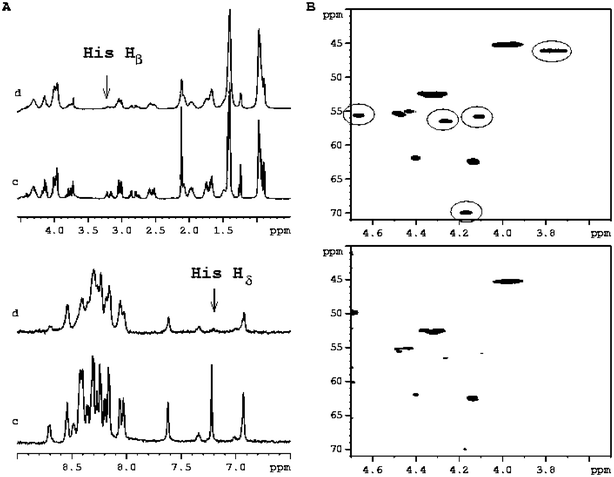 | ||
| Fig. 2 (A) Selected regions of 1H-NMR spectra of PrP106–126 1.8 mM in H2O–D2O at pH 5.7 before (c) and after the addition of 0.2 equivalents of Cu(II) (d). The arrows indicate the most affected His protons. (B) 1H–13C HSQC spectra of PrP106–126 1.8 mM in H2O–D2O pH = 5.7 T = 298 before (upper) and after the addition of 0.2 equivalents of Cu(II) (bottom). The most affected circled cross-peaks belongs to Gly-126, His-111, Lys-106, Lys-110 and Thr-107. | ||
Whenever required, the exchange off-rate can be estimated in two different ways:2
(i) The temperature dependent paramagnetic spin-lattice relaxation rates (R1p) can be fitted by introducing the Eyring equation into eqn (2):
 | (4) |
 | ||
| Fig. 3 Temperature dependence of the lnR1p values measured for Hδ His-111 of PrP106-126 1.8 mM in D2O–H2O at pH 5.7 after the addition of 0.05 copper equivalents. | ||
(ii) The R1b can be calculated for a proton at fixed distance from the metal and inserted into eqn (2) to obtain k−1off. This approach is particularly suitable for His-containing peptides interacting with Cu(II), where the distance of the imidazole Hε from copper is fixed at 0.31 nm independently of Nπ or Nτ acting as nitrogen donors. Once determined, the distance Cu(II)–Hδ can be obtained, thus enabling Nπ or Nτ to be distinguished as the copper bound nitrogen atom. The data relative to peptide–copper complexes are reported in Table 2, where it is apparent that Nπ is the preferential binding donor in most cases. Only for the prion octarepeat dimer, involvement of both nitrogens could be suggested, namely Nπ of His-62 and Nτ of His-70. In fact, since the Hδ signals are superimposed, what is measured is just the mean value of the Cu–Hδ distances in the two cases.
| PrP Monomer | PrP Dimer | PrP 106–113 | PrP 106–126 | APP 145–155 | |
|---|---|---|---|---|---|
| No. His residues | 1 | 2 | 1 | 1 | 3 |
| Hε–Cu2+ distance/nm | 0.31 | 0.31 | 0.31 | 0.31 | 0.31 |
| 0.31 | 0.31 | ||||
| 0.31 | |||||
| Hδ–Cu2+distance/nm | 0.50 | 0.43 | 0.50 | 0.50 | 0.50 |
| 0.43 | 0.50 | ||||
| 0.50 | |||||
| N-types involved in Cu2+ binding | Nπ | Nπ, Nτ | Nπ | Nπ | Nπ, Nπ, Nπ |
The calculated exchange rates of the considered systems are reported in Table 3. Since a slower exchange rate obviously identifies a stronger metal–ligand association, it follows that the appearance of NMR spectra and the evaluation of k−1off allow immediate estimation of the strength of metal binding. From this point of view, among the considered sequences, the PrP106–113 and PrP106–126 peptides exhibit the largest affinity for Cu(II) and such affinity increases with rising pH. On the other hand, the single octarepeat unit exchanges with the fastest rate from the copper coordination sphere, revealing, as expected, an association weaker than the double octarepeat unit. The same difference in exchange rates between the monomer and the dimer is also evidence for the diverse behaviour of the ln(R1p) vs. 1/T plots of the His-Hε proton, shown in Fig. 4.
 | ||
| Fig. 4 Temperature dependence of the lnR1p values measured for His aromatic protons in the copper(II) complexes of PrP monomer (left) and PrP dimer (right). | ||
| PrP monomer | 4.5 × 10−4 |
| PrP dimer | 1.6 × 10−3 |
| APP(145–155) | 5.0 × 10−3 |
| PrP106–126 | 9.2 × 10−3 |
| PrP106–113 | 1.1 × 10−2 |
As further support for the interplay between exchange rate and paramagnetic effects, the normalized values of the His Hε proton are reported in Fig. 5 for all the investigated peptides. Since the Cu2+–Hε distance, as already observed, is fixed at 0.31 nm in any case, the different values are exclusively determined by the exchange rate. It can be observed immediately that the PrP monomer exchanges from the copper coordination sphere at the fastest rate, in contrast with the rather slow rate of exchange of PrP(106–113) and PrP(106–126); as already documented, the consequence is, in these latter cases, a large amount of the metal ion must be added, before any paramagnetic effect on signal diverse from imidazole protons becomes detectable.

It can be therefore concluded that: (i) an estimate of the exchange rate must be obtained whenever structural details of paramagnetic metal complexes are sought; (ii) the exchange rate may generally be measured by the temperature dependence of the paramagnetic spin-lattice relaxation rates; (iii) when dealing with His-containing peptides and copper, the anchoring features of the imidazole ring towards this metal ion allow simple evaluatation of the exchange rate from the paramagnetic contribution to the His Hε proton.
The structure of the complex is obtained by restraining molecular mechanics and dynamics calculations with metal–nucleus distances obtained from eqn 3. It may be observed that such calculations do not require small errors in input parameters and also that the r−6 dependence of R1b allows for relatively large uncertainties in evaluated exchange rates. As examples, the structures obtained with copper bound to PrP(106–113), PrP octarepeat dimer and APP(145–155) are shown in Fig. 6, and the effects of using lower and upper limits for the exchange rate upon evaluated copper–proton distances calculated in the complex with the PrP dimer are displayed in Fig. 7, where it is evident that errors as large as 20% in the exchange rates only poorly affect the calculated distances.
 | ||
| Fig. 6 Structure of Cu(II)–PrP106–113 (left), Cu(II)–APP(145–155) (middle), and of the Cu(II)–PrP dimer (right). Structure calculations were based on obtained metal proton distances and were performed by restrained molecular dynamics with simulated annealing in the torsional angle space. The figures were created with MOLMOL 2K.1.0. | ||
 | ||
| Fig. 7 Copper–protons distances determined by lower and upper limits of calculated τM values for the Cu(II)–PrP dimer complex. | ||
Since it is becoming progressively clear that several proteins involved in neurodegenerative disorders play a major role in maintaining copper homeostasis and also that oxidative stress in neurons is a driving factor in initiating protein aggregation that leads to disease-specific inclusion bodies, handy and reliable tools are required for delineation of structural features of adducts involving redox-sensitive metal ions potentially undergoing Fenton chemistry. From this point of view, paramagnetic NMR of copper complexes may play a central role provided the slow exchange kinetics is interpreted and its limiting effects are overcome.
Acknowledgements
The 600 MHz 1H–15N and 1H–13C HSQC spectra were recorded at the UNIFRA Large Scale Facility in Frankfurt; the support of the European Community (Access to Research Infrastructure action of the Improving Human Potential Program) is kindly acknowledged.References
- I. Bertini and C. Luchinat, Coord. Chem. Rev., 1998, 150, 1–296.
- E. Gaggelli, N. D'Amelio, D. Valensin and G. Valensin, Magn. Reson. Chem., 2003, 41, 877–883 CrossRef CAS.
- C. Conato, W. Kamysz, H. Kozlowski, M. Luczkowski, Z. Mackiewicz, F. Mancini, P. Mlynarz, M. Remelli, D. Valensin and G. Valensin, Eur. J. Inorg. Chem., 2003, 1694–1702 CrossRef CAS.
- C. Conato, W. Kamysz, H. Kozlowski, M. Luczkowski, Z. Mackiewicz, P. Mlynarz, M. Remelli, D. Valensin and G. Valensin, J. Chem. Soc., Dalton Trans., 2002, 3939–3944 RSC.
- M. Remelli, M. Luczkowski, A. M. Bonna, Z. Mackiewicz, C. Conato and H. Kozlowski, New J. Chem., 2003, 27, 245–250 RSC.
- M. Łuczkowski, H. Kozlowski, M. Stawikowski, K. Rolka, E. Gaggelli, D. Valensin and G. Valensin, J. Chem. Soc., Dalton Trans., 2002, 2269–2274 RSC.
- B. Belosi, E. Gaggelli, R. Guerrini, H. Kozlowski, M. Łuczkowski, F. M. Mancini, M. Remelli, D. Valensin and G. Valensin, ChemBioChem, 2004, 5, 349–359 CrossRef CAS.
- H. Kozlowski, W. Bal, M. Dyba and T. Kowalik-Jankowska, Coord. Chem. Rev., 1999, 184, 319–346 CrossRef CAS.
- D. Valensin, F. M. Mancini, M. Łuczkowski, A. Janicka, K. Wiśniewska, E. Gaggelli, G. Valensin, L. Łankiewicz and H. Kozlowski, J. Chem. Soc., Dalton Trans., 2004, 16–22 RSC.
- K. J. Barnham, W. J. McKinstry, G. Multhaup, D. Galatis, C. J. Morton, C. C. Curtain, N. A. Williamson, A. R. White, M. G. Hinds, R. S. Norton, K. Beyreuther, C. L. Masters, M. W. Parker and R. Cappai, J. Biol. Chem., 2003, 278, 17401–17407 CrossRef CAS.
- D. R. Brown, K. Qin, J. W. Herms, A. Madlung, J. Manson, R. Strome, P. E. Fraser, T. Kruck, A. von Bohlen, W. Schulz-Schaeffer, A. Giese, D. Westway and H. Kretzschmar, Nature, 1997, 390, 684–687 CrossRef CAS.
- E. Aronoff-Spencer, C. S. Burns, N. I. Avdievich, G. J. Gerfen, J. Peisach, W. E. Antholine, H. L. Ball, F. E. Cohen, S. B. Prusiner and G. L. Millhauser, Biochemistry, 2000, 39, 13760–13771 CrossRef CAS.
- C. S. Burns, E. Aronoff-Spencer, C. M. Dunham, P. Lario, N. I. Avdievich, W. E. Antholine, M. M. Olmstead, A. Vrielink, G. J. Gerfen, J. Peisach, W. G. Scott and G. L. Millhauser, Biochemistry, 2002, 41, 3991–4001 CrossRef CAS.
- J. M. Steward and J. D. Young, Solid Phase Peptide Synthesis, 2nd ed., Pierce Chemical Company, Rockford, Illinois, 1984 Search PubMed.
- N. A. Sole and G. Barany, J. Org. Chem., 1992, 57, 5399–5403 CrossRef CAS.
- T. L. Hwang and A. J. Shaka, J. Magn. Reson., Ser. A, 1995, 112, 275–279 CrossRef CAS.
- J. G. Huber, J. M. Moulis and J. Gaillard, Biochemistry, 1996, 35, 12705–12711 CrossRef CAS.
- P. Güntert, C. Mumenthaler and K. Wüthrich, J. Mol. Biol., 1997, 273, 283–298 CrossRef CAS.
- HYPERCHEM, Hypercube release 5.1 Pro for Windows ( 1997), Hypercube Inc.Waterloo, Canada.
- I. Solomon, Phys. Rev., 1955, 99, 559–565 CrossRef CAS.
- L. D. Hall and H. D. W. Hill, J. Am. Chem. Soc., 1976, 98, 1269–1270 CrossRef CAS.
- R. Freeman, H. D. W. Hill, L. D. Hall and B. L. Tomlinson, J. Chem. Phys, 1974, 61, 4466–4473 CrossRef CAS.
Footnote |
| † XWINNMR 2.6 is software provided by Bruker for managing NMR spectra. |
| This journal is © The Royal Society of Chemistry 2005 |