Identification of the F1F0 mitochondrial ATPase as a target for modulating skin pigmentation by screening a tagged triazine library in zebrafish
Da-Woon
Jung†
b,
Darren
Williams†
a,
Sonya M.
Khersonsky
b,
Tae-Wook
Kang
b,
Noushin
Heidary
a,
Young-Tae
Chang
b and
Seth J.
Orlow
*c
aThe Ronald O. Perelman Department of Dermatology and the Department of Cell Biology, New York University School of Medicine, New York, NY10016, USA
bDepartment of Chemistry, New York University, New York, NY10003, USA
cSeth Orlow, NYU School of Medicine, Department of Dermatology, 560 First Avenue, Room H-100, New York, NY 10016, USA. E-mail: seth.orlow@med.nyu.edu
First published on 8th March 2005
Abstract
A triazine-based combinatorial library of small molecules was screened in zebrafish to identify compounds that produced interesting phenotypes. One compound (of 1536 screened) induced a dramatic increase in the pigmentation of early stage zebrafish embryos. This compound, PPA, was also found to increase pigmentation in cultured mammalian melanocytes. The cellular target was identified as the mitochondrial F1F0-ATP synthase (ATPase) by affinity chromatography. Oligomycin, a small molecule known to inhibit the mitochondrial ATPase, competed with PPA for its cellular target in melanocytes. In addition, PPA was shown to alter the membrane potential of mitochondria, consistent with inhibition of the mitochondrial ATPase. Thus, PPA has been successfully used as a chemical probe in a forward chemical genetic approach to establish a link between the phenotype and the protein. The results attest to the power of screening small molecule libraries in zebrafish as a means of identifying mammalian targets and suggest the mitochondrial ATPase as a target for modulating pigmentation in both melanocytes and melanoma cells.
Introduction
Melanin pigments the hair, skin, and iris, and helps to decrease the risk of skin cancer and protect against photoaging.1 Discovering drugs that increase skin pigmentation is desirable for a number of reasons. For example, increased melanin synthesis will enhance protection from UV radiation without exposing the skin to the deleterious effects of UV light. Drugs increasing pigmentation might also be useful in treating disorders of hypopigmentation including postinflammatory hypopigmentation and albinism. In addition, achieving a safe tan or darkening hair color may be desirable from a cosmetic point of view.2 At present, molecules capable of increasing skin pigmentation in the absence of ultraviolet light stimulation are unavailable for use in humans.2Chemical genetics is a relatively new field of research that has the potential to provide powerful tools for identifying novel drug candidates and their cellular target(s).3–10 Small molecules replace the mutation-inducing agents or X-ray irradiation employed by classical genetics. Combinatorial techniques11–16 allow the rapid screening of large numbers of small molecules that can be identified by producing novel phenotypes in a cellular or embryonic system. An affinity matrix made of the immobilized active compound is used to identify biological targets.
The zebrafish (Danio rerio) has received much attention from the scientific community as a vertebrate organism amenable to high throughput screening protocols for discovering biologically active small molecules.17–19 The small size and transparency of zebrafish embryos have been utilized in previous reports of small molecule screening. In the current study we employed a tagged library approach to accelerate the conversion of a hit compound to an efficient affinity matrix.6 A pigmentation-inducing small molecule, obtained from library screening in zebrafish and tested in a number of mammalian melanocyte cell lines, demonstrates that the F1F0 mitochondrial ATPase is a target for modulating pigmentation in normal and albino mammalian melanocytes and in melanoma cells.
Results
Identification of triazine-based compounds that increase pigmentation in zebrafish
One compound from the library of 1536 compounds was found to produce an interesting pigmentation phenotype in zebrafish embryos. This compound, termed PPA, produced a marked increase in pigmentation throughout the embryo (Fig. 1). A version of PPA with the linker removed (termed ADA) produced a similar effect on embryo pigmentation at a lower range of concentrations. A structurally related compound, termed PPA06 (Fig. 1a), which failed to affect pigmentation, was chosen as a negative control for melanocyte studies. Higher magnification views of the dorsal lateral region of PPA- or ADA-treated embryos showed the melanophores to be markedly increased in size. (Fig. 1c). PPA began to produce marked general effects on embryo development at concentrations above 10 µM. | ||
| Fig. 1 Structures of compounds used in this study and their screening in the zebrafish system. (a) Chemical structure and designation of the triazine compounds used in this study. PPA06 is a negative control for PPA and ADA is a version of PPA with the linker structure removed. (b) Screening of a tagged triazine library for 16 h in zebrafish embryos at the 10 somite stage of treatment. Treatment with PPA or ADA caused a general increase in pigmentation throughout the embryo and defects in the development of the notochord, brain and eyes. An embryo treated with DMSO alone is included for comparison (magnification ×30). (c) Higher magnification view of the dorsal lateral region showing the increased area of pigmentation caused by drug treatment (magnification ×100). | ||
PPA and its analogues increase pigmentation in mammalian melanocytes
Treatment with PPA and ADA (a version of PPA lacking the linker side chain, see Fig. 1a) caused dose dependent increases in melanin levels in the murine melanocyte cell line melan-a and in B16–F10 melanoma cells (Fig. 2b–c). An increase in pigmentation could be seen visually using light microscopy (Fig. 2a). | ||
| Fig. 2 (a) The compound PPA increases melanocyte pigmentation. The increase in melan-a melanocyte pigmentation can be seen by light microscopy of living cells. 3-Isobutyl-1-methylxanthine (IBMX) is an inhibitor of phosphodiesterases that increases pigmentation by raising intracellular cAMP. Melanocytes were grown in a 24-well tissue culture plate and treated with drug for 72 h before microscopy (magnification ×100). (b)–(d) PPA increases pigmentation in melan-a melanocytes and induces pigmentation in non-pigmented B16–F10 melanoma cells and albino melan-p melanocytes. Cells were treated with drug for 72 h before melanin assay. Each data point for melan-a is the average of three wells of a 24-well tissue culture plate (error bars = standard deviation). Each data point for B16–F10 and melan-p is the average of two wells of a 24-well tissue culture plate. ADA is identical in structure to PPA, except that it lacks the linker portion of the drug. ADA retains the ability to increase the pigmentation of melan-a melanocytes. PPA06 is a negative control compound and has a similar structure to PPA. | ||
PPA and its analogues increase pigmentation in albino melanocytes
We have previously reported the discovery by screening of triazine-based compounds that correct the pigmentary defect in albino melan-p melanocytes and were found to bind the mitochondrial ATPase.20 While PPA could also induce pigmentation in melan-p cells (Fig. 2d), the concentration of PPA needed to induce pigmentation in melan-p cells (20–50 µM) was typically far higher than for those compounds we previously identified by directly screening for albinism-correcting compounds (effective at 1–5 µM).Identification of the mitochondrial F1F0-ATPase as a cellular target
Eluted proteins from affinity matrix-conjugated PPA exposed to an extract of melan-a melanocytes included a 55 kD doublet and a 30 kD protein that were specific for pigment stimulating compounds and not seen with related compounds inactive in the pigmentation screen (Fig. 3a). In addition a number of proteins were seen to bind non-specifically with respect to pigmentation, inasmuch as they bound to analogues of PPA that lacked pigment-stimulating activity. Pre-incubating the cell lysate with free PPA diminished the presence of these proteins in the affinity-matrix eluate (Fig. 3b). The free PPA was an effective competitor of target binding to the matrix-conjugated compound. | ||
| Fig. 3 Identifying the cellular target of compound PPA. (a) Drug target study for the compound PPA in melan-a cells. Eluted proteins were separated by 12% SDS-PAGE. Affinity matrix, ethanolamine-conjugated (Eth.), PPA06 (inactive compound)-conjugated (PPA06) and PPA-conjugated (PPA). The prominent 55 kD doublet and 30 kD bands that are specific for PPA are indicated by an asterisk. (b) Competition studies: extract of melan-a cells was allowed to interact with affinity matrix as described in Materials and methods. PPA-conjugated affinity matrix with 100, 200 and 300 µM PPA as competitor. The reduction in binding of the 55 kD doublet to the PPA-conjugated affinity matrix is indicated by an asterisk. (c) Sequencing results for the 55 kD doublet and 30 kD protein bands specific for immobilized PPA. The peptides detected by Ion Trap mass spectrometry are shown in red. (d) Oligomycin competition study for the compound PPA in melan-a cells. PPA-conjugated affinity matrix with 25, 50 and 100 µM oligomycin as competitor. The reduction in binding of the PPA-conjugated affinity matrix to the 55 kD doublet is indicated by an asterisk. (e) Two competition studies for the compound PPA in melan-a melanocytes. Eluted proteins were blotted and probed with antibodies against alpha and beta subunits the mitochondrial ATPase. Eth. = ethanolamine matrix + 2% DMSO (negative control); DMSO = PPA-conjugated affinity matrix + 2% DMSO (positive control); +oligomycin = PPA-conjugated affinity matrix with 100, 50 and 25 µM oligomycin as competitor; +PPA = PPA-conjugated affinity matrix with 400, 200 and 100 µM PPA as competitor; +ADA = PPA-conjugated affinity matrix with 400, 200 and 100 µM ADA as competitor; (f) Competition studies for the compound PPA in SKMel19 melanocytes. Eluted proteins were blotted and probed with antibodies against the beta subunit, OSCP and d subunits of mitochondrial ATPase. PPA6 = PPA6 (inactive compound) conjugated affinity matrix + 2% DMSO (negative control); PPA = PPA (active compound) conjugated affinity matrix + 2% DMSO (positive control); 100 µM oligo = PPA-conjugated affinity matrix with 100 µM oligomycin as competitor; Eth. = ethanolamine matrix + 2% DMSO (negative control). | ||
Ion-Trap mass spectrometry identified the three pigmentation-specific proteins as subunits of the mitochondrial F1F0-ATPase. The 55 kD doublet was identified as the alpha and beta subunits. (Fig. 3c). The 30 kD band was identified as the gamma subunit. An inhibitor of the mitochondrial ATPase, oligomycin, competed with the binding of the alpha and beta ATPase subunits to the matrix-conjugated compounds (Fig. 3d). Close examination of the 30 kD subunit band shows that 100 µM oligomycin can compete with the binding of the gamma subunit to the agarose-bound PPA, although the decrease in binding is much less dramatic than that seen for the alpha and beta mitochondrial ATPase subunits.
To further demonstrate that the cellular target for PPA's pigmentary effects is indeed the mitochondrial ATPase, proteins eluted from the matrix-conjugated compounds were blotted and probed with antibodies against subunits of the mitochondrial ATPase (Fig. 3e–f). Proteins were prepared from both murine (melan-a melanocytes) and human (SKMel19 melanoma) cells. PPA was shown to bind to the d- and OSCP (oligomycin sensitivity-conferring protein) subunits of the mitochondrial ATPase. Oligomycin could compete with PPA for binding to the subunits of the ATPase. We reasoned that if the ATPase were truly the target for pigment induction, then oligomycin and/or aurovertin (a small molecule inhibitor of mitochondrial ATPase that binds the beta subunit) might also affect pigmentation. As shown in Fig. 4, both oligomycin and aurovertin indeed increased pigmentation in cultured melanocytes. The increase in pigmentation could be seen as soon as 24 h after addition of the drug.
 | ||
| Fig. 4 Other small molecule inhibitors of the mitochondrial ATPase enhance melanocyte pigmentation. Oligomycin and aurovertin increase pigmentation in melan-a melanocytes. Bafilomycin, a known pigmentation enhancer that inhibits the vacuolar ATPase, is included for comparison. Each data point is the average of two wells of a 24-well tissue culture plate. Abbreviations: Oligo = oligomycin, Auro = aurovertin, Baf = bafilomycin. | ||
PPA increases pigmentation in melanocytes cells by inhibition of the mitochondrial ATPase
MitoFluor594™ is a mitochondrial membrane potential-sensing dye. Staining of melanocytes with MitoFluor594 demonstrated that PPA influenced mitochondrial membrane potential in a manner similar to that observed with the mitochondrial F1F0–ATPase inhibitor, oligomycin. This effect was seen as soon as one hour after drug treatment. At relatively low magnification, the melanocytes treated with PPA or oligomycin show a markedly decreased intensity of MitoFluor594 staining, demonstrating that PPA, like oligomycin, is an inhibitor of the mitochondrial ATPase (Fig. 5a). The reduced MitoFluor594 staining of the mitochondria of PPA-treated melanocytes could be seen at higher magnification (Fig. 5b).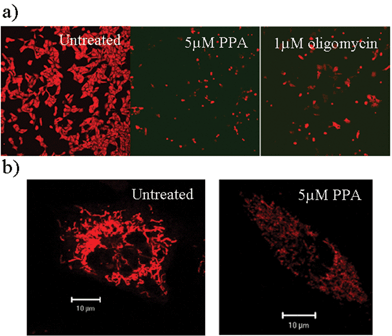 | ||
| Fig. 5 The compound PPA alters mitochondrial membrane potential. (a) Melan-a melanocyte mitochondria show altered membrane potential 1 h after treatment with PPA, a pigmentation-inducing drug that binds the mitochondrial ATPase. The effect is similar to that produced by the mitochondrial ATPase inhibitor, oligomycin. Cells were seeded at 50% confluency 12 h before exposure to drug (magnification ×100). (b) The effect of PPA on mitochondrial membrane potential can be seen within individual melanocytes (magnification ×600). | ||
Discussion
We have demonstrated by screening a tagged triazine-based small molecule library that the mitochondrial F1F0–ATPase is a target protein whose inhibition can increase the pigmentation of normal melanocytes and melanoma cells. In addition, we report that oligomycin and aurovertin B, compounds that also target the mitochondrial ATPase, both increase melanin synthesis in normal melanocytes. New pigment-enhancing compounds such as PPA could lead to new products that increase pigmentation in the skin, hair and eyes for medical and/or cosmetic purposes without the need for UV exposure (a so-called “safe tan”).Traditionally, active molecules selected and modified after biological screening are fitted with a linker, to provide an attachment point to the affinity bead. This modification has the potential to cause a loss of activity, requiring time-consuming and laborious structure-activity relationship (SAR) studies. We have incorporated the linkers before biological screening to allow a straightforward method of isolation of the target protein which avoids potential compromise of the lead compound's activity. The triazine scaffold was chosen due to its ease of manipulation and structural similarity to purine and pyrimidine, since purine and pyrimidine binding sites are common in proteins.21 Purine and pyrimidine-based libraries have been shown to be active in various biological systems.22–26
Zebrafish pigmentation research has focused on identifying and characterizing mutations that alter the production of melanin or influence the development or subsequent migration of pigment precursor cells in the neural crest.27–29 Attention has now turned to the use of zebrafish as a tool for functional genomics approaches to pigment analysis and the high-throughput screening of chemical libraries for drug discovery.17–19,30 These new applications are supported by this work, which uses the zebrafish as a tool for identifying a chemical probe successfully used in a forward chemical genetic approach to establish a link between the phenotype and the protein.
Previous research has shown that a variety of compounds are capable of increasing pigmentation in melanocyte cultures. For example, a marked and rapid increase in the pigmentation of normal human melanocyte cultures can be produced by ion gradient disruptors (such as monensin or nigericin) and inhibitors of the vacuolar-type (H+) ATPase (such as bafilomycin A1 or concanamycin A).27–31 Vacuolar-type ATPases transport H+ ions into various endomembrane systems and cellular organelles, and are responsible for organelle acidification.28,32,33 We hypothesize that the compounds we have identified increase melanin synthesis by inhibiting mitochondrial ATPase-mediated transport of H+ ions, resulting in the alkalinization of cellular endomembrane systems. This hypothesis is supported by the pigmenting effects of two known inhibitors of the mitochondrial ATPase, oligomycin and aurovertin B.
We have previously reported the identification of compounds that induce pigmentation in albino melanocytes deleted for the p gene, a model for human oculocutaneous albinism type 2.20 Those compounds were discovered by screening in a mammalian cellular system (inducing pigmentation in albino melanocytes) rather than screening in zebrafish. Thus, it is interesting to compare the pigmenting compounds identified by these two screening systems. The compounds identified using albino melanocytes are toxic when applied to healthy melanocytes and melanoma cells (data not shown). In contrast, PPA can induce pigmentation in a wide variety of melanocyte cultures. Overcoming a genetic defect in albino melanocytes is fundamentally different from making normal melamocytes darker, and thus it was somewhat surprising that the same target was identified in these 2 different studies.Moreover, even though the target appears to be the same, the molecules that correct the pigmentary defect in albino melan-p cells are not very effective at increasing pigmentatation in melan-a cells and vice versa. This is a testament to the advantage of screening in zebrafish, because it allowed us to identify a compound that can pigment a range of mammalian melanocyte cell lines. However, although the zebrafish is attractive for screening compounds in a whole-organism system, it is more difficult for drug target studies and producing reliable dose–response data. Additionally, the compounds we previously identified from this library that pigment solely albino melanocytes were toxic for the developing embryos and, therefore, were not detected in the zebrafish screen.
It is interesting to compare the induction of pigmentation by bafilomycin, an inhibitor of the vacuolar-type-ATPase, and that produced by inhibitors of the mitochondrial ATPase. Bafilomycin produces an increase in pigmentation similar to that brought about by treatment with PPA, oligomycin and aurovertin (Fig. 4). Oligomycin routinely induced pigmentation at significantly lower doses than aurovertin. A number of studies of melanocyte pigmentation were completed to investigate whether PPA could act synergistically with oligomycin or aurovertin or other known inducers of pigmentation. However, no synergistic effects were observed (data not shown).
Further studies of the novel melanocyte pigmenting compounds that we have identified include the detailed characterization of binding to the mitochondrial ATPase. The mitochondrial ATPase consists of two parts, F1 and F0. F1 is the catalytic portion and consists of five subunits. F0 consists of 10 subunits in mitochondria and is arranged as a membrane-embedded proton-conducting channel.34 The ability of PPA to bind the d subunit in the F0 portion of the mitochondrial ATPase shows that it binds to the intact mitochondrial ATPase (Fig. 3f). More detailed biochemical studies will be needed to decipher which mitochondrial ATPase subunit(s) is the precise molecular target for our triazine-based pigmenting compounds.
In conclusion, we have successfully demonstrated the power of a tagged library approach for efficient forward chemical genetics in zebrafish, in this case demonstrating a role for the mitochondrial ATPase in increasing the pigmentation of healthy melanocytes and inducing pigmentation in albino melanocytes and melanoma cells. Our small molecule library facilitates the connection of a hit compound to the affinity matrix by incorporating a linker directly to the compounds before their phenotypic screening. The same tagged library approach can be readily applied to other cellular and whole organism screens.
Materials and methods
Development of the small molecule library
The design and production of the tagged triazine library has been reported previously.35 Briefly, a solid-phase method was used to construct a tagged triazine library, where three building blocks were prepared separately and assembled orthogonally to yield 1536 highly pure compounds. Each library compound contained one of a variety of triethyleneglycol (TG) linkers at one of the diversity sites of the triazine scaffold.Small molecule screening in zebrafish
Zebrafish experiments were performed in compliance with the relevant laws and institutional guidelines of New York University, and the University's Institutional Animal Care and Use Committee approved the experimental protocol. Zebrafish eggs were collected at 8 cell- and 10 somite-stages, and arrayed in 96-well plates (three embryos/well) containing test compound in 1% DMSO/Hanks-derived buffer. During the course of 30 h for 8 cell stage and 16 h for 10 somite stage of treatment, at 28.5 °C, phenotypic changes were observed using a high magnification dissecting microscope.Embryos were individually screened for visible changes in pigmentation at several time points (every 2 h for the first day, every 4 h for day 2, and twice for day 3). Only compounds that caused the same phenotype over a range of concentrations were selected for follow-up studies. Compounds that showed no activity or led to non-specific toxicity were not considered further.
Cell culture
Melan-a (a/a, P/P) is an immortalized melanocyte line derived from C57BL/6J mice. These cells produce black melanin pigment. Melan-p1 is an immortalized melanocyte line from albino C57BL/6J mice lacking p gene transcripts due to overlapping deletions.36 Melan-p1 cells are hypopigmented. SKMel19 is a hypopigmented human melanoma cell line.37 B16–F10 is a melanoma line derived from wild-type C57BL/6J mice.38Melan-a cells were maintained in DMEM culture medium containing 2 mM L-glutamine, 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 IU per L penicillin, 100 mg L−1 streptomycin, 5% fetal bovine serum and 200 nM tetradecanoyl phorbol acetate at 37 °C and 5% CO2. Melan-p cells were maintained in the same culture conditions as melan-a cells, except for the substitution of RPMI for DMEM culture medium. B16 and SKMel19 cells were maintained in the same culture conditions as melan-a cells, except for the absence of tetradecanoyl phorbol acetate.
000 IU per L penicillin, 100 mg L−1 streptomycin, 5% fetal bovine serum and 200 nM tetradecanoyl phorbol acetate at 37 °C and 5% CO2. Melan-p cells were maintained in the same culture conditions as melan-a cells, except for the substitution of RPMI for DMEM culture medium. B16 and SKMel19 cells were maintained in the same culture conditions as melan-a cells, except for the absence of tetradecanoyl phorbol acetate.
Melanin assay
Cells were rinsed with phosphate buffered saline (PBS), and lysed with an extraction buffer [50 mM Tris, pH 7.5; 2 mM ethylenediamine tetraacetic acid (EDTA) pH 7.8; 150 mM NaCl; 1% Triton X-100] with protease inhibitor cocktail at 4 °C. Cell extracts were then spun at 12000 rpm for 10 min at 4 °C. The remaining pellet was assayed for melanin by rinsing twice with ethanol–ether (1 ∶ 1) and dissolving in 200 µL of 2 N NaOH in 20% DMSO at 60 °C. A 100 µL aliquot of the resulting solution was then measured for absorbance at 490 nm.
Isolation and identification of cellular target(s)
Protein was extracted from cells by incubation with extraction buffer (1 mM CaCl2; 150 mM NaCl; 10 mM Tris, pH 7.4; 1% Triton X-100; 1 mM PMSF plus one tablet of protease inhibitor cocktail (Roche, NJ, USA) per 20 mL buffer) for 5 min on ice. Crude lysate was centrifuged at 13000 rpm for 10 min. The protein concentration of the supernatant was measured by the Bradford assay (Bio-Rad; CA, USA) and adjusted to a final concentration of 1 µg µL−1 prior to affinity chromatography.
25–50 µL of agarose affinity matrix conjugated compound was washed with 1 mL bead buffer (10 mM Tris, pH 7.4; 5 mM NaF; 250 mM NaCl; 5 mM EDTA; 5 mM EGTA; 0.1% Triton X-100 plus protease inhibitor cocktail (Roche, NJ, USA)). Matrices were incubated with 50–200 µg of protein extract plus an identical volume of bead buffer at 4 °C or 30 °C. For studies of competition of drug binding to a cellular target, the competitor was added to the mixture of protein extract/bead buffer and incubated at 4 °C for 30 min prior to incubation with the matrix. The supernatant containing unbound proteins was removed by centrifugation and the matrices were washed seven times with 1 mL bead buffer. Proteins bound to the matrices were eluted by incubation with 50 µL Laemmli buffer (Bio-Rad; CA, USA) at 94 °C for 3 min.
Eluted proteins were separated by 7.5% or 10% SDS-PAGE and visualized by silver staining (Amersham, NJ, USA). Prominent protein bands specific to active matrices were excised from each gel and identified by Ion Trap mass spectrometry (NYU Protein Analysis Facility, Skirball Institute of Biomolecular Medicine, NY, USA).
Antibodies and reagents
Monoclonal antibodies against the α-, β-, d- and OSCP (oligomycin sensitivity-conferring protein) subunits of the ATPase (F1F0) were purchased from Molecular Probes Inc. (Eugene, OR).Oligomycin and Aurovertin B were purchased from Sigma-Aldrich, MO, USA. Bafilomycin A1 was purchased from Wako Pure Chemical Industries Ltd, Osaka, Japan.
Western blot analysis
Proteins were separated by 7.5% or 10% SDS-PAGE and transferred onto membranes (Immobilon-P; Millipore, Waltham, MA)Microscopy
Cells were grown in 6-well tissue culture plates for 48 h and treated with the drug of interest. Melanin content in live cells was assessed using phase-contrast microscopy (LSM510; Carl Zeiss, Thornwood, NY) All data were processed with Adobe Photoshop 7.0 (Adobe Systems, Mountain View, CA).Measurement of mitochondrial membrane potential
MitoFluor 594™ (Molecular Probes Inc., Eugene, OR) is a mitochondrial membrane potential-sensing dye. Live cells, grown in 4-well chamber slides (Nalge Nunc Int., IL), were incubated with 500 nM MitoFluor 594™ in growth medium for 30 min in a tissue culture incubator at 37 °C and 5% CO2. The slides were analyzed using a digital fluorescence microscope (LSM510; Carl Zeiss, Thornwood, NY). All data were analyzed with a 4× lens and processed with Adobe Photoshop 7.0 (Adobe Systems, Mountain View, CA).Acknowledgements
This work was supported in part by the PHS grant AR41880 (SJO).References
- B. Gilchrest, T. Fitzpatrick, R. Anderson and J. Parrish, Br. J. Dermatol., 1977, 96, 245–248 CAS.
- J. Hoeyberghs, Br. Med. J., 1999, 318, 512–516 CAS.
- N.S. Gray, Curr. Opin. Neurobiol., 2001, 11, 608–614 CrossRef CAS.
- S. M. Khersonsky and Y. T. Chang, ChemBioChem, 2004, 5, 903–908 CrossRef CAS.
- R. S. Lokey, Curr. Opin. Chem. Biol., 2003, 7, 91–96 CrossRef CAS.
- G. Mitsopoulos, D. P. Walsh and Y. T. Chang, Curr. Opin. Chem. Biol., 2004, 8, 26–32 CrossRef CAS.
- S. L. Schreiber, Bioorg. Med. Chem., 1998, 6, 1127–1152 CrossRef CAS.
- S. L. Schreiber, Chem. Eng. News, 2003, 81, 51–61.
- D. S. Tan, Nat. Biotechnol., 2002, 20, 561–563 CrossRef CAS.
- G. E. Ward, K. L. Carey and N. J. Westwood, Cell. Microbiol., 2002, 4, 471–482 CrossRef CAS.
- S. L. Schreiber, Bioorg. Med. Chem., 1998, 6, 1127–1152 CrossRef CAS.
- S. L. Schreiber, Chem. Eng. News, 2003, 81, 51–61.
- D. S. Tan, Nat. Biotechnol., 2002, 20, 561–563 CrossRef CAS.
- G. E. Ward, K. L. Carey and N. J. Westwood, Cell. Microbiol., 2002, 4, 471–482 CrossRef CAS.
- G. Jung, Combinatorial Chemistry: Synthesis, Analysis, Screening, Wiley-VCH, Cambridge, 1999 Search PubMed.
- S. Miertus and G. Fassina, Combinatorial Chemistry and Technology. Principles, Methods, and Applications, Marcel Dekker, New York, 1999 Search PubMed.
- C. A. MacRae and R. T. Peterson, Chem. Biol., 2003, 10, 901–908 CrossRef CAS.
- R. T. Peterson, B. A. Link, J. E. Dowling and S. L. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 12965–12969 CrossRef CAS.
- H. S. Moon, E. M. Jacobson, S. M. Khersonsky, M. R. Luzung, D. P. Walsh, W. Xiong, J. W. Lee, P. B. Parikh, J. C. Lam, T. W. Kang, G. R. Rosania, A. F. Schier and Y. T. Chang, J. Am. Chem. Soc., 2002, 124, 11608–11609 CrossRef CAS.
- D. Williams, D. W. Jung, S. M. Khersonsky, N. Heidary, Y. T. Chang and S. J. Orlow, Chem. Biol., 2004, 11, 1251–1259 CrossRef CAS.
- N. S. Gray, Curr. Opin. Neurobiol., 2001, 11, 608–614 CrossRef CAS.
- P. G. Baraldi, B. Cacciari, R. Romagnoli, G. Spalluto, A. Monopoli, E. Ongini, K. Varani and P. A. Borea, J. Med. Chem., 2002, 45, 115–126 CrossRef CAS.
- Y. T. Chang, N. S. Gray, G. R. Rosania, D. P. Sutherlin, S. Kwon, T. C. Norman, R. Sarohia, M. Leost, L. Meijer and P. G. Schultz, Chem. Biol., 1999, 6, 361–375 CrossRef CAS.
- Y. T. Chang, G. Choi, Y. S. Bae, M. Burdett, H. S. Moon, J. W. Lee, N. S. Gray, P. G. Schultz, L. Meijer, S. K. Chung, K. Y. Choi, P. G. Suh and S. H. Ryu, Chembiochem., 2002, 3, 897–901 CrossRef CAS.
- A. Gangjee, J. Yu, R. L. Kisliuk, W. H. Haile, G. Sobrero and J. J. McGuire, J. Med. Chem., 2003, 46, 591–600 CrossRef CAS.
- D. E. Verdugo, M. T. Cancilla, X. Ge, N. S. Gray, Y. T. Chang, P. G. Schultz, M. Negishi, J. A. Leary and C. R. Bertozzi, J. Med. Chem., 2001, 44, 2683–2686 CrossRef CAS.
- R. N. Kelsh, M. Brand, Y. J. Jiang, C. P. Heisenberg, S. Lin, P. Haffter, J. Odenthal, M. C. Mullins, F. J. van Eeden, M. Furutani-Seiki, M. Granato, M. Hammerschmidt, D. A. Kane, R. M. Warga, D. Beuchle, L. Vogelsang and C. Nusslein-Volhard, Development, 1996, 123, 369–389 Search PubMed.
- D. W. Raible and J. S. Eisen, Development, 1994, 120, 495–503 Search PubMed.
- I. Ziegler, Pigment Cell Res., 2003, 16, 172–182 CrossRef CAS.
- M. A. Pickart, S. Sivasubbu, A. L. Nielsen, S. Shriram, R. A. King and S. C. Ekker, Pigment Cell Res., 2004, 17, 461–470 CrossRef CAS.
- K. Chen, P. Manga and S. J. Orlow, Mol. Biol Cell., 2002, 13, 1953–1964 CrossRef CAS.
- K. Chen, L. Minwalla, L. Ni and S. J. Orlow, Pigment Cell Res., 2004, 17, 36–42 CrossRef CAS.
- B. B. Fuller, D. T. Spaulding and D. R. Smith, Exp. Cell Res., 2001, 262, 197–208 CrossRef CAS.
- J. Ancans, D. J. Tobin, M. J. Hoogduijn, N. P. Smit, K. Wakamatsu and A. J. Thody, Exp. Cell Res., 2001, 268, 26–35 CrossRef CAS.
- S. Drose and A. K. Altendorf, J. Exp. Biol., 1997, 200, 1–8 Search PubMed.
- R. Halaban, R. S. Patton, E. Cheng, S. Svedine, E. S. Trombetta, M. L. Wahl, S. Ariyan and D. N. Hebert, J. Biol. Chem., 2002, 277, 14821–14828 CrossRef CAS.
- D. R. Smith, D. T. Spaulding, H. M. Glenn and B. B. Fuller, Exp. Cell Res., 2004, 298, 521–534 CrossRef CAS.
- L. Ernster, Molecular Mechanism in Bioenergetics, Elsevier, Amsterdam, 1992 Search PubMed.
- S. M. Khersonsky, D. W. Jung, T. W. Kang, D. P. Walsh, H. S. Moon, H. Jo, E. M. Jacobson, V. Shetty, T. A. Neubert and Y. T. Chang, J. Am. Chem. Soc., 2003, 125, 11804–11805 CrossRef CAS.
- E. V. Sviderskaya, D. C. Bennett, L. Ho, T. Bailin, S. T. Lee and R. A. Spritz, J. Invest. Dermatol., 1997, 108, 30–34 CrossRef CAS.
- A. N. Houghton, F. X. Real, L. F. Davis, C. Cordon-Cardo and L. J. Old, J. Exp. Med., 1987, 165, 812–829 CrossRef CAS.
- I. J. Fidler, Cancer Res., 1975, 35, 218–224 CAS.
Footnote |
| † Both authors contributed equally to this work |
| This journal is © The Royal Society of Chemistry 2005 |