Effect of contamination by oxygen at trace level in miniature flame hydride atomizers
Alessandro
D’Ulivo
*a,
Jiři
Dědina
b and
Leonardo
Lampugnani
a
aC.N.R., Istituto per i processi chimico-fisici, Laboratorio di Chimica Analitica Strumentale, Area della ricerca di Pisa, Via G. Moruzzi, 1, 56124 Pisa, Italy. E-mail: dulivo@ipcf.cnr.it; Fax: +39-050-3152555
bAcademy of Sciences of the Czech Republic, Institute of Analytical Chemistry, Laboratory of Trace Element Analysis, Videňska 1083, 14220 Prague 4, Czech Republic
First published on 23rd November 2004
Abstract
A new type of atomization interference, caused by traces of oxygen in gases supplied to the atomizer, has been identified in three different types of miniature flame hydride atomizer for atomic fluorescence spectrometry, namely argon–hydrogen diffusion flame (MDF), flame-in-gas shield (FIGS) and double flame (DF), which is a combination of MDF and FIGS. The interference magnitude is dramatically dependent on the analyte element (Se ≪ As ≤ Sb), on the type of atomizer and on the atomizer operating parameters. The thresholds of interference could be as low as a few parts per million (μl l−1) of oxygen in the carrier gas. The main sources of interfering oxygen are, in order, the oxygen stripped out from sample and reagent solutions in the hydride generation step followed by the oxygen diffusing inside the gas circuit of hydride generation apparatus.
Introduction
Miniature diffusion flames (MDF) are widely employed as hydride atomizers in atomic fluorescence spectrometry (AFS) for trace element determination in both commercial1 and laboratory assembled apparatus.2 More recently, the flame-in-gas-shield atomizer (FIGS)3 has been developed. The fundamental processes taking place in MDF, FIGS and also in a hybrid of these two atomizers, termed the double flame atomizer (DF), were studied by employing atomic absorption spectrometry (AAS).4–6 It has been shown that all these three atomizers, MDF, FIGS and DF, are fairly resistant to atomization interferences due to other hydride forming elements, notably in comparison with externally heated quartz tube atomizers.6,7In the course of the past years, during the investigations performed in the authors’ laboratories on miniature flame hydride atomizers, several effects of apparently unexplainable nature have been observed, notably the irreproducibility of sensitivity observed in a FIGS atomizer as a function of the oxygen flow rate for microflame, and the curvature observed in the plots of sensitivity versus sample flow rate. It appeared that these effects were due to erratic levels of oxygen contamination of gases supplied to the atomizer. The aim of the present work was to understand this new type of atomization interference.
Experimental
Instrumentation
A PerkinElmer 503 atomic absorption spectrometer was used for all measurements. Selenium, antimony and arsenic electrodeless discharge lamps (PerkinElmer type II) were employed at the current setting recommended by manufacturer. The optical beam cross section was reduced in dimensions by a rectangular window down to size of 1.5 mm (width) × 2 mm (height); it was positioned just above the atomizer top in the axial position (observation height = 1 mm, lateral coordinate = 0 mm).4,5 Absorbance measurements were performed at the 196.0 nm line for selenium (2 nm bandpass), at 217.6 nm line for antimony (0.2 nm bandpass) and at the 193.7 nm line for arsenic (0.7 nm bandpass).The concentrations of trace oxygen in the gas stream flowing through the hydride generation (HG) system and the atomizers were measured by an Ox-detector (Mecanalyse, Analyseur d’Oxygene, W.O.M. type), with a limit of detection of 0.1 μl l−1. For these measurements, hydrogen cannot be used and it was replaced by argon.
Hydride generation
Hydrogen selenide, arsine and stibine were chemically generated from aqueous solution using NaBH4 reduction in a continuous flow system. A schematic representation of the apparatus is reported in Fig. 1. | ||
| Fig. 1 Schematic representation of the experimental apparatus employed for hydride generation. Points A, B and C represent the locations for the measurements of oxygen contamination in the flowing gases. | ||
Working solutions of 7–10 g l−1 NaBH4 in 6 g l−1 NaOH were employed and they were prepared daily from an 80 g l−1 NaBH4 in 60 g l−1 NaOH stock solution (microfiltered, stored frozen). Sample and reductant flow rate could be varied in the range of 0.7–5 and 0.3–2.3 ml min−1, respectively, by varying the speed of the peristaltic pump (Ismatec, Reglo Dig MS-4/12 V1.13) in the range of 13–100 rpm. For most of the experiments 52 rpm was employed, corresponding to 2.6 and 1.3 ml min−1 sample and reductant flow rates, unless otherwise specified.
Gas lines downstream of the flow control units were made of Teflon or Tygon tubing. T-junctions were equipped with miniature polypropylene barbed fittings (Cole Parmer).
Se(IV), As(III) and Sb(III) sample solutions were obtained by dilution of 1000 mg l−1 stock standard solution with 1 mol l−1 HCl.
In the case of HG experiments performed in order to verify the effect of sample flow rates on sensitivity, either nitrogen or air was bubbled through the solution according to the scheme reported in Fig. 2. Five different flow rates were checked (Table 1) and for each of them absorbance measurements with solutions saturated either with air or nitrogen were performed. The experiments required a quite long time (up to 60 min) and during this long period of time the risk of losses from HCl media due to volatile chloride generation8 could be a problem. For this reason the sample solutions were prepared in 0.5 mol l−1 H2SO4 for Sb(III) and Se(IV), and in 0.1 mol l−1 H2SO4 for As(III). However, the efficiency of hydrogen selenide generation can be lower in sulfuric acid medium compared with hydrochloric acid generation media.9 In particular, the hydrogen selenide generation is catalysed by halogen ions in the order iodide > bromide > chloride.9,10 Therefore, in order to maintain the optimum hydrogen selenide generation efficiency, 0.5 mol l−1 KI has been added to the NaBH4 reductant solution.10
 | ||
| Fig. 2 Schematic representation of apparatus employed to measure the effect of oxygen dissolved in solution on signal in HG-AAS experiments. | ||
| Pump speed/rpm | F a/ml min−1 | C Ox b/μl l−1 |
|---|---|---|
| a Total flow rate, sample + reductant solutions. b Oxygen concentration in 1000 ml min−1 argon carrier gas. c Oxygen concentration measured with oxygen detector at the point B of experimental apparatus (see Fig. 1). d Estimated by COx = 8.5 + (6 × F), assuming quantitative release of oxygen from reaction solution. | ||
| 0 | 0 | 8.5 ± 1c |
| 13 | 1.0 | (14)d |
| 26 | 2.0 | (21)d |
| 52 | 3.9 | 29 ± 2c (32)d |
| 65 | 4.8 | (37)d |
| 100 | 7.3 | (52)d |
The total gas flow rate leaving the HG apparatus and entering the atomizer was 1000 ml min−1. It consisted of 180 ml min−1 argon added to the mixing cross, and 520 ml min−1 argon and 300 ml min−1 hydrogen, the last two being added after the gas–liquid separator (see Fig. 1).
Atomizers
The atomizer is the same as was employed previously and it is schematically represented in Fig. 3. It consists of a vertical quartz tube (inside diameter 6.25 ± 0.05 mm, outside diameter 8.0 mm) surrounded by a removable, cylindrical, glass shielding unit serving to produce vertical outer concentric argon shielding flow at the rate of 2 l min−1. A non-deactivated gas chromatographic fused silica capillary (0.53 mm id, Supelco, Bellefonte, LA) was axially positioned in the vertical quartz tube, serving to introduce the oxygen (1.0–50 ml min−1) supporting a microflame. The capillary can be moved along the tube axis and could be fixed at any desired distance from the atomizer top in the range of 0–45 mm. In the present work all the measurements were performed with the capillary aligned with the atomizer top.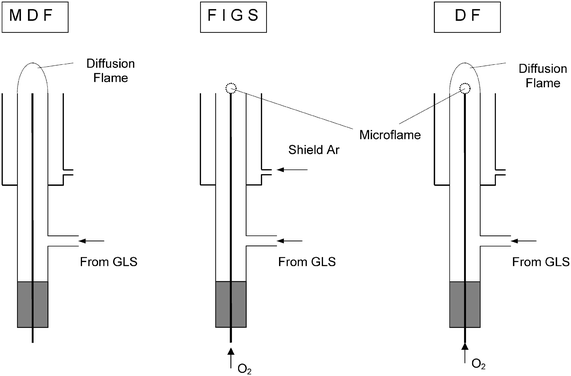 | ||
| Fig. 3 Schematic representation of the atomizer and the different atomization modes employed in the present study. | ||
The gas mixture from the HG apparatus was introduced into the side arm of the vertical quartz tube. The atomizer was operated in three different modes (see Fig. 3).
In the FIGS mode, oxygen was introduced to the fused silica capillary so that a microflame was burning at its end while argon was allowed to flow through the glass-shielding unit.
In MDF mode an argon–hydrogen diffusion flame burned at the end of the vertical quartz tube. Both oxygen flow to the microflame and argon flow to the shielding unit were stopped.
In double flame mode the microflame and MDF burned simultaneously while argon flow for the shielding unit was stopped.
The control of all the gas flow rates (see Fig. 1) was performed by ball rotameters, except the microflame oxygen flow rate, which was controlled by a mass flow meter (Aalborg) downstream to a ball rotameter fitted with a high accuracy valve.
Controlled contamination of gases by trace oxygen
In order to contaminate the gas flow reaching the atomizer with a know concentration of oxygen, a flow of air was introduced just upstream of the side arm of the atomizer through a T-junction (see Fig. 1). The flow rate of air was controlled in the range of 0.1–4.3 ml min−1 by a peristaltic pump equipped with Tygon tubing (0.89 mm id). Calibration of the peristaltic pump was achieved by bubble flow meter measurements. This allowed us to achieve an oxygen contamination level in the carrier gas (1 l min−1 total flow rate) in the range of 20–860 μl l−1, in addition to the background contamination level (see discussion). Linear regression of the expected oxygen concentration at a 1.0 l min−1 carrier gas flow rate, C°ox (μl l−1), as a function of pump speed, ω (rpm), gave C°ox = (−4 ± 1.7) + (8.38 ± 0.05) ω, r2 = 0.999![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 82. Measurements with the oxygen detector (at point C, see Fig. 1) gave a linear regression for experimental oxygen concentration, Cox
=
(9 ± 2)
+
(8.36 ± 0.06)
ω, with r2
= 0.999 78, in good agreement with C°ox except for the intercept values. The difference between the intercepts, about 13 μl l−1, was due to background contamination arising from oxygen diffusion through tubings and junctions, which is in agreement with experimental measurements (see Results and Discussion). The use of air introduced by a peristaltic pump was then considered a suitable method for the addition of known concentration of oxygen contaminat to the carrier gas.
82. Measurements with the oxygen detector (at point C, see Fig. 1) gave a linear regression for experimental oxygen concentration, Cox
=
(9 ± 2)
+
(8.36 ± 0.06)
ω, with r2
= 0.999 78, in good agreement with C°ox except for the intercept values. The difference between the intercepts, about 13 μl l−1, was due to background contamination arising from oxygen diffusion through tubings and junctions, which is in agreement with experimental measurements (see Results and Discussion). The use of air introduced by a peristaltic pump was then considered a suitable method for the addition of known concentration of oxygen contaminat to the carrier gas.
Presentation of results
The results are given in graphs and tables. The precision estimated from range of the steady state signals was better than 2% (RSD). The accuracy was verified by independent measurements.Results and discussion
Identification of the problem
Obviously, the temperature in the observed volume of FIGS and DF is controlled by the microflame oxygen flow rate. Spatial distributions of temperature were investigated in previous work for both MDF4 and FIGS5 (at a microflame oxygen flow rate of 20 ml min−1) using the same apparatus as is employed in the present work. For MDF at an observation height of 1 mm, the temperature spans from about 100 °C, close to the atomizer axis, to about 800 °C, 3 mm out of axis. In the same positions, for FIGS at an oxygen flow rate of 20 ml min−1, temperatures were about 1600 °C and 150 °C, respectively. There are no data on temperature in DF: however, the increase of the temperature close to the atomizer axis up to 1600 °C should be expected when increasing the oxygen flow rate to 20 ml min−1. In the case of FIGS, a decrease of the oxygen flow rate from 20 to 2 ml min−1 is expected to reduce the temperature close to the atomizer axis substantially.Fig. 4 shows the dependence of relative FIGS and DF signals (related to MDF for the actual analyte) on microflame oxygen flow rate. The behaviour is different for the three tested elements. For selenium, both FIGS and DF at low oxygen flow rates for microflame give similar signal to MDF, while at higher oxygen flow rates the signal decreases. It seems therefore that in the case of Se the concentration of free atoms in the observation volume is controlled mainly by thermal expansion caused by temperature variation in FIGS and DF.
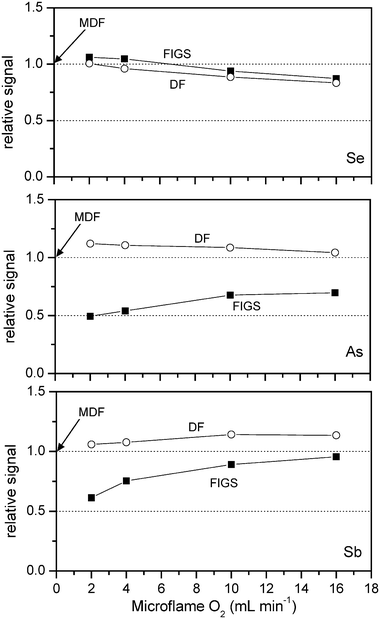 | ||
| Fig. 4 Effect of microflame oxygen flow rates in FIGS and DF atomization modes. Signals are related to those observed in MDF atomization mode. 0.2 μg ml−1 Se(IV) and As(III) in 1 mol l−1 HCl solution. 0.5 Sb(III) μg ml−1 1 mol l−1 HCl solution. | ||
The situation appears quite different for antimony and arsenic. The DF signal is always higher than for MDF, while for FIGS the signal is always lower, especially at the lower microflame oxygen flow rates. In the case of As and Sb the concentration of free atoms in the observation volume should be controlled by other effects besides the thermal expansion. Among these effects, an insufficient atomization efficiency at the lower microflame oxygen flow rates seems unlikely because for FIGS linear calibration graphs are obtained in the analyte concentration range of 0.1–1.0 μg ml−1 even for microflame oxygen flow rate as low as 2.0 ml min−1. Other effects should play a role in determining the shape of the plot reported in Fig. 4 for As and Sb.
It has been observed that the signal depression for FIGS at the lower microflame oxygen flow rates (<4 ml min−1) is much more pronounced than shown in Fig. 4 in the first runs of the HG apparatus after a long inactivity period (e.g., in the morning after the night stop). Then the signal at lower oxygen supply rates gradually increases to merge with that shown in Fig. 4 after about 60 min. The preliminary hypothesis was that the observed effect is generated by atmospheric oxygen, which could diffuse inside the HG apparatus, flowmeters, tubings and gas lines during the long inactivity periods of the HG apparatus. This was confirmed by measurements performed with the oxygen detector. Indeed, after the night stop only argon at 1 l min−1 was flushed through the HG apparatus. The oxygen concentration, COx, monitored for 60 min at the flowmeter outlet (point A, Fig. 1), decreased dramatically with time. For example COx was measured to be 27 and 1.5 μl l−1 O2 after argon was allowed to flow through the line for 20 and 120 min, respectively. After 120 min COx seemed to be stable at 1.5 μl l−1, which is the lowest level of contamination achievable with the present experimental setup.
The contamination by oxygen was suspected to affect the free atom concentration in the observation volume of the atomizer. This was proved by experiments reported in following sections.
Background concentration of oxygen in the apparatus
A certain concentration of oxygen is always present in the gas flowing through the experimental apparatus. It comes mainly from diffusion through junctions, tubings and from sample and reagent solutions. Downstream the HG apparatus, COx, was measured at the output of a gas–liquid separator: it increased from 1.5 μl l−1, measured at point A (see Fig. 1), to 8.5 μl l−1 measured at point B (see Fig. 1), in the absence of solutions pumped by peristaltic pump. With water pumped through the reductant (1.3 ml min−1) and sample (2.6 ml min−1) channels of HG apparatus, COx increased to about 29 μl l−1. This increase corresponds quite well to the amount of the oxygen dissolved in water at 25°C,11 about 6 μl ml−1, and indicates the almost quantitative stripping of dissolved gases by argon bubbling into solution. This is in line with a previous finding11 that a prolonged flushing with standard purity argon reduces the oxygen concentration in water from 6 μl ml−1 to 0.12 μl ml−1. Downstream from the T-junction for the introduction of air contamination (point C, see Fig. 1), the level of oxygen increased further by 2–3 μl l−1. This additional contamination is obviously due to the above mentioned air diffusion through gas connections.In conclusion, under the conditions employed for hydride generation in the present work, the background concentration level of O2 present in the gas flow reaching the atomizer could be estimated around 31 ± 3 μl l−1. A summary of oxygen contamination due to the HG apparatus is reported in Table 1.
Effect of oxygen dissolved in aqueous solution
Since a considerable fraction of contaminating oxygen arises from reactant solutions the role of dissolved oxygen concentration was investigated. The measurements were performed after either air or nitrogen (1.5 l min−1) had flowed for 5 min through solutions (see Experimental). To check the signal stability with time, several runs were performed at regular intervals, while gas was continuously flowing through the solutions. The absobance difference between air- and nitrogen-bubbled solutions for a given solution flow rate (Fig. 5) is due to the different amount of oxygen which is present in the solution. The behaviour of the three analytes is quite different: Se determination is, in contrast to As and Sb determinations, insensitive to oxygen dissolved in solution in the range of reactant solution flow rate investigated. The interference of dissolved oxygen cannot take place in the liquid phase (during the hydride generation step). The reason is that molar ratios of oxygen/analyte and oxygen/reductant are independent of the pump speed, so that any interference taking place in the liquid phase would have also to be independent of the pump speed. Additionally, oxygen dissolved in the solution cannot oxidize the tetrahydroborate(III) reagent, even at reductant concentration below the nanomolar level where the oxygen/reductant molar ratio is greater than five–six orders of magnitude.12 | ||
| Fig. 5 Effect of pump speed on signal obtained with MDF atomization mode using either air saturated or nitrogen saturated sample and reductant solutions. Conditions: 0.2 μg ml−1 Se(IV) in 0.5 mol l−1 H2SO4; 0.2 μg ml−1 As(III) in 0.1 mol l−1 H2SO4; 0.5 Sb(III) μg ml−1 in 0.5 mol l−1 H2SO4. | ||
Consequently, the only reasonable explanation of the dissolved oxygen interference is that oxygen stripped out from solutions enhances the oxygen contamination of gases introduced to the atomizer which, in turn, reduces the free atom concentration in the observation volume of the atomizer.
Oxygen interference in different atomizers
The effect of contamination of gases introduced to the atomizer in MDF, FIGS and DF modes by air added with a peristaltic pump just upstream of the atomizer inlet side arm (see Fig. 1) is quantified in Table 2. The data reported confirm that the interference caused by traces of oxygen in gases takes place in the atomisation step. The resistance to oxygen interference depends dramatically on both type of atomizer and the analyte identity. The best resistance to this interference is given by DF followed by MDF and then by FIGS, the latter being very sensitive to this kind of interference, especially at the lower oxygen flow rates for microflames. Concerning the analytes, selenium determination is relatively resistant to this kind of interference while it resulted much more pronounced for arsenic and antimony. These results explain satisfactorily the different behaviour of Se, As and Sb in different atomizers reported in Fig. 4.| Atomizer mode | ||||||
|---|---|---|---|---|---|---|
| MDF | FIGS | DF | ||||
| Microflame oxygen | Microflame oxygen | |||||
| Element | Tolerance or critical limit/μl l−1 | 2.0 (ml min−1) | 16 (ml min−1) | 2.0 (ml min−1) | 16 (ml min−1) | |
| a Concentration of oxygen giving 10% signal depression. b Concentration of oxygen giving 50% signal depression. | ||||||
| Sb | T.L.a | 20 | <15 | <15 | 20 | 60 |
| C.L.b | 70 | 60 | >900 | 100 | >900 | |
| As | T.L. | 48 | 45 | 45 | 65 | 110 |
| C.L. | 195 | 70 | 240 | 530 | >900 | |
| Se | T.L. | 210 | 110 | 480 | >900 | >900 |
| C.L. | >900 | 530 | >900 | >900 | >900 | |
The above results indicate the potential problems arising from oxygen interference in hydride generation techniques using MDF, as for example in HG-AFS. An additional level of contamination could be introduced by an inappropriate realization of connections and by the use of materials more permeable to oxygen diffusion. For example, it has been verified that 80 cm of silicone tubing (4 mm id, 2 mm wall thickness) introduces about 60 μl l−1 of oxygen in an argon stream flowing at 1 l min−1 flow rate inside the tube.
However, considering the only contribution of the oxygen dissolved in solution, it must be underlined that the use of solution total flow rates (sample + reductant flow rates) much higher than 5 ml min−1 has often been used in order to improve concentration detection limits.1,13 If such a sample flow rate is combined with the total gas flow rate reaching the atomizer, about 300–500 ml min−1, which is usual in analytical HG-AFS,1,2 the resulting oxygen contamination level is around 100–200 μl l−1, which could be the origin of severe signal depression according to tolerance and critical limits reported in Table 2.
The results of HG-AAS measurements reported in this work demonstrate that oxygen contamination of gases supplied to atomizer decreases the free analyte atom population, whatever is the mechanism of interference. Consequently, the oxygen interference must be operative also in HG-AFS, where additional signal depression could be caused by quenching effects. The interference arising from carrier gas contamination by oxygen has not yet been observed in HG-AFS, probably because it is difficult to recognize it. For example, oxygen interference affects only the slope of calibration curves in the concentration range 0.1–1.0 μg ml−1 of analyte. On the contrary, atomization interferences between hydride forming elements affect both the shape and the slope of calibration curves.7
An investigation of the mechanism of oxygen interference was not the aim of the present work. However, some reasonable hypotheses could be put forward, based on the present knowledge about the highly inhomogeneous spatial distribution of free atoms and temperatures in MDF and FIGS.4,5 Interfering oxygen is not completely burned in the atomizer and it can react with free atoms and/or with free hydrogen radicals, causing a decay of the free atom population and/or a depletion in atomization efficiency. Experiments designed to shed some light on the mechanism are in progress.
Acknowledgements
The support of this work by the grants of GACR no. 203/01/0453 by the Institute of Analytical Chemistry and by the agreement between Academy of Science of the Czech Republic and National Research Council of Italy is hereby greatly acknowledged.References
- L. Rahman, W. T. Corns, D. W. Bryce and P. B. Stockwell, Talanta, 2000, 52, 833 CrossRef CAS.
- A. D’Ulivo, E. Bramanti, L. Lampugnani and R. Zamboni, Spectrochim. Acta, Part B, 2001, 56, 1893 CrossRef.
- J. Dědina and A. D’Ulivo, Spectrochim. Acta, Part B, 1997, 51, 1737 CrossRef.
- J. Dědina, A. D’Ulivo, L. Lampugnani, T. Matoušek and R. Zamboni, Spectrochim. Acta, Part B, 1998, 53, 1777 CrossRef.
- A. D’Ulivo, J. Dědina, L. Lampugnani and T. Matoušek, J. Anal. At. Spectrom., 2002, 17, 253 RSC.
- A. D’Ulivo and J. Dědina, Spectrochim. Acta, Part B, 2002, 57, 2069 CrossRef.
- A. D’Ulivo and J. Dědina, Spectrochim. Acta, Part B, 1996, 51, 481 CrossRef.
- P. Smichowski and S. Farias, Microchem. J., 2000, 67, 147 CrossRef CAS.
- J. Agterdenbos, J. T. van Elteren, D. Bax and J. P. Ter Heege, Spectrochim. Acta, Part B, 1986, 41, 303 CrossRef.
- A. D’Ulivo, L. Gianfranceschi, L. Lampugnani and R. Zamboni, Spectrochim. Acta, Part B, 2002, 57, 2081 CrossRef.
- J. Dědina and B. Welz, J. Anal. At. Spectrom., 1992, 7, 307 RSC.
- E. Bramanti, A. D’Ulivo, L. Lampugnani, G. Raspi and R. Zamboni, J. Anal. At. Spectrom., 1999, 14, 179 RSC.
- T.-L. Deng, Y.-W. Chen and N. Belzile, Anal. Chim. Acta, 2001, 432, 293 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |