Cold vapour generation and on-line trapping of cadmium species on quartz surface prior to detection by atomic absorption spectrometry
Deniz
Korkmaz
a,
Cevdet
Demir
b,
Fırat
Aydın†
a and
O. Yavuz
Ataman
*a
aDepartment of Chemistry, Middle East Technical University, 06531 Ankara, Turkey
bDepartment of Chemistry, Uludağ University, 16059 Bursa, Turkey
First published on 9th December 2004
Abstract
A quartz trap for on-line preconcentration of Cd species was designed. The cold vapour generation technique was used for the generation of Cd species. The trapping medium was formed by external heating of the inlet arm of a quartz T-tube. The generated analyte species were trapped on a quartz surface heated to the collection temperature, 350 °C, and the collected species were revolatilized when the trap was heated further to revolatilization temperature, 1000 °C, and hydrogen gas was introduced in the trapping medium. Two-level fractional factorial design and central composite design were used to optimize generation conditions in the flow injection mode. The results of the fractional factorial design demonstrated that the factors and their interactions were statistically significant. Three factors, length of reaction coil, carrier HCl concentration and NaBH4 concentration, were considered to be the most significant parameters in the optimization and their optimum values were found to be 30 cm, 0.3 M and 3% (m/v), respectively. Sea-water (BCR), tomato leaves (NIST 1573a) and oyster tissue (NIST 1566b) standard reference materials were analyzed to assess the accuracy of the proposed method. For a collection period of 3.0 min, i.e., 6.0 ml sample volume, the 3σ limit of detection was 1.8 pg ml−1; the enhancement factor for LOD was found to be 90 as compared with FI-HGAAS. The sample throughput rate was 12 h−1.
Introduction
Chemical vapour generation techniques are frequently used for trace element detection.1 Volatile Cd species were first generated by ethylation2 and by using NaBH4 in an organic medium,3 and determined by atomization in flame and externally heated quartz tube atomizers, respectively. A highly sensitive determination of Cd by the use of cold vapour generation technique was introduced by Sanz-Medel et al.,4 in which the formation of an unstable intermediate, CdH2, was suggested which then dissociated into atomic Cd. On the other hand, generation of atomic and molecular Cd species from aqueous media upon reaction with NaBH4 was recently investigated by Feng et al.5 It was demonstrated that generated atomic species were stable in water, having a half-life of 2.2 min. The authors suggested that the initial products of the reduction reaction appeared to be free atoms, not the hydride, and subsequent interaction with nascent hydrogen gave rise to the molecular hydride. The process was found to be moderated by the chemistry of surface groups exposed to the reaction products.5 On the other hand, the generation of nascent hydrogen by acid decomposition of NaBH4 was not found to be feasible, as reported by recent investigations.6,7 The mechanism of formation/atomization of Cd species remains unsolved at this point.An excellent overview of literature data on Cd determination by atomic spectrometric techniques with vapour generation was recently reported by Lampugnani et al.8
The use of a cylindrical quartz tube as an on-line preconcentration device for Pb9 and Sb10 hydrides has been reported previously. A quartz T-tube and multiple microflame quartz tube11,12 were employed as atomizers, respectively. In these studies,9,10 PbH4 and SbH3 were trapped on a quartz surface heated to the collection temperature and the collected species were revolatilized when the trap was heated further to revolatilization temperature and hydrogen gas was introduced in the trapping medium.
In this study, use of a quartz surface for on-line preconcentration of Cd species generated by cold vapour technique was investigated. The inlet arm of the simple quartz T-tube was employed as the trapping region. Literature data exhibits variations and inconsistency for the determination of Cd by cold vapour generation because of difficulties related to generation, stripping of solution and transportation of the generated unstable species.8 Therefore, experimental design methods were used for the evaluation of the effective factors and to optimize the significant factors which may influence the analytical signal.13–16 A two-level fractional factorial design was employed to sort out the most significant parameters and a central composite experimental design was used for their further optimizations.
Experimental
Reagents
Working solutions were prepared from a 1000 mg l−1 stock Cd solution (Aldrich) in 0.4 M HCl. NaBH4 (Merck) solutions were prepared in 0.1% (w/v) NaOH (Carlo Erba). All the solutions were prepared daily. Deionized water was obtained from a Milli-Q Water Purification System. Ar, H2 and 1% O2 + 99% Ar gases were supplied from Habas Industrial and Medical Gas Products, Ankara. For interference studies, the solutions containing chloride and nitrate were prepared by dissolving solid CaCl2 (Riedel) and solid Ca(NO3)2·4H2O (Riedel) in water, respectively. For other interference studies, standard solutions were diluted using water; 100 mg l−1 Ni(II) in 2% HNO3 (Leeman ABS Inc.), 100 mg l−1 Pb(II) in 2% HNO3 (Leeman ABS Inc.), 1005 mg l−1 Cu(II) in 1% HNO3 (Aldrich), 1000 mg l−1 As(III) in 0.5 M HNO3 (Merck), 100 mg l−1 Se(IV) in 2% HNO3 (Leeman ABS Inc.), 1010 mg l−1 Sn(II) in 8% HCl (Aldrich), 100 mg l−1 Co(II) in 2% HNO3 (Leeman ABS Inc.), 1000 mg l−1 Fe(III) in 0.5 M HNO3 (Merck) and 100 mg l−1 Au(III) (Spectrosol AAS standard, BDH Chemicals Ltd.) were used.Instrumentation
A Pye Unicam PU9200 atomic absorption spectrometer equipped with deuterium background correction was used. A Cd hollow cathode lamp (Unicam) was employed as the light source, operated at 6 mA, and set to 228.8 nm. The slit width was 0.5 nm. Integrated absorbances were measured.The flow system used for both optimization of cold vapour generation and trapping of cadmium species is depicted in Fig. 1. Both flow injection (FI) and continuous flow (CF) modes were employed. During optimization of the vapour generation step, a 6-port injection valve (Rheodyne, Model 5020) was used in sample stream. For these optimizations, no attempt was made to trap the analyte. After optimizing generation conditions in FI mode, trapping procedures were optimized. For this purpose, sample solution was fed continuously for a fixed time period and therefore injection valve was not employed.
 | ||
| Fig. 1 Analytical set-up for hydride generation, trapping and atomization. | ||
Two peristaltic pumps (Gilson Minipuls 3) were used: one for sample/carrier and reductant solutions and the other to drain the gas–liquid separator (GLS). Sample/carrier and reductant flow rates were 2.0 ml min−1 and 1.2 ml min−1, respectively. In the case of FI experiments, a 100 μl sample loop was used. All the coils and transfer lines were made of 0.8 mm id PTFE tubing. The two streams were mixed in a chemifold and then along the 30.0 cm reaction coil. Volatilized cadmium species were then purged from the liquid mixture along a 20.0 cm stripping coil with an argon stream added from a T-piece, before their entrance to the GLS. A GLS with a 3.0 ml inner volume was used: the details can be found elsewhere.17 The gas phase was introduced directly to the inlet arm of the quartz T-tube by 185 mm long PTFE tubing. A T-piece was inserted between the GLS and quartz T-tube in order to introduce H2 whenever necessary. Gas flow rates were controlled by calibrated flowmeters (Cole-Parmer).
The quartz T-tube had an optical arm length of 115 mm, an id of 8 mm and an od of 10 mm. The inlet arm was 75 mm in length and 3.0 and 4.0 mm in inner and outer diameters, respectively.
Trap
The trapping medium used was the inlet arm of the quartz T-tube. The trapping zone was selected as the closest possible position to the junction point of inlet and optical arms (Fig. 1), taking advantage of the fact that Cd was determined by cold vapour generation technique so that the quartz T-tube was unheated. In this way transport losses were minimized. Six or seven pieces of broken quartz were inserted in the trapping region as filling material to increase trapping efficiency. Broken quartz pieces were produced by crushing a quartz tube of 4 mm id and 6 mm od; among the pieces produced, those with a longest dimension of about 3 mm were selected. In case of significant sensitivity drop, the quartz T-tube was cleaned in a mixture of concentrated HNO3 ∶ HF (7 ∶ 3) for 10 min, and devitrified filling material was discarded and replaced with the unused ones. Devitrification of quartz can be identified by a hazy film caused by fine micro-cracks.The trapping medium was resistively heated with a 0.31 mm diamater Ni–Cr wire, 330 mm long, having a resistance of 1 Ω, coiled around the inlet arm of the quartz T-tube. The resultant Ni–Cr coil covered a 20 mm portion of the inlet arm. The voltage difference applied to the wire was controlled by a variable potential power supply and a 750 W transformer. The input voltage of the variable potential power supply was 220 V. The inner wall temperature of the trap was measured with a thermocouple and calibrated with respect to the applied voltage (Table 1). Temperature was measured by means of a nickel–chromium thermocouple placed at the middle section of the trap.
| Voltage/V | Temperature/°C |
|---|---|
| 0.7 | 155 |
| 1.4 | 351 |
| 2 | 623 |
| 2.7 | 793 |
| 3.4 | 941 |
| 4.1 | 1030 |
The temperature measurements were carried out by keeping the Ar flow rate constant at 170 ml min−1. Introduction of H2 for the revolatilization cycle did not increase the temperature values. In fact, H2 decreases the temperature inside the trap slightly owing to the higher thermal conductivity of this gas as compared with Ar. Upon increasing the voltage difference from 1.4 V to 4.1 V, the temperature increased linearly with elapsed time up to 1000 °C, which was the maximum feasible temperature, and stayed constant thereafter. The maximum feasible temperature was reached within 30 s with an unfilled inlet arm.
Procedure
The collection of generated volatile Cd species was realized upon heating the trap to 350 °C (1.4 V). After a certain preconcentration time in continuous flow mode, the peristaltic pump was stopped and the voltage difference was increased to 4.1 V. Sixty seconds were needed in order to obtain the highest atomization signals; this was attributed to the fact that the trapping medium was filled with quartz pieces and additional time was required to bring all the pieces to the same temperature. In addition to the H2 from the decomposition of NaBH4 (approximately 90 ml min−1), only Ar, at a flow rate of 170 ml min−1, was present in the medium as the carrier gas. For the revolatilization of collected species, a H2 flow was switched on and H2 was sent to the trap from a T-piece inserted downstream of the GLS. The flow rate of H2 was 200 ml min−1, which made the total gas flow rate 370 ml min−1 during the revolatilization step, bearing in mind that the pump was off at this stage. After obtaining the signal, H2 flow was switched off, the voltage difference was brought to 1.1 V and another 60 s were required to bring the temperature down to the collection level. Another collection cycle was then started.Accuracy check
SRMs 1573a Tomato Leaves and 1566b Oyster Tissue (Freeze-Dried), obtained from NIST, and Trace Elements in Seawater SRM, obtained from BCR, were used to validate the proposed method. Tomato leaves and oyster tissue samples were digested by adding 5.0 ml of concentrated HNO3 on 0.250 g sample in each case. An additional 0.5 ml of concentrated HF was added on tomato leaves samples. A Milestone Ethos Plus microwave oven was used for digestion. A 20 min microwave program was applied for both sample types as follows: the vessels were brought from ambient temperature to 180 °C in 5 min and were kept at 180 °C for 5 min. Then they were brought to 200 °C in 5 min and were kept at this temperature for another 5 min. The resulting solutions were diluted to 50.0 ml with deionized water.Trace Elements in Seawater SRM was acidified so that the final sample was in 0.40 M HCl. No other treatment was required. Tomato leaves and oyster tissue samples were further diluted 150 and 250 times, respectively, with 0.40 M HCl before analysis.
Results and discussion
Cd species were generated and detected at room temperature throughout the studies. Use of a flame heated quartz tube caused a 50% reduction in the analytical signals which was in line with the observations of Sanz Medel et al.4 In their work, the reduction was explained by the expansion of gases on heating, diluting the analyte and shortening its residence time in the optical path. Trapping of some of the species on the heated inlet arm before they enter the optical path may also contribute to the decrease in the signal with an externally heated quartz tube. Nevertheless, this should only be making a minor contribution, since with a bare inlet arm, trapping of Cd species was fairly inefficient.Optimization of conditions for FI-cold vapour generation of Cd
A two-level fractional factorial design was first employed for exploratory purposes. The aim was to screen the number of factors which could be affecting the analytical response and interactions between factors. The most significant factors which should be studied in detail later were sorted out with this design. Five factors, length of reaction coil (x1), length of stripping coil (x2), carrier HCl concentration (x3), sample HCl concentration (x4), and concentration of the reductant, NaBH4 (x5), were involved in fractional factorial design. In this case we needed to perform 25–1 experiments. Four additional experiments at the central point (0 level) were also added to the design. Levels were coded as +1 “high” and −1 “low”. Real levels were selected on the basis of some preliminary experiments. The factors and their levels which were used in the fractional factorial design are shown in Table 2. The measurements were performed in random order. The experiments in the experimental design and measurements were replicated three times. The coefficients and calculated t-values using a coded design matrix are given in Table 3. The significance of factors is determined by the Student t-test as follows:where si is the standard deviation of the coefficient bi. The tabulated t-value at 95% confidence for 4 degree of freedom (20–16) is 2.78. All coefficients exhibited t-values greater than 2.78, implying that all factors were significant (Table 3). The factors which had the most significant effects on the integrated absorbance values were the length of reaction coil (x1), concentration of carrier HCl (x3), and concentration of NaBH4 (x5).

| Factor | Unit | −1 | 0 | +1 |
|---|---|---|---|---|
| a Factors: x1, length of reaction coil; x2 , length of stripping coil; x3, carrier HCl concentration; x4, sample HCl concentration; x5, NaBH4 concentration. | ||||
| x 1 | cm | 10 | 20 | 30 |
| x 2 | cm | 10 | 15 | 20 |
| x 3 | M | 0.10 | 0.20 | 0.30 |
| x 4 | M | 0.05 | 0.15 | 0.25 |
| x 5 | %, w/v | 1.0 | 2.0 | 3.0 |
| Coefficients | t-values | |
|---|---|---|
| b0 | 0.754 | 34.744 |
| b1 | 0.149 | 24.214 |
| b2 | 0.014 | 11.413 |
| b3 | 0.284 | 36.590 |
| b4 | 0.240 | 19.468 |
| b5 | 0.391 | 31.228 |
| b12 | 0.123 | 51.525 |
| b13 | 0.067 | 12.570 |
| b14 | 0.022 | 12.401 |
| b15 | 0.074 | 110.477 |
| b23 | 0.014 | 5.446 |
| b24 | 0.075 | 26.782 |
| b25 | −0.055 | −11.915 |
| b34 | −0.113 | −27.720 |
| b35 | 0.372 | 34.142 |
| b45 | 0.136 | 21.189 |
The three significant factors were optimized by means of the central composite design at fixed stripping coil length, 20 cm, Ar flow rate, 170 ml min−1, and sample acidity of 0.4 M. The sample concentration was 5 ng ml−1, passed through a 100 μl loop. The real values corresponding to the coded levels for the rotatable central composite design are given in Table 4. Using a model of the form
| y = b0 + b1x1 + b2x2 + b3x3 + b11x12 + b22x22 + b33x32 + b12x12 + b13x13 + b23x23 |
| Level | |||||
|---|---|---|---|---|---|
| Factor | −1.682 (α) | −1 | 0 | 1 | + 1.682 (α) |
| Factors: x1, length of reaction coil; x2, carrier HCl concentration; x3, NaBH4 concentration. | |||||
| x 1 | 3.18 | 10 | 20 | 30 | 36.82 |
| x 2 | 0.13 | 0.20 | 0.30 | 0.40 | 0.47 |
| x 3 | 2.16 | 2.50 | 3.00 | 3.50 | 3.84 |
By considering the coded levels, expression for the model was obtained as:
| y = 2.265 + 0.169x1 + 0.003x2 − 0.022x3 − 0.081x12 − 0.119x22 − 0.051x32 − 0.007x12 + 0.015x13 + 0.124x23 |
 | ||
| Fig. 2 Estimated response surface for length of reaction coil (x1) versus NaBH4 concentration (x3). | ||
Double peaks in FI cold vapour generation of Cd
Generation conditions for volatile Cd species are rather critical and require careful control of the parameters. Unlike the other hydrides, sample and carrier acid concentrations may vary and are critical. The experiments showed that higher acid concentrations in the carrier as compared with sample (as an example, 0.2 M for sample and 0.3 M for carrier) resulted in double peaks in FI mode using 100 μl sample loop, as displayed in Fig. 3. Lampugnani et al.8 suggested that because of substantial differences in the electronegativity of Cd and H, the covalent character of chemical bonds in CdH2 should be less pronounced than with other, more electronegative hydride forming elements; therefore, this hydride could expected to be intrinsically less stable as well as in an acidic medium. | ||
| Fig. 3 Double peaks observed; 5.0 ng ml−1, no trap, 0.40 M HCl in carrier flow, 0.20 M HCl in sample. | ||
The resulting double peaks in our case might be explained by the incomplete generation or decomposition of hydride; Cd2+ ions remaining in the reaction mixture or on the walls of the GLS may have been converted to the hydride with an additional hydrogen radical front. Therefore, the acidity of the sample solution should be high enough to convert essentially all the Cd2+ ions to the hydride.
Optimization of collection and revolatilization
The decrease in integrated absorbance values for Cd as a function of temperature of the trapping region was depicted in Fig. 4. A lower signal indicates better trapping. The trapped analyte species were subsequently volatilized by heating the trap up to 1000 °C prior to the next experiment. The peak area values were reduced by 90% when the inlet arm of the quartz T-tube was heated to 350 °C, indicating retention of Cd species on the quartz surface. It should be mentioned that the inlet arm was filled with 6–7 pieces of quartz in this configuration: with a bare inlet arm or with fewer pieces of quartz in the inlet arm, the reduction in the signal never exceeded 50%, indicating incomplete trapping. Even with this configuration 100% trapping was not achieved, but no further attempt was made to increase the amount of filling material in the trapping region since this would result in broader revolatilization peaks. | ||
| Fig. 4 Variations of signal with trap temperature during FI operation of 100 μl of 1.0 ng ml−1 Cd. | ||
The results for theoptimization of collection and revolatilization temperatures are given in Fig. 5. The optimum trapping temperature found, 350 °C, was relatively low as compared with PbH49 and SbH3,10 where 500 °C and 650 °C were found to be optimum values, respectively. The decrease in the signal for collection temperatures higher than 350 °C was due to the release of some of the analyte species from the quartz surface during the trapping stage. The optimum revolatilization temperature was reached 60 s after increasing the voltage difference from 1.4 to 4.1 V.
 | ||
| Fig. 5 Influence of collection temperature (squares, volatilization temperature of 1000 °C) and revolatilization temperature (triangles, collection temperature of 350 °C) on the observed signal; 0.3 ng Cd. | ||
Effect of carrier and revolatilization gases
Use of a mixture of 1% O2 + 99% Ar as the carrier gas reduced the analytical response of Cd by 25%, even for FI mode experiments in which no trapping was applied. This might be due to the formation of CdO, which may be remaining in the reaction mixture or may not be atomizing even if it reaches the optical path. When a mixture of Ar and O2 was used in trap experiments, the reduction in the atomization signal was 50% as compared with Ar alone. Here, the presence of O2 should be affecting not only generation but also the revolatilization step. Although these experiments were not direct evidence, the negative effect of O2 may suggest that both the collected and revolatilized species were atomic Cd.Use of a higher flow of Ar for the revolatilization step was also tried to see if it would have any beneficial effect on the analytical signal. However, flows higher than 170 ml min−1 decreased the integrated absorbance values due to a dilution effect. Therefore, Ar gas was kept at a flow of 170 ml min−1 for both collection and revolatilization steps.
In order to revolatilize the collected species, simply heating the trap to the revolatization temperature was not sufficient. The presence of H2 gas was essential, which was in accordance with the observations for Pb9 and Sb.10 As for Sb, H2 gas was introduced after reaching the revolatilization temperature; introduction of H2 at the beginning of the revolatilization step caused broad signals. 200 ml min−1 was chosen as optimum for H2 flow.
Effect of filling materials
Apart from the quartz pieces, quartz frit (porous quartz disc) pieces were also tried as filling material to understand if they would act as a better trapping medium for Cd. Unfortunately, it was not possible to revolatilize the analyte from quartz frit, possibly due to the diffusion of analyte species into the pores, resulting in a better but not reversible analyte trapping.Use of 6–7 pieces of quartz was necessary to efficiently trap the hydride.
Analytical figures of merit
The typical analytical signal shape observed with this configuration is shown in Fig. 6. The full width at half maximum is approximately 0.9 s: use of more filling material caused broader peaks. | ||
| Fig. 6 The typical analytical signal. Collection temperature: 350 °C; revolatilization temperature: 1000 °C; 20 pg ml−1 Cd, 3 min collection. Peak height: 0.057; peak area: 0.051. Seven pieces of quartz as filling material in the trapping arm. | ||
3σ LODs, characteristic masses (m0) and characteristic concentrations (C0), calculated by using integrated absorbances, were summarized in Table 5. The enhancement factor for the LOD for a collection time of 180 s (corresponding to sample volume of 6.0 ml) was found to be 90 as compared with FI-HGAAS. The precision of measurements was 5% RSD (n = 10). For a collection time of 3 min, the calibration plot was linear between 20–100 pg ml−1 with a best line equation and correlation coefficient of A = 0.0025C−0.0001 and 0.999, respectively, where C was the concentration of Cd in pg ml−1. No purification of the used reagents were required owing to the low blank values.
| C 0/pg ml−1 | m o/pg | LOD/pg ml−1 | |
|---|---|---|---|
| FI-HGAAS (100 μl loop) | 12.2 | 1.2 | 164 |
| Quartz trap | 1.7 | 10.2 | 1.8 |
Collection periods higher than 180 s were not used for practical reasons. The sample throughput rate for a collection of 180 s was 12 h−1. Better rates can be obtained by using lower collection times. The lifetime of a filling material was not less than 100 measurements. The proposed method was applied to the analysis of standard reference materials for accuracy assessment. The results are presented in Table 6. Good agreement with the certified values is evident. For the analysis of sea-water SRM, direct calibration was applied. Calibration by standard additions was used for the analysis of tomato leaves and oyster tissue SRMs.
| SRM | Certified | Found |
|---|---|---|
| Trace Elements in Seawater/nmol kg−1 | 0.175 ± 0.018 | 0.180 ± 0.009 |
| 1573a Tomato Leaves/mg kg−1 | 1.52 ± 0.04 | 1.50 ± 0.06 |
| 1566b Oyster Tissue/mg kg−1 | 2.48 ± 0.08 | 2.45 ± 0.15 |
The LOD obtained is the same as the best attained, according to the literature, with vapour generation in situ trapping in graphite furnace; in pg ml−1, 10,18,19 5,20 721 and 2.8 The corresponding sample volumes in ml were 1.4,18,20 1.0,19 0.521 and 1.8.8
Interference studies
Interference studies have been carried out using optimized conditions without and with a trap. The effects of some hydride forming species, some transition elements and calcium salts of chloride and nitrate are given in terms of percent relative signals in Fig. 7, Fig. 8 and Fig. 9, respectively. In addition to the enhancement in sensitivity, employment of a trap resulted in a reduced interference effect in most cases when the interferent to analyte ratio was the same. The trap’s advantage is more prominent in the case of transition elements. This advantage is valid also to some extent in the case of hydride forming elements; arsenic has a positive (enhancing) interference both without and with a trap. Regarding the effects of calcium salts of nitrate and chloride, use of a trap practically provides an interference free operation.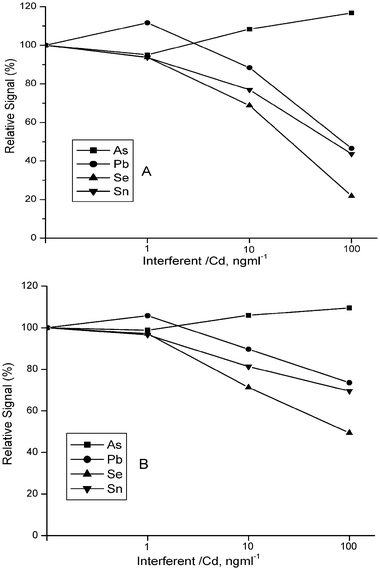 | ||
| Fig. 7 Interference effect of some hydride forming species, As(III), Pb(II), Se(IV), Sn(II), without (A) and with (B) trap. Analyte concentrations are 1.0 and 0.1 ng ml−1 for measurements without and with trap, respectively. Optimized conditions were used. | ||
 | ||
| Fig. 8 Interference effect of some transition elements, Ni(II), Cu(II), Co(II), Fe(III), Au(IV), without (A) and with (B) trap. Analyte concentrations are 1.0 and 0.1 ng ml−1 for measurements without and with trap, respectively. Optimized conditions were used. | ||
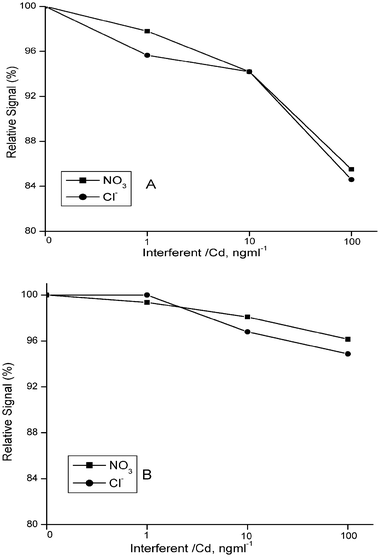 | ||
| Fig. 9 Interference effect of nitrate and chloride ions as calcium salts, without (A) and with (B) trap. Analyte concentrations are 1.0 and 0.1 ng ml−1 for measurements without and with trap, respectively. Optimized conditions were used. | ||
Conclusions
A novel technique has been proposed for on-line preconcentration of Cd species generated by the cold vapour technique. Using a central composite design, the most critical parameters which affect the generation of Cd species in FI mode, using a 100 μl sample loop, were found to be the length of the reaction coil, the concentration of carrier HCl and concentration of NaBH4. Since interactions were present between the factors, using the one factor at a time approach would not have been convenient for optimizations. The negative effect of oxygen on both FI and trap signals of Cd suggests that Cd species were both collected and revolatilized in atomic form. Further studies are required to clearly understand the nature of the species collected on a quartz surface. For Cd, the detection limits provided are the same as the best detection limits attained with in situ trapping in a graphite furnace.This study, at present, is the last one in a series performed in our laboratory. The previous studies involved determination of Pb9 and Sb.10 The idea of using the quartz surface as a trap material has originated from our previous studies where a slotted quartz tube was used for in situ trapping using flame AAS.22–24 It seems that a quartz surface has potential as a trap material for analytes that can be converted into gaseous species, such as hydrides.
Acknowledgements
The financial support of Middle East Technical University Research Fund through grant BAP-2001-07-02-00-08 is gratefully acknowledged.References
- J. Dědina and D. L. Tsalev, Hydride Generation Atomic Absorption Spectrometry, Wiley, Chichester, 1995 Search PubMed.
- A. D’Ulivo and Y. Chen, J. Anal. At. Spectrom., 1989, 4, 319–322 RSC.
- J. Cacho, I. Beltran and C. Nerin, J. Anal. At. Spectrom., 1989, 4, 661–663 RSC.
- A. Sanz-Medel, M. C. Valdes-Hevia y Temprano, N. Bordel Garcia and M. R. Fernandez de la Campa, Anal. Chem., 1995, 67, 2216–2223 CrossRef CAS.
- Y.-L. Feng, R. E. Sturgeon and J. W. Lam, Anal. Chem., 2003, 75, 635–640 CrossRef CAS.
- A. D’Ulivo, C. Baiocchi, E. Pitzalis, M. Onor and R. Zamboni, Spectrochim. Acta, Part B, 2004, 59, 471–486 CrossRef.
- A. D’Ulivo, Spectrochim. Acta, Part B, 2004, 59, 793–825 CrossRef.
- L. Lampugnani, C. Salvetti and D. L. Tsalev, Talanta, 2003, 61, 683–698 CrossRef CAS.
- D. Karadeniz Korkmaz, N. Ertaş and O. Y. Ataman, Spectrochim. Acta, Part B, 2002, 57, 571–580 CrossRef.
- D. Korkmaz, J. Dědina and O. Y. Ataman, J. Anal. At. Spectrom., 2004, 19, 255–259 RSC.
- J. Dědina and T. Matoušek, J. Anal. At. Spectrom., 2000, 15, 301–304 RSC.
- T. Matoušek, J. Dědina and A. Selecká, Spectrochim. Acta, Part B, 2002, 57, 451–462 CrossRef.
- B. İzgi, C. Demir and Ş. Güçer, Spectrochim. Acta, Part B, 2000, 55, 971–977 CrossRef.
- S. Cerutti, J. A. Salonia, S. L. C. Ferreira, R. A. Olsina and L. D. Martinez, Talanta, 2004, 63, 1077–1082 CAS.
- T. Lundstedt, E. Seifert, L. Abramo, B. Thelin, A. Nystrom, J. Pettersen and R. Bergman, Chemom. Intell. Lab. Syst., 1998, 42, 3–40 CrossRef CAS.
- A. G. Gonzalez, Anal. Chim. Acta, 1998, 360, 227–241 CrossRef CAS.
- T. Matoušek, M. Johansson, J. Dědina and W. Frech, Spectrochim. Acta, Part B, 1999, 54, 631–643 CrossRef.
- H. Goenaga Infante, M. L. Fernandez Sanchez and A. Sanz-Medel, J. Anal. At. Spectrom., 1998, 13, 899–903 RSC.
- H. Matusiewicz, M. Kopras and R. E. Sturgeon, Analyst, 1997, 122, 331–336 RSC.
- H. Goenaga Infante, M. L. Fernandez Sanchez and A. Sanz-Medel, J. Anal. At. Spectrom., 1997, 12, 1333–1336 RSC.
- P. Bermejo-Barrera, J. Moreda-Piñeiro, A. Moreda-Piñeiro and A. Bermejo-Barrera, At. Spectrosc., 1998, 19, 100–106 CAS.
- N. Ertaş, D. Karadeniz Korkmaz, S. Kumser and O. Y. Ataman, J. Anal. At. Spectrom., 2002, 17, 1415–1420 RSC.
- D. Korkmaz, S. Kumser, N. Ertaş, M. Mahmut and O. Y. Ataman, J. Anal. At. Spectrom., 2002, 17, 1610–1614 RSC.
- D. Korkmaz, M. Mahmut, R. Helles, N. Ertaş and O. Y. Ataman, J. Anal. At. Spectrom., 2003, 18, 99–104 RSC.
Footnote |
| † On leave from Department of Chemistry, Dicle University, 21280 Diyarbakır, Turkey. |
| This journal is © The Royal Society of Chemistry 2005 |