Flow-injection analysis with fluorescence detection for the determination of trace levels of ammonium in seawater
Roslyn J.
Watson
*a,
Edward C. V.
Butler
ab,
Lesley A.
Clementson
a and
Kate M.
Berry
a
aCSIRO Marine Research, GPO Box 1538, Hobart, Tasmania 7001, Australia. E-mail: Ros.Watson@csiro.au
bAntarctic Climate and Ecosystems CRC, University of Tasmania, Private Bag 80, Hobart, Tasmania 7001, Australia
First published on 1st December 2004
Abstract
A method using flow-injection, gas-diffusion, derivatisation and then fluorescent detection has been established for ammonium ion determination in seawater. The fluorescent derivative formed by reacting ortho-phthaldialdehyde (OPA) and sulfite with ammonia gives high sensitivity while removing potential interferences. This is required to measure the low concentrations of ammonium often seen in the open ocean. The experimental conditions (flow-rate, reagent concentrations, membrane configurations, etc.) were manipulated to improve performance. For a sample throughput of 30 samples h−1, the limit of detection was 7 nM, the coefficient of variation was 5.7% at 800 nM, and the calibration curve was linear to at least 4 μmol L−1. Interferences were minimised by a gaseous diffusion step. Volatile small molecular-weight amines as interferents were discriminated against by this method. They neither passed through the membrane as efficiently as ammonia, nor reacted as readily with OPA when sulfite was the reductant. Contamination by ammonia from laboratory and shipboard sources complicates application of the method to natural waters, especially measurement of low concentrations (<100 nM) in open-ocean waters. Steps to overcome contamination are described in detail. Some results are presented for ammonium determination in Southern Ocean and Huon Estuary (Tasmania) waters.
Introduction
Analysis of ammonium present in estuarine, coastal and open-ocean waters is fundamental to understanding biogeochemical cycling of nitrogen and its influence upon marine ecosystems. Ammonium is involved in many biogeochemical processes. It fuels ‘regenerated’ production by marine phytoplankton,1 is produced by heterotrophic bacteria, and is involved in other microbial processes such as nitrification and denitrification.2 The net result of these processes is that ammonium is frequently present at concentrations lower than 1 μmol L−1. Such low concentrations are difficult to analyse as they are close to or below the detection limit for many methods and the risk of contamination is high.1Many of the analytical methods currently used are modifications of the indophenol blue (or Berthelot) reaction, using spectrophotometry to measure the products.3–7 These techniques generally use phenol, which can be difficult to work with in confined areas such as shipboard laboratories. Several of these methods are time consuming and sensitive to changes in reaction conditions and contamination.8,9
Ortho-phthaldialdehyde (OPA) is used routinely to determine a range of amino acids by HPLC with fluorescence detection.10–13 The derivatisation reaction uses a thiol, usually 2-mercaptoethanol, as a mild reductant.14 The resulting 1-alkylthio-2-alkyl-substituted isoindoles are intensely fluorescent. Several authors have used the 2-mercaptoethanol-OPA reagent in flow-injection analysis (FIA) for ammonia determination.15–17 In 1991, Jones18 also described a method that incorporated the 2-mercaptoethanol-OPA reaction into a gas-diffusion FIA system for analysis of marine waters. He included procedures for purifying and removing interference in reagents by ammonia or volatile amines.
OPA has been used in the analysis of various thiols by HPLC.19 Sulfite was also detectable by this method. Genfa and Dasgupta20 used sulfite in their FIA method for analysing ammonium in fresh water. Sulfite was more stable and acceptable to work with than 2-mercaptoethanol but was less reactive. Kérouel and Aminot21 developed this method further for use with brackish and saline water samples, as did Holmes et al.22 who developed a highly sensitive manual method. Aminot et al.23 also developed a method suitable for in situ analyses. The method described here combines the advantages of the gas-diffusion method of Jones18 with the sulfite-OPA reaction. It optimises the method for ultra-trace level measurement, avoids the use of phenol, and guards against common sources of interference and contamination in shore-based and shipboard laboratories.
Experimental
Reagents
Reagents used were AnalaR grade (BDH, Kilsyth, VIC, Australia) unless specified. All solutions were made up in water that was distilled then freshly de-ionised using a Milli-Q water system (Millipore, Bedford, MA, USA). Filters, glassware and plasticware were soaked overnight in 10% v/v HCl and rinsed with Milli-Q water before use. Nitrogen gas used was 4.0 grade (99.99% purity) N2 (Linde gas, Yennora, NSW, Australia).The carrier reagent was 37.5 mmol L−1 H2SO4 (0.2% v/v solution).
The NaOH reagent was 0.45 mol L−1. In early experiments, the NaOH reagent also contained 20% w/v trisodium citrate. The trisodium citrate was originally included to prevent hydroxide precipitation when the pH of the seawater samples increased.
The OPA reagent was first diluted to 0.37 mol L−1 in HPLC-grade methanol (Waters, Milford, MA, USA) (0.1 g of OPA in 2 ml of methanol). 2 ml of this solution was then further diluted to 500 ml using a buffer solution containing 0.188 mol L−1 disodium hydrogen phosphate and 0.45 mmol L−1 sodium sulfite. The final pH was 9.3. The solution was filtered through a 0.45 μm mixed cellulose esters filter (Millipore, Bedford, MA, USA) and then sonicated under vacuum for 30 min. Both HPLC-grade OPA (BioChemika, Fluka, Buchs, Switzerland) and reagent grade OPA (97%, Sigma, St Louis, MO, USA and ICN, Costa Mesa, CA, USA) were tried with no difference in either the analytical performance or the storage qualities of the reagents. This confirms the results of Genfa and Dasgupta.20 The OPA reagent was stored in an amber glass bottle to prevent the reagent deteriorating.
The NaOH solution was bubbled with N2 for at least 20 min.
The scrubbing solution for the NaOH reagent was 10% v/v H2SO4.
When the FIA system was not in use, cleaning solutions were flushed through it. The cleaning solutions were 0.2% v/v H2SO4 for the carrier and the NaOH flow lines and 0.5% v/v methanol for the OPA flow line.
All solutions, except the OPA solution, were prepared fresh each day. The OPA solution could be made in advance and stored in the dark until required. However, it needed to be filtered and sonicated immediately before use.
Standard solutions
Standards were made from ammonium sulfate, which was dried for at least 1 h at 110 °C and stored in a vacuum desiccator. A stock standard of 100 mmol L−1 NH4+ was made up in Milli-Q water. The stock was diluted into 2 mmol L−1 and then 40 μmol L−1 intermediate standards. A series of working standards, typically up to 1 μM, were made in 35 g L−1 NaCl solution. Working standards were prepared from freshly made stock and intermediate standards in volumetric glassware. The working standards were either analysed fresh or stored frozen in polypropylene tubes until required.Apparatus
A schematic diagram of the FIA system used is shown in Fig. 1. To prevent contamination from laboratory air, all reagent solutions and samples were kept and processed in a 150 L glove cabinet (Mecaplex, Grenchen, Switzerland) that was flushed with N2. The peristaltic pump used was an Alitea MXV (Stockholm, Sweden) 8-channel pump. Flow rates were achieved by setting the pump rate to 9.9 rev min−1 and using Tygon pump tubing (TACS Australia, North Sydney, NSW, Australia) metered to 1.0, 0.1 and 0.6 ml min−1 for the carrier, NaOH and OPA solutions, respectively. Elsewhere in the system, 0.8 mm i.d. low-pressure PTFE tubing and polypropylene Cheminert fittings (Supelco, Bellefonte, PA, USA) were used. The PTFE tubing lengths were kept to the minimum lengths needed to connect components. Ammonia contamination was removed from the NaOH solution by pumping the solution through a 24 cm long diffusion cell with the scrubbing solution pumped in parallel on the other side. Samples were injected manually into an injection port from within the N2-purged chamber. The injection port was connected to a Cheminert (LDC, Riviera Beach, FL, USA) 6-port PTFE injector, with a 1 ml sample loop, via ∼70 cm of PTFE tubing. The injector was fitted inside the door of an ICI (Dandenong, VIC, Australia) TC1800 temperature controller that was set to 70 °C. The temperature controller also contained the gas-diffusion unit consisting of two polymethylmethacrylate (PMMA) gas-diffusion cells connected in series. The first diffusion cell in series was a 10 cm long Tecator (Höganäs, Sweden) cell, while the second cell was made in-house with similar channel dimensions. The membranes used in the cells were prepared from commercial PTFE tape (“plumbers’ tape”, 12 mm × 0.075 mm). The PFTE tubing in and out of the cells were connected so that the donor (Carrier/NaOH reagents) stream and acceptor (OPA reagent) streams were flowing in parallel through the cells. The cells were connected, via 80 cm of PFTE tubing, to a GBC (Dandenong, VIC, Australia) LC1250 fluorescence detector with a 2 mm flow through cell set to an excitation wavelength of 310 nm and an emission wavelength of 390 nm. A Yew (Tokyo, Japan) 6032 chart recorder recorded the signal from the detector.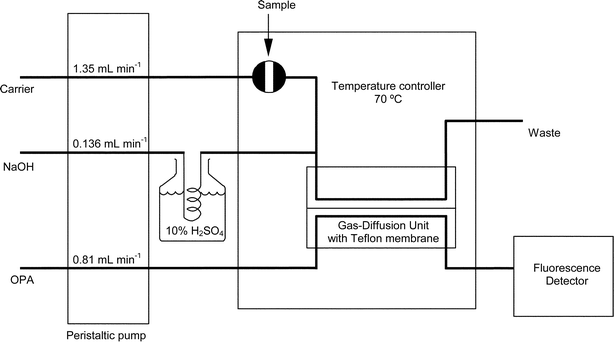 | ||
| Fig. 1 Schematic diagram of the FIA system, using gas-diffusion, for the determination of ammonia. | ||
Sample analysis
Samples were collected in triplicate in 10 ml polypropylene tubes (Sarstedt, Mawsons Lake, SA, Australia). They were either analysed in duplicate or triplicate immediately after collection or stored frozen for later analysis. An acid-cleaned 5 ml plastic syringe (Terumo, Tokyo, Japan), with 10 cm of 1/8′′ o.d. PTFE tubing attached, was used to draw up water samples. For each sample, the syringe was rinsed with 2–3 ml and then filled with 5 ml of sample. The PFTE tubing was then removed, and the sample injected into the system via the switching valve and sample loop (Fig. 1). A single syringe was used for each sample set comprising: standards (lowest to highest concentration)–samples–standards–samples (repeats)–standards. It was changed for a new syringe if a sample with a high ammonia concentration was encountered.When the system was not in use overnight or on weekends, all of the flow lines were flushed with cleaning solutions. The pump speed for all flow lines was reduced to 1 rev min−1, both to reduce stress on the membrane and to conserve the cleaning solutions. For long periods of shutdown all the lines were flushed with cleaning solutions for a day before allowing all of the lines to run dry. To restart after a shutdown the cleaning solutions were run through the lines for at least 16 h.
Results and discussion
Optimisation of FIA system
Genfa and Dasgupta20 suggested that the phosphate buffer should be kept as a separate reagent from the OPA and sulfite and mixed within the FIA system. The baseline was noisier when the reagents were kept separate compared with a single buffer–OPA–sulfite reagent mixture. This reagent was stable when stored in the dark at room temperature for several weeks.
A simple test of doubling the concentration of OPA and sulfite, both individually and together, did not increase the response from the fluorescence detector.
Initially the buffer used for the OPA reagent was a 0.25 mol L−1 H3BO3 solution that was adjusted to pH 9.5 with 10 mol L−1 NaOH solution. High variation in the background fluorescence was seen with this buffer due to slight changes in the pH. The buffer was changed to disodium hydrogen phosphate alone. This produced a buffered pH of 9.3 in the mixed reagent, similar to the pH of 9.5 indicated by Jones.18 This buffer had the advantage of being simpler to prepare than similar OPA methods,18,20 where the buffers were adjusted with sodium hydroxide solution to obtain a selected pH. It also reduces the risk of sensitivity changes due to small changes in pH and the risk of contamination from the additional handling required in pH adjustment.
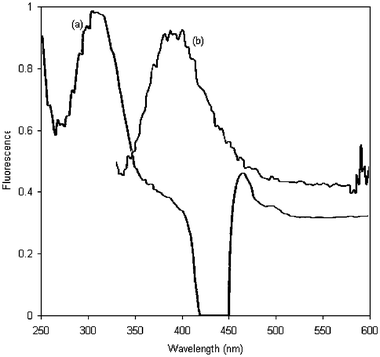 | ||
| Fig. 2 Excitation (a) and emission (b) spectra of the fluorogenic compound. | ||
Increasing the membrane surface area was tested using a single longer cell. Initially this was done with a cell that was made of two PMMA blocks that sandwiched two PTFE gaskets separated by a membrane (similar to Nakata et al.24). Each of the gaskets in this cell were 0.5 mm thick PFTE sheets that had channels (2 mm wide × 235 mm long) cut out of them. The gaskets allowed for changes in the cell chamber dimensions that might improve diffusion characteristics. Unfortunately the cell was prone to leaking. While attempting to stop the leaking, the cell was tightened to the point where the gaskets were compressed to 0.3 mm thick and the channels widened. The new dimensions of the chambers meant that the membranes puckered up against the PMMA blocks.
A second long cell, with channels etched into the two PMMA blocks, was made and tested. While this cell did not leak, the peaks were wider rather than higher compared to the peaks from the dual cell set-up. Peak area was increased, but at the expense of sample throughput. The baseline was noisier than with the dual cell set-up with the signal-to-noise ratio going from 25 in the dual cell system to 18 in the single large cell set-up.
Although parallel flow of donor and acceptor streams through the gas-diffusion cells was the normal configuration, the flow direction of these streams was changed to opposing flow. Opposing flow increases the concentration gradient between the donor and acceptor streams and thus can increase gas-diffusion across the membrane. When opposing flow was attempted, the reagent streams ruptured the membrane so further testing of this orientation was abandoned.
The final cell configuration chosen was the dual cells in series with the donor and acceptor streams passing through the cells in parallel.
The tape was fitted in a dust-free setting, since it could hold a static charge that attracted dust and lint. When these dust particles were present, they moved through the system and were deposited at connections or in the detector flow cell and created blockages.
Bubbles also damaged the membrane and appeared for several reasons. The more common ones were faulty connections or lines that had been allowed to run dry. Bubbles also appeared at hot spots in the cell or lines. These occurred where the diffusion cell or surrounding tubing came in direct contact with the hot metal of the temperature controller. The simple insulation of the cell (e.g. by wrapping it with a low-linting wiper) was sufficient to prevent this. The hot diffusion cell was also allowed to cool for a few minutes before stopping the reagent flow.
The pump speed was altered gradually to the required flow rate to help prolong the life of the membrane.
Cleaning solutions were pumped though the lines at an operating speed of 1 rev min−1 overnight. This helped to reduce the incidence of blockages. Although the cause of the blockages was unknown, potential sources were from hydroxide precipitates and sample particulates from seawater samples and the recrystallisation of the disodium hydrogen phosphate.
With care the membrane generally lasted for 2–3 d of use and at times up to a week. Short lifetimes for the membrane were an indicator of the FIA system not performing at its optimum.
To increase sample throughput the pump speed was raised to 9.9 rev min−1 (equating to 1.36 ml min−1, 0.81 ml min−1 and 0.136 ml min−1 for the respective reagent streams). Although the increase in pump speed increased the sample throughput by ∼35% sensitivity decreased by ∼23%. The diminished sensitivity arose from a combination of decreased opportunity for gaseous diffusion of ammonia and decreased reaction time for the diffused ammonia with OPA. Increasing the length of the diffusion cell to compensate would also decrease the sample throughput, thus defeating the original purpose of increasing the pump speed. However, by increasing the temperature in the controller the reaction rate of ammonia and OPA increased, and presumably gaseous diffusion as well. The temperature was increased to 70 °C. This restored sensitivity to ∼91% of the original value. At 70 °C bubbles began to appear in the detector. Preventing direct contact between the diffusion cell and the hot metal of the temperature controller, as mentioned above, removed this interference. Since raising the temperature would promote greater bubble formation, temperatures above 70 °C were not attempted.
Standard additions of ammonium were made with estuarine waters representing salinities of 0, 5, 8, 19 and 33 as well as Milli-Q and 35 g L−1 NaCl solutions. Four standard additions were analysed, in triplicate, for each sample (1, 2, 3 and 4 μmol L−1). The data from these experiments are shown in Table 1. The estuarine samples were similar to the Milli-Q and NaCl solutions. Typical results from the analysis of estuarine samples are shown in Fig. 3. The Huon Estuary is a salt-wedge estuary in southern Tasmania. The salinity ranged from 0.0 to 34.8, while the ammonium concentrations ranged from 40 nmol L−1, in surface waters in the lower section of the estuary, to 10 μmol L−1 at a mid-estuary site immediately downstream of a sewage treatment plant.
 | ||
| Fig. 3 Results from samples collected on 2–4/12/97 from the Huon Estuary in southern Tasmania. Samples were collected in the top 0.5 m (surface), fluorescence maximum (mid) and from within the salt wedge (bottom). | ||
| Estuarine samples with standard addition | Fluorescence response/FU μM−1 |
|---|---|
| Milli-Q | 1.29 |
| 35 g NaCl L−1 | 1.27 |
| Salinity 33 | 1.26 |
| Salinity 19 | 1.29 |
| Salinity 8 | 1.29 |
| Salinity 5 | 1.29 |
| Salinity 0 | 1.27 |
Control of contamination
Boiling the reagent to drive off ammonia was investigated as a technique for contamination removal. However, this was not effective as there was no decrease in the blank concentrations.
An alternative for purifying the NaOH reagent was an in-line diffusion cell. A 240 mm long diffusion cell was placed between the reagent reservoir and the peristaltic pump. The NaOH reagent was pumped through the cell on one side of a PTFE-tape membrane, while on the other side in parallel, 10% H2SO4 was pumped at 0.135 ml min−1. This was as effective in removing ammonia contamination from the NaOH reagent as the microporous tubing.
After one batch of trisodium citrate was so highly contaminated that all clean-up procedures were inadequate, the use of a NaOH reagent without citrate was investigated. Small changes in pH resulted from removing the citrate. Although some hydroxide precipitates, probably magnesium salt, were formed with marine water samples, these did not appear to interfere with analysis. There was no noticeable effect on the lifetime of the membrane, or in response between NaCl standards and standards made in seawater (Fig. 4). This result may arise from several factors. The membrane would prevent any of the precipitate from interfering with the detector response. The time from when the sample was mixed with hydroxide until it leaves the diffusion cell was short, less than 10 s. The acid in the carrier may help to remove any precipitate between sample injections.
 | ||
| Fig. 4 Comparison of standards made up in 35 g L−1 NaCl (×) and seawater (•). Standards were analysed without citrate in the hydroxide reagent. | ||
As it appeared the land-based laboratory had a contaminated atmosphere, a 150 L glove box (Mecaplex, Grenchen, Switzerland) was subsequently used for the storage of reagents and for the handling of samples. The glove box was flushed with N2 for 3–4 h before analysis at approximately 2 L min−1 and the N2 flow was maintained throughout analysis. Although handling of samples was more awkward, results improved with 14% coefficient of variation for a 100 nmol L−1 standard.
The glove box also allowed us to perform analyses at sea in general laboratories, where the airborne contamination may be high. For example, this method has been used on a Southern Ocean voyage in a laboratory that had been regularly cleaned with ammonia-based cleaners and where fish samples were normally processed. Using the glove box in this environment samples with ammonium concentrations below 50 nmol L−1 could be analysed (Fig. 5).
 | ||
| Fig. 5 Results from low concentration samples collected in December 1995 at 50S 145E in the Southern Ocean. | ||
The analysis of duplicates at sea showed that there was still potential for contamination through handling. Powder-free gloves were used when collecting or handling samples. Initially the polypropylene tubes were cleaned in 10% HCl but there was little difference between acid-cleaned and new tubes stored with Milli-Q and 35 g L−1 NaCl solution. If anything the acid-cleaned tubes were occasionally high, possibly owing to contamination from the additional handling during cleaning. Samples were collected in triplicate in single-use sample tubes. Using single-use tubes reduced the risk of contaminating the sample between repeat injections. Samples were injected inside the glove box because this helped to eliminate atmospheric contamination. There was little difference between samples analysed fresh compared to those stored frozen for up to 50 d.
Figures of merit
Ammonia concentrations were calculated using peak height data. Although the upper limit of linearity was not determined, ammonia standards were linear to at least 4 μmol L−1 as established by Fowlis–Scott nonlinear function and linearity plots.26,27 The correlation coefficient for the usual working range of standards, up to 1 μM, was R2 = ∼0.994 and the limit of detection was 7 nM, determined from three times the standard deviation of the analysis of six Mill-Q water blanks and six 35 g NaCl L−1 blanks. Precision, determined as the standard deviation of the analysis of 24 × 800 nmol L−1 standards, was 5.7% or 46 nmol L−1. The sample injection rate was 30 samples h−1.Conclusions
Through rigorous testing under actual operating conditions and using representative samples a method has been developed that is suitable for the trace level analysis of ammonium in seawater, and is free of salt error for measurements in estuarine waters. The method relies on OPA-sulfite chemistry combined with gas-diffusion to be selective for ammonium with little interference. The FIA system has been optimised using diffusion cell design, membrane materials, flow rates and temperature to maximise sensitivity, while maintaining high sample throughput and system robustness. High sensitivity requires stringent contamination control and we have recommended procedures for sample handling, reagent cleaning and enclosure of part of the instrument in a nitrogen atmosphere. The resultant FIA system is suitable for the difficult conditions often experienced with shipboard analysis.Acknowledgements
The authors thank Linda Kalnejais, Emily Hilder, Jacqueline Arthur and Perran Cook who contributed to testing of this method through CSIRO vacation scholarships and fieldwork. Andy Revill and Rhys Leeming for their useful comments on this manuscript. We are also grateful to Denis Mackey, John Parslow and the captains and crew of RV Franklin and RV Southern Surveyor for allowing us to participate on their voyages to test this method at sea. Thanks to the members of the Huon Estuary Study for their help with the collection of estuarine samples. The Huon Estuary Study was partially funded by the Fisheries Research and Development Corporation (project no. 96/284).References
- J. J. McCarthy and E. J. Carpenter, in Nitrogen in the marine environment, ed. D. G. Capone and E. J. Carpenter, Academic Press Inc., New York, 1983, pp. 487–512 Search PubMed.
- J. P. Zehr and B. B. Ward, Appl. Environ. Microbiol., 2002, 68, 1015 CrossRef CAS.
- K. Grasshoff and H. Johannsen, J. Cons. Int. Explor. Mer., 1972, 34, 516 Search PubMed.
- R. F. C. Mantoura and E. M. S. Woodward, Estuar. Coast. Shelf Sci., 1983, 17, 219 CAS.
- M. A. Brzezinski, Mar. Chem., 1987, 20, 277 CrossRef CAS.
- H. Verdouw, C. J. A. van Echteld and E. M. J. Dekkers, Water Res., 1978, 12, 399 CrossRef CAS.
- J. Kanda, Water Res., 1995, 29, 2746 CrossRef CAS.
- C. J. Patton and S. R. Crouch, Anal. Chem., 1977, 49, 464 CrossRef CAS.
- A. Aminot, D. S. Kirkwood and R. Kérouel, Mar. Chem., 1997, 56, 59 CrossRef.
- M. Roth, Anal. Chem., 1971, 43, 880 CrossRef CAS.
- P. Lindroth and K. Mopper, Anal. Chem., 1979, 51, 1667 CrossRef CAS.
- N. Jørgensen and R. E. Jensen, Mar. Chem., 1997, 57, 287 CrossRef.
- I. Molnár-Perl and A. Vasanits, J. Chromatogr., A, 1999, 835, 73 CrossRef CAS.
- S. S. Simons and D. F. Johnson, J. Org. Chem., 1978, 43, 2886 CrossRef CAS.
- A. Ríos, M. D. Luque de Castro and M. Valcarcel, Anal. Chim. Acta, 1986, 187, 139 CrossRef CAS.
- S. S. Goyal, D. W. Rains and R. C. Huffaker, Anal. Chem., 1988, 60, 175 CrossRef CAS.
- T. Aoki, S. Uemura and M. Munemori, Anal. Chem., 1983, 55, 1620 CAS.
- R. D. Jones, Limnol. Oceanogr., 1991, 36, 814 CAS.
- K. Mopper and D. Delmas, Anal. Chem., 1984, 56, 2557 CrossRef CAS.
- Z. Genfa and P. K. Dasgupta, Anal. Chem., 1989, 61, 408 CrossRef CAS.
- R. Kérouel and A. Aminot, Mar. Chem., 1997, 57, 265 CrossRef.
- R. M. Holmes, A. Aminot, R. Kerouel, B. A. Hooker and B. J. Peterson, Can. J. Fish. Aquat. Sci., 1999, 56, 1801 CrossRef CAS.
- A. Aminot, R. Kerouel and D. Birot, Water Res., 2001, 35, 1777 CrossRef CAS.
- R. Nakata, T. Kawamura, H. Sakashita and A. Nitta, Anal. Chim. Acta, 1988, 208, 81 CrossRef CAS.
- P. Cook, personal communication.
- R. Cassidy and M. Janoski, LC-GC, 1992, 10, 692 CAS.
- C. A. Dorschel, J. L. Ekmanis, J. E. Oberholtzer, F. V. Warren and B. A. Bidlingmeyer, Anal. Chem., 1989, 61, 951A CAS.
| This journal is © The Royal Society of Chemistry 2005 |