Determination of pollution trends in an abandoned mining site by application of a multivariate statistical analysis to heavy metals fractionation using SM&T-SES†
G. Pérez and M. Valiente*
Centre GTS, Unitat de Química Analítica, Departament de Química, Universitat Autònoma de Barcelona, Facultat de Ciències, Edifici CN, 08193, Bellaterra, Barcelona, Spain. E-mail: Manuel.Valiente@.uab.es
First published on 3rd December 2004
Abstract
The mobility, availability and persistence of Heavy Metals (HMs), As, Cd, Cu, Ni, Pb and Zn, in contaminated soils of a former abandoned mining area were evaluated by means of a sequential extraction scheme (SES) and applying a multivariate statistical analysis to the obtained data. Chemical partitioning of HMs in each sample was determined in four fractions (acid-soluble, reducible, oxidable and residual) following the Standard Measurements and Testing (SM&T) SES, formerly BCR-SES. Statistical evaluation of results by pattern recognition techniques allowed identification of groups of samples with similar characteristics and observations of correlations between variables, determining the pollution trends and distribution of HMs within the studied area. Typical metal-fraction association and metal availability characteristics of heavy metals have been depicted. The obtained results indicate an urgent need to attenuate the hazard in that area posed by high concentrations of toxic metals, which exceed the limits specified by different European legislations on soil reclamation.
Introduction
Different pollution sources that have been topics of recent interest include improper waste dumping, incidental accumulation, agricultural chemicals, abandoned industrial activities and atmospheric fallout, among the most cited.1 In particular, for mining and industrial abandoned sites, prior to evaluating the recovery of the polluted area, an evaluation of the extent and distribution of contamination is required in order to identify the area to be treated and the type of treatment that should be considered based upon the observed pollution trends. In these sources, heavy metals, HMs, frequently are the main pollutants and their mobilization due to weathering of solid inorganic materials under exogenic conditions is favoured, leading to environmental chemical pollution.For risk assessment purposes, the HMs mobility and their related availability is of a primal importance since toxicity is directly related to such characteristics.2 Moreover, as is well known, pseudototal HMs content does not provide real information on available amounts of HMs and it represents the worst possible situation, overestimating the real hazard. Consequently, there is a need for a methodology able to provide information about reactivity or mobility of pollutants. In this sense, sequential extraction schemes (SES), became a commonly used evaluative and informative tool by providing details on the distribution or partitioning of HMs in soils and sediments, which is directly related to the prediction of their mobility.1
This methodology is based on the process known as fractionation,3 where a sequential series of selective extractant reagents with an increasing extractant power is employed. The goal of this procedure is to selectively dissolve or solubilise the different solid phases or mineralogical fractions.4–6 By this methodology, knowledge of how HMs partition among the various geochemical phases is obtained. Such knowledge allows for a better insight into the mechanisms of HMs retention and release involved in the process of migration and decontamination, thus providing an evaluation of availability, mobility or persistence.
Two decades ago, Tessier proposed a five-step SES, which is still widely used,7 often with modifications in order to fit better to the target sample.8,9 SES have been widely used to assess the mobile fraction of different HMs of environmental impact and to evaluate the HMs distribution between the different phases of a variety of samples such as industrially contaminated soils,10–19 river sediments,20–25 sewage sludge,26–30etc. The wide variety of SES and the related lack of comparability between results, led to the harmonisation of SES under the auspices of the former Community Bureau of Reference (BCR), now Standards Measurements and Testing (SM&T), producing a certified reference material for a three-step SES.31–33
The main drawbacks of SES have been identified as readsorption and redistribution of metals.34–37 Also, SES applications have been mostly limited to low contamination sites. However, SES are still very useful to identify trace element partitioning into the various solid phases of soil and to determine labile fractions of trace elements in a verifiable manner. On the other hand, SES data can provide additional valuable knowledge by a proper exploratory data analysis of the experimental information. For instance, a systematic correlation of the different fractionation data, normally absent, would help the process characterization of a particular contaminated area.
Taking into account the mentioned limitations, the present study has been addressed to reveal the potential of SES application to a highly polluted site in overcoming the indicated boundaries. In this context, the present investigation is concerned with the fractionation of the HMs As, Cd, Cu, Ni, Pb and Zn in soils of a ditch network system designed to confine, control and monitor flows of water at a former abandoned mining area at Salsigne (France). Although As has not been considered in the SM&T-SES reference materials and applications (except in a recent work),38 we have analysed the As content in the different fractions because it is the main toxic contaminant of the target soils. We are aware of the limitations of such results on As for a possible contribution to risk evaluation due to its particular chemical behaviour as an oxoanion. In this sense, values can be taken as the minimum mobility of this element under the given conditions. Furthermore, to best characterize the polluted site, a correlation of sample content was carried out by multivariate statistical analysis of SES data including latent factors responsible for the data set structure and apportioning of pollutant sources. A comparison of the obtained data with current regulation limits has been carried out and can be of use for risk assessment purposes.
Experimental
Sampling site
The polluted site of Salsigne is located in southern France, in the Orbiel river basin, 13 km north of Carcassone. Geologically, the site mainly includes accumulations of sulfide minerals containing various metals such as iron, copper, gold, as well as arsenic and bismuth. Large surfaces present a significant pollution problem due to improper waste storage, although it is difficult to complete a related inventory because important masses of waste are not visible. Pollution of the locations around the treatment installations and waste storage areas is very high and is also important under these installations due to the percolation of pollutants in the ground. Broader pollution has been driven by rainwater leaching of pollutants, which flow into the Orbiel River and to some of its effluents. Appropriate measurements of pollutants are essential to assess suitable actions and thus prevent serious harmful problems. Because of the frequent strong winds in this area, additional pollution is produced from dust emitted to the atmosphere by the old pyrometallurgical installations or take-offs from deposits of very fine residues of arsenic oxides. Currently, there are approximately 5000 tons of these wastes, but demolition of buildings around furnaces will probably uncover a few hundred additional tons. The total contaminated zone covers an estimated area of 40 hectares.Fig. 1 presents the locations of single sampling points within the studied area. The sampling points were selected in a random manner to cover both the whole ditches network, which displays varying degrees of pollution, and relevant locations within the polluted area. A total of 21 surface samples were collected. 11 samples were collected along the ditches network and 10 from polluted surfaces exposed to significant flow of water during rain events, close to ditches.
 | ||
| Fig. 1 Location of sampling points on the polluted site of Salsigne. Black lines represent the ditches network. | ||
Sample preparation and soil properties
Composite samples were made of 10 unit samples mixed together to provide a better representation of a selected surface by obtaining the composition average. Surface unit samples (10 cm) were collected with a trowel (after removing the top 2 cm layer in contact with the atmosphere). Soil samples were air dried and ground to below 100 μm grain size with a tungsten steel bite grinder, which was cleaned with inert material between samples. Such a particle size was selected to accomplish a similarity with the employed CRM (BCR 701) to assess the traceability of the applied SM&T-SES. Once prepared, samples were placed in plastic bags or polypropylene bottles and stored at 4 °C to prevent possible changes in metal fractionation. Soil characteristics, hereafter called edaphological parameters, such as pH (total and potential), conductivity and organic matter content, were determined according to the official methods of soil analysis envisaged by local governmental regulations (Junta de Residus, Generalitat de Catalunya).39 Total and potential soil pH were determined in a soil ∶ water (1.0 ∶ 2.5) and soil ∶ KCl 0.1 mol−1 (1.0 ∶ 2.5) ratio suspension, respectively, at room temperature. Organic matter was evaluated by loss on ignition at 600 °C after 4 h. Conductivity was evaluated after extracting a soil ∶ water (1 ∶ 10) ratio suspension for 2 h. Major components were determined by X-ray fluorescence spectrometry (XRF) using 56 geological international reference samples for calibration. Samples were diluted (1 ∶ 40) with lithium tetraborate and melted in a radio-frequency inductive oven to obtain 30 mm diameter pearls. Ranges and average values of sample characteristics are given in Table 1.| Range | Mean | |
|---|---|---|
| pH (potential/total) | 6.4–8.6/5.3–7.9 | 7.6/7.2 |
| Organic matter (%) | 0.01–0.53 | 0.22 |
| Conductivity/mS dm−1 | 74–1591 | 324 |
| Major components (%) | ||
| SiO2 | 69.3–28.2 | 47.5 |
| Al2O3 | 13.8–5.4 | 8.8 |
| Fe2O3 | 37.3–2.5 | 12.5 |
| CaO | 28.5–0.6 | 11.0 |
| MgO | 8.0–1.4 | 3.3 |
| K2O | 3.2–0.8 | 2.0 |
| MnO | 0.28–0.04 | 0.13 |
| TiO2 | 0.80–0.24 | 0.49 |
| P2O5 | 0.28–0.05 | 0.13 |
Procedure for sequential extraction
The applied SES corresponds to the Standard Measurements and Testing procedure (SM&T-SES), including the updated modifications to improve the reproducibility of results.40 The corresponding four fractions, including the recommended pseudototal digestion of the remaining residue (F4) after the three steps of the SM&T-SES, are described in Table 2. Microwave digestion treatment was applied to determine both pseudototal metal determination in the original samples and residual fraction (F4). For quality control, a mass balance was evaluated by comparison of the pseudototal metal content determined in the original samples with the sum of extracted metal percentages in the four steps. Prior to application to real samples, the SM&T-SES was validated by means of the BCR 701 reference material, obtaining a good traceability for corresponding HMs in each fraction as can be deduced from Table 3. After each SES step, the suspension was centrifuged and the supernatant separated from the solid phase by filtering through a 0.22-micron filter Millex-GS (Millipore, Ireland) to avoid the nebulizer fouling when using ICP-MS or ICP-OES. The resulting extracts were placed in polypropylene bottles and stored at 4 °C prior to analysis, except extracts from the second step, which were analysed immediately due to instability and degradation of the extracting reagent. All experiments were performed in triplicate, including the control samples for the vessel, reagent and procedural blanks. Relative standard deviations of the results were typically below 12%. Higher deviations were observed for some samples due to both very low concentrations of the measured metal and heterogeneity of soil samples.| Nominal target phase | Reagent and conditions | Comments |
|---|---|---|
| Exchangeable + acid- and water-soluble | Shaking for 16 h with 0.11 mol l−1 acetic acid | Weakly-bounded metals retained on soil surface by relatively weak electrostatic interactions, which can be released by changes in ionic composition, modifications of adsorption or desorption of metals on sediment constituents or affected by production or consumption of protons. Often considered representative of available amounts. |
| Reducible | Shaking for 16 h with 0.5 mol l−1 hydroxilammonium chloride, pH = 1.5 | Trace elements contained on iron and manganese (hydr)oxides are released, because of their thermodynamic instability under anoxic conditions and dissolution of metal-oxide phases under controlled Eh and pH conditions. |
| Oxidable | Digestion with hydrogen peroxide at room temperature, evaporation, redigestion and evaporation, then shaking for 16 h with 1.0 mol l−1 ammonium acetate | Trace elements bounded to various forms of organic matter as biotic detritus, organic coatings on inorganic particles or living organisms. The degradation of organic matter under oxidizing conditions is responsible for releasing trace elements. |
| Residual | Digestion with Aqua Regia (ISO 11466 protocol) | Trace elements in the lattice of primary and secondary minerals. In this case possible changes in environmental conditions would have no effect on the release of metals from this fraction on a time-scale of several years. |
| F1 (Acid soluble fraction) | F2 (Reducible fraction) | F3 (Oxidable fraction) | (Pseudototal content) | |||||
|---|---|---|---|---|---|---|---|---|
| Determineda | Certified | Determined | Certified | Determined | Certified | Determined | Indicativeb | |
| a Results are expressed as the mean of four determinations ± standard deviation.b Indicative values obtained from BCR 701 certificate. | ||||||||
| Cd | 7.0 ± 1.1 | 7.3 ± 0.4 | 3.1 ± 0.2 | 3.8 ± 0.3 | 0.24 ± 0.04 | 0.27 ± 0.06 | 11.4 ± 0.8 | 11.7 ± 1.0 |
| Cu | 49 ± 3 | 49.3 ± 1.7 | 116 ± 4 | 124 ± 3 | 55 ± 7 | 55 ± 4 | 272 ± 5 | 275 ± 13 |
| Ni | 16.7 ± 0.5 | 15.4 ± 0.9 | 27.9 ± 0.7 | 26.6 ± 1.3 | 15.8 ± 0.8 | 15.3 ± 0.9 | 101 ± 2 | 103 ± 4 |
| Pb | 3.6 ± 0.4 | 3.2 ± 0.2 | 119 ± 3 | 126 ± 3 | 11.0 ± 1.5 | 9 ± 2 | 141 ± 3 | 143 ± 6 |
| Zn | 202 ± 7 | 205 ± 6 | 112 ± 2 | 114 ± 5 | 45 ± 2 | 46 ± 4 | 450 ± 8 | 454 ± 19 |
Apparatus and reagents
Major components were determined using a Philips PW2400 X-ray spectrophotometer with Rh excitation tubes and a Philips radio-frequency inductive oven (model PERL’X2, Holland). HMs were determined in both the three steps of SES and aqua regia digests, using a ARL minitorch inductively coupled plasma optical emission spectrometer (ICP-OES) (model 3410, Valencia, CA, USA). For trace elements below ICP-OES detection limits, a ThermoElemental inductively coupled plasma mass spectrometer (ICP-MS) (model PQExcell, Windsford, UK) was employed. Quantification of HMs was with respect to reagent-matched standard solutions, obtained by serial dilution of commercial stock solutions (Merck Darmstadt, Germany and J. T. Baker, North Kingstown, RI, USA). Multi-element standard solutions were used for ICP-OES and ICP-MS. Analytical grade reagents, supplied by Panreac, Barcelona, Spain, J. T. Baker, Phillipsburg, NJ, USA, or Merck, Darmstadt, Germany, were used throughout. All glassware and plastic containers were previously soaked overnight in 25% nitric acid and rinsed with double distilled water. Sample digestions for pseudototal determination were performed in perfluoroalcoxy (PFA) vessels, with a CEM Corporation microwave laboratory unit (CEM Mars X, Mathews, NC, USA). Conventional sequential extraction was performed using a SBS end-over-end mechanical shaker (model ABT-4, Barcelona, Spain). Extracts were separated from solid residues using a Pacisa centrifuge (model C-5, Barcelona, Spain).Multivariate statistical analysis
Chemometric techniques for pattern recognition were applied to the analytical SES data obtained from the 21 samples analysed.41 The combined function of scores and loadings identifies both the groups of samples with similar behaviour and the existing correlations between the original variables. The corresponding data matrix includes the six metal concentrations in each of the four fractions plus the edaphological parameters of samples. Data was processed by applying Principal Component Analysis (PCA) as well as Hierarchical Cluster Analysis (HCA) in order to gain knowledge on the distribution of pollutants by detecting similarities and differences between the data. Factor analysis was performed by evaluation of principal components and computing the eigenvectors higher than 1 (Kaiser Criterion). Afterwards, the rotation of principal components was carried out by Varimax normalized algorithm which allows an easier interpretation of the principal component by both maximizing the variance of the extracted factors and reducing uncertainties that accompany initial unrotated factor loadings. Varimax Rotation of the matrix was applied after factor analysis, using those principal components that contribute more than 5% of the total variance of the data set. In addition, HCA was applied using Ward’s method of agglomeration and squared Euclidean distance as the measurement of similarity. Correlation between parameters, PCA and HCA were applied using SPSS v10.0 and XlStat v5.2 statistical computer programs.Results and discussion
Five metals, Cd, Cu, Ni, Pb and Zn and the metalloid As, were determined in the different soil samples extracts, both those obtained from the SES application and those resulting from digestion procedures. Corresponding concentration values related to dry matter weight are statistically summarized in Table 4 and the whole dataset is shown in Table ESI-1.† The data obtained correspond to the areas close to the ditches network of the mining site, so the pollution assessment will be limited to this area.| F1 (Acid soluble fraction) | F2 (Reducible fraction) | F3 (Oxidable fraction) | (Pseudototal content) | |
|---|---|---|---|---|
| a First value represents the arithmetic average of 21 samples, second value represent the median of 21 values.b Minimum and maximum values, respectively. | ||||
| As | 384a, 26 (0.8–3840)b | 2899, 1211 (40–13873) | 344, 228 (96–837) | 11333, 5820 (643–53104) |
| Cd | 5.6, 1.9 (0.2–45) | 6.6, 3.3 (0.01–48) | 1.7, 0.9 (0.01–12) | 38, 29 (2–145) |
| Cu | 616, 87 (0.01–3706) | 449, 152 (0.01–2715) | 509, 230 (23–2044) | 2345, 1134 (80–9790) |
| Ni | 12, 9.6 (1.1–47) | 16, 8.6 (0.01–68) | 25, 25 (10–46) | 86, 55 (21–522) |
| Pb | 20, 2.6 (0.01–338) | 246, 118 (0.01–1362) | 49, 26 (2.9–255) | 730, 564 (69–3566) |
| Zn | 414, 103 (0.01–3779) | 344, 195 (6.6–2888) | 230, 111 (0.01–2469) | 1678, 769 (69–15639) |
A basic consideration refers to the background level of metals in soils as a result of natural phenomena,42 such as the contribution of parent material, common anthropogenic activities, agriculture, traffic, etc. Then, pollution is confirmed when metal concentrations are higher than typical values for soils found in the literature and exceed the levels present in nearby areas. To this extent, the results of pseudototal content were compared with maximum acceptable concentrations in soils, reported for reclamation of contaminated sites following the Dutch intervention values43 as well as the French legislation44 values given in Table 5. On the other hand, it is known that high pollutant concentrations accompanied by high standard deviations suggest anthropogenic sources, while homogeneous distribution across the site and therefore lower standard deviations, indicate a major natural source or lithogenic character.45
| Range | Common valuesa | Earth’s crust | Dutch legislation | French legislation | |||
|---|---|---|---|---|---|---|---|
| Target values | Intervention values | Sensitive use | Non-sensitive use | ||||
| a Values for agricultural soils. | |||||||
| As | 29 | 55 | 37 | 120 | |||
| Cd | 0.01–2.0 | 0.2–1 | 0.15 | 0.8 | 12 | 20 | 60 |
| Cu | 2–250 | 20–30 | 70 | 36 | 190 | — | — |
| Ni | 2–750 | 50 | 80 | 35 | 210 | 140 | 900 |
| Pb | 2–300 | 10–30 (rural) | 16 | 85 | 530 | 400 | 2000 |
| 30–100 (urban) | |||||||
| Zn | 1–900 | 50 | 220 | 140 | 720 | 9000 | — |
In the case of the Salsigne site, all samples exceed the Dutch intervention values for As, Cd and Cu (except sample 24), for Ni only sample 9, Pb (except samples 7, 8, 13, 19, 23, 24) and Zn (except samples 4–8, 13, 16, 19, 24), indicating that the pollution has anthropogenic sources. Although high levels of heavy metals have been reported in sedimentation areas of contaminated soils in the vicinity of metalworking factories and mining areas, Salsigne values are exceptionally high, so, the site can be considered as “critically polluted”. In particular, while As is the major pollutant, Cu, Pb and Zn content also has to be considered to properly assess the pollution level at the site. In addition, when considering the data from the first step of the SES (mobile fraction), a considerable number of samples also surpass the mentioned Dutch levels.
Data transformation
Because of the high positive and negative skewness of some of the primary data, appropriate data transformation is needed for a suitable statistical treatment. Furthermore, possible outliers must also be identified because strongly skewed distributions and outliers can contribute to biased conclusions in statistical analyses. In our case, the obtained data are both skewed and sharply distributed. Skewness is due to particularly high values of As and Zn in some of the samples. On the other hand, kurtosis is observed because of data clustering at low values of target variables for the majority of samples. Such behaviour reveals a lack of normal distribution of the data. For this reason, a logarithmic transformation was applied that resulted in a general smaller skewness and kurtosis of most variables (except Ni in F2 and F4, Pb in F2 and Zn in F3). This transformation is widely applied in order to normalise positively skewed data sets.However, such a transformation of environmental data sets does not always follow lognormal distributions. In such cases, a different transformation is needed, and Box–Cox transformation is one of the most frequently used.46,47 This transformation is given by the equation listed below

Treatment of pseudototal metal content data
From the available dataset, after the application of PCA treatment to the pseudototal metal concentration transformed data, a significant correlation (above 0.7 for a 0.05 level of significance) is observed between some variables, being most significant for Ni and Zn or Cu and Pb. The contribution to total variance of the first three PCs is 48%, 20% and 12%, respectively (80% in all). Cu, Ni, Pb and Zn concentrations mainly describe the first component, while the second component depends strongly on pH and the third component increases with organic matter content, O.M.%. The combined plot of scores and loadings of PC1 vs. PC2 shown in Fig. 2(a) observes that PC1 accounts for the pollution of the site and classifies samples with similar pollutant content in two different groups and two outlier samples (S9 and S15) at the opposite extremes of the site. Within the first group, samples S4, S6–8, S13, S21–24 are included. Their common characteristics are concerned with a low level of contamination and their location on the site at the beginning of ditches. Taking into account the pseudototal concentration, these samples could be considered as representative samples of the lithogenic background. The other group of samples containing S1–3, S5, S10–12, S16–19, are those samples located at the intermedious and final segments of the ditches network, the high Cd, Cu, Ni, Pb and Zn content being the main variables contributing to PC1 (see Fig. 2(a)). These samples are related to the continuous input of HMs to the catchment areas associated with these sampling points. In this area, close to the warehouse and storages neighbourhood is where the continuous erosion and deposition of waste have led to the accumulation of such a huge amount of heavy metals. Samples S9 and S15 could be catalogued as outliers in Fig. 2(b) due to a high content of As (improper storage of AsO3 waste), and high amounts of Zn (because of an enriched Zn-slag treatment area), respectively. | ||
| Fig. 2 (a) Combined plot of scores and loadings for the released pseudototal amount of metals in the digestion of soils after Varimax rotation. (b) Dendogram obtained by cluster analysis of pseudototal metal concentrations. | ||
Statistical data processing (SM&T-SES fraction contents)
For the SM&T-SES first fraction, PCA is able to extract three PCs that contribute to 79% of the total variance; PC1 (42%) is dominated by HMs contribution, while PC2 (21%) will explain pH and conductivity behaviour and PC3 (13%) is loaded with O.M.%. From the selected plot of Fig. 3(a), where PC1 vs. PC2 are represented, the available content of samples increases with PC1 being S1, 2, 9, 10; those samples that represent the most hazardous area at the end of the ditches network. Less polluted samples, in terms of HMs release under acidic conditions, S13, 15, 23, 24, are inversely correlated to this PC but in a more widely spread distribution. The rest of the samples with an intermediate available HMs content lie between previously indicated groups. Results from the dendogram in Fig. 3(b) confirm the observed three groups differentiated by the available content when a 20% dissimilarity is selected. | ||
| Fig. 3 (a) Combined plot of scores and loadings for the extracted amounts of metals in the first fraction of SM&T-SES after Varimax rotation. (b) Dendogram obtained by cluster analysis of the extracted metals in the first fraction of SM&T-SES. | ||
For the second fraction of SM&T-SES, again three PCs contribute to a total variance of 71%. In this case, the loading of the extracted PCs is different from that obtained when considering the first fraction content. PC1 (47%) is mainly related to all HMs except Zn which loads PC2 together with pH (12%) and finally O.M.% in PC3 (11%). Accordingly, by plotting PC1 vs. PC2, the discrimination depending on the amount of HMs available under reducing environmental conditions and their distribution on the site can be interpreted similarly to the distribution of pseudototal and first fraction content. However, samples (e.g. S1) will be classified in different groups because of the selected fraction. Samples with a high available amount under reducing conditions show a positive score (S2, 3, 9, 10 and 17). Within the low level polluted samples, widely distributed along PC1 with negative scores, and considering the obtained dendogram in Fig. 4(b), two subgroups (S13, 15 and S7, 8, 23, 24) can be observed because of the high differences in available Zn. Specifically, S15 owes its high Zn content to enriched Zn-slag deposits. In this respect, S13 receives the influence of the S15 area, while the S7, S8, S23 and S24 area observes a minor influence of mentioned deposits due to natural diffusion barriers. Classification of the rest of the samples, having intermediate concentrations available, is less clear since target parameters of pollution have no specific differentiation (i.e. variables contributing to PC1 are not relevant enough to show a similar discrimination to that observed in high and low level polluted samples).
 | ||
| Fig. 4 (a) Combined plot of scores and loadings for the extracted amounts of metals in the second fraction of SM&T-SES after Varimax rotation. (b) Dendogram obtained by cluster analysis of the extracted metals in the second fraction of SM&T-SES. | ||
In the third fraction of SM&T-SES, discrimination between samples is more difficult that in previous fractions, which may be linked to the low-extracted amounts of HMs, now representing the available amount of HMs under oxidizing conditions (bound to O.M.). Four components are extracted contributing to a total variance of 78%, with PC1 (32%) having high loadings of all HMs except As while O.M.% dominates PC2 (21%), pH mainly contributes to PC3 (13%) and As loads PC4 (11%). The correlations among samples, observing the plot of PC1 vs. PC2 in Fig. 5(a) and dendogram in Fig. 5(b), have to be understood in terms of pH, Cu and Zn content, with samples S1, 2, 9, 10, 12, 20, 21 being those with a high amount of available HMs under oxidizing conditions. The rest of the samples present lower available amounts, with S4, S8 and S19 being less polluted. Intermediate polluted samples are grouped into two clusters at each side of highly polluted samples and are differentiated by their common pH. Despite a few differences in some samples, a similar spatial distribution is observed as those previously extracted in the other fractions when considering the available amount of HMs under oxidizing conditions.
 | ||
| Fig. 5 (a) Combined plot of scores and loadings for the extracted amounts of metals in the third fraction of SM&T-SES after Varimax rotation. (b) Dendogram obtained by cluster analysis of the extracted metals in the third fraction of SM&T-SES. | ||
Interpretation of sequential extraction results
From the statistical analysis and considering the anthropogenically-induced pollution, there are three main groups of samples that can be clearly differentiated by metal mobility, which belong to different segments of the ditch network. In general terms an increase in the available content is observed when the pseudototal content increases.48 A representative average value of the fractionation pattern for each element in each group is shown in Fig. 6 which illustrates the specific mentioned differences. Also, significant correlations between HMs fraction contents and major matrix components were determined using the non-parametric Spearman correlation coefficient (rs), which is based on ranks, is insensitive to outliers and does not requires data normality.49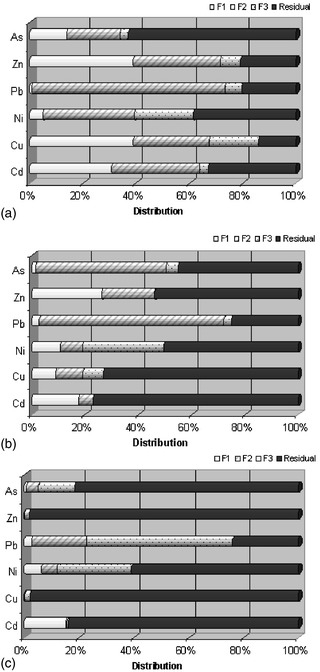 | ||
| Fig. 6 Representative fractionation patterns of HMs resulting from the statistical analysis of the whole data set. (a) Group I, (b) Group II and (c) Group III. | ||
A common observed trend in the highly polluted samples (final segment of ditches network) that we classify as group I, Fig. 6(a), includes a high availability of Cd and Zn, indicated by the data of the first fraction content. The available Cu amount also has to be remarked upon. Although this element is frequently associated with the oxidable fraction,50 the observed Cu in the first fraction may result from the overloading of possible organic binding sites and the characteristics of anthropogenic surface pollution which displays less adsorption and is preferably linked by weak interactions of contaminants with phase components. It is also interesting to note that almost 70% of the pseudototal Pb amount could be released under reducing conditions, F2, representing a high exposure risk. It is in these samples that the preferred association of Pb with iron and manganese oxides is well illustrated.51 However, the reducible Pb contents are significantly anticorrelated (α = 0.05) with the MnO content (rs = −0.962). This value can be understood as an oversaturation of this component in the reducible fraction. The relatively low proportion of As in the first fraction (10%) may lead to a misinterpretation of the pollution impact, since the high As content actually means that the first fraction exceeds by a factor of forty the maximum level allowed for intervention purposes. Concerning As distribution, the major residual fraction can be as a result of both the lithogenic characteristics of the site and the ancient mining activity that provided a continuous input of arsenic to soils. For Ni distribution, most characteristic is the 40% release under reducing conditions that follows the trend of Pb. The samples classified within this group represent the areas surrounding the final segment of the ditches network; the most polluted area, due to both surfaces with the highest amount of disposed waste related to the warehouse area and the accumulation process from the rest of the ditches network.
In samples from the middle stretch of ditches, group II, Fig. 6(b), similar trends and associations as those previously commented on in highly polluted samples are depicted. However, a net reduction is observed in the degree of availability as well as an increase of HMs content in the residual fraction, indicating a general reduction in HMs mobility that can be due to a higher lithogenic nature of these samples. Accordingly, a remarkable reduction of Cu mobility and availability, against group I, is observed. Thus, the sum of the first three fractions does not exceed 25% of the pseudototal amount. The significant anticorrelation (α = 0.05) of the Cu oxidable content with SiO2 and Al2O3 (rs = −0.744 and −0.678, respectively) reveals the affinity of Cu to the residual fraction. On the other hand, Cd and Zn present the highest mobility by considering their first fraction content, that together with the absence of these metals in the oxidable fraction indicate the possible anthropogenic origin of these metals. Pb shows almost the same distribution as in highly polluted samples except a slight increase in the residual fraction. Therefore a considerable Pb hazard is still observed under reducing conditions. The residual Pb increase correlates significantly (α = 0.05) with SiO2, Al2O3 and Fe2O3 content (rs = −0.638, −0.689, 0.870, respectively), denoting the preferred association of Pb with the crystalline Fe2O3 component. Conversely, a clear modification of the fractionation pattern is observed for As distribution since the reducible fraction increases in this group. This fact is corroborated by a significant correlation (α = 0.05) of As reducible content with Fe2O3 content (rs = 0.863). It is important to note that a similar misinterpretation as in heavily polluted samples can be observed, in this case, for the second fraction content (almost 40% of the pseudototal amount is available). In this group, the Ni distribution along the oxidable fraction has increased, probably due to the Ni affinity for the less leacheable fractions (organic matter and residual matrix).
In group III, (samples of lowest pollution) Fig. 6(c), there is a net reduction of the mobility and a general major residual distribution of some elements such as Zn and Cu, in contrast with previous groups. These aspects indicate a main lithogenic origin of these elements, such as Zn occlusion into stable structures such as crystalline Fe2O3, rather than anthropogenic pollution. It is interesting to note that for Zn the released contents in every fraction are significantly correlated (α = 0.05) to Fe2O3 content (rs = 0.952 F1, 0.973 F2, 0.986 F3, 0.940 Residual). However, Cd and Ni distributions are quite similar to those present in the highly polluted samples from group II, although the pseudototal content of these samples is lower than group II samples, but still above intervention values. For Pb, the main association is observed with the oxidable fraction, which can be understood to be more of lithogenic rather than anthropogenic origin. This is corroborated by a significant correlation (α = 0.05) in the reducible fraction between Pb content and Fe2O3 and MnO (rs 0.855 for Fe2O3 and 0.864 for MnO). The residual phase also presents a significant correlation of Pb content with Fe2O3 (rs 0.966) suggesting the occlusion of this element within crystalline structures resistant to leaching conditions. Arsenic also presents a major residual content, and the corresponding concentrations are lower than in groups I and II. These observations are related to samples from the initial segment of the ditches, where catchment areas contributing to ditches are less polluted than in the final area. Also, the steeped topography of the area avoids the accumulation process, thus reducing the pollution observed in these samples.
Conclusions
Application of suitable statistical analysis to SES raw data of HMs concentration in mining polluted soils requires a previous Box–Cox transformation of strongly skewed data to effectively normalise such data and diminish the negative effect of outliers. Such analysis reveals similarities or differences among samples as well as correlations or anticorrelations between variables, illustrating the extension and distribution trends of HMs in the studied area. These polluted soils show a high level of As, Cu, Cd, Pb and Cu contamination, anthropogenically enhanced by mining activity. Along the studied areas surrounding the ditches network, it is possible to distinguish three groups of samples having a general increase of pollution that enhances the mobility and availability of the studied HMs. However, some areas at the middle of the ditches network can be identified as localised hot spots with an abnormal amount of a single element related to former waste and slag disposal areas.From partitioning data, it is possible to conclude that there are significant differences in the distribution of the studied HMs, with Cd and Zn being the most mobile HMs. Consequently, an easier exchange of these elements between the soil and the water column is expected, independently of sampling points. The main hazard of other HMs such as As and Pb is related to their availability under reducing conditions.
In spite of the limitations of sequential extraction procedures, the value of the information obtained by using the statistical analysis of data presented here can be appreciated in the risk assessment of such polluted sites as well as in addressing specific solutions for their appropriate remediation.
Acknowledgements
The authors thank IRH (France) for their help in collection, preparation and supplying samples from the Salsigne contaminated site. Financial support has been provided by DIMDESMOTOM European project (EVK1-CT-1999-00002). Universitat Autònoma of Barcelona is also acknowledged for providing a scholarship grant to G. Pérez.References
- A. V. Filgueiras, I. Lavilla and C. Bendicho, J. Environ. Monit., 2002, 4, 823 RSC.
- M. Kersten and U. Förstner, in Trace Element Speciation: Analytical Methods and Problems, ed. G. E. Battley, CRC Press, Boca Raton, FL, 1989, pp. 245–317 Search PubMed.
- D. M. Templeton, F. Ariese, R. Cornelis, L. G. Danielsson, H. Mutuau, H. P. Van Leeuwen and R. Lobinski, Pure Appl. Chem., 2000, 72(8), 1453 CrossRef CAS.
- A. K. Das, R. Chakraborty, M. L. Cervera and M. De La Guardia, Talanta, 1995, 42, 1007 CrossRef CAS.
- G. Rauret, Talanta, 1998, 46, 449 CrossRef CAS.
- F. M. G. Tack and N. G. Verloo, Int. J. Environ. Anal. Chem., 1995, 59, 225 CAS.
- A. Tessier, P. G. C. Campbell and M. Bisson, Anal. Chem., 1979, 51(7), 844 CrossRef CAS.
- G. Rauret, R. Rubio and J. F. López-Sánchez, Int. J. Environ. Anal. Chem., 1989, 36, 69 CAS.
- J. L. Gómez-Ariza, I. Giráldez, D. Sánchez-Rodas and E. Morales, Sci. Total Environ., 2000, 246, 271 CrossRef CAS.
- A. Barona, I. Aranguiz and A. Elías, Chemosphere, 1999, 39(11), 1911 CrossRef CAS.
- C. M. Davidson, A. L. Duncan, D. Littlejohn, A. M. Ure and A. L. Garden, Anal. Chim. Acta, 1998, 363, 45 CrossRef.
- L. Fanfani, P. Zuddas and A. Chessa, J. Geochem. Explor., 1997, 58, 241 CrossRef CAS.
- E. C. Fonseca and E. F. Da Silva, J. Geochem. Explor., 1998, 61, 203 CrossRef.
- C. Gommy, E. Perdrix, J. C. Galloo and R. Guillermo, Int. J. Environ. Anal. Chem., 1998, 72(1), 27 CAS.
- S. O. Kim and K. W. Kim, J. Hazard. Mater., 2001, B85, 195 CrossRef CAS.
- I. M. C. Lo and X. Y. Yang, Waste Manage., 1998, 18, 1 CrossRef CAS.
- I. Maiz, I. Arambarri, R. Garcia and E. Millan, Environ. Pollut., 2000, 110, 3 CrossRef CAS.
- J. E. Maskall and I. Thornton, Water, Air Soil Pollut., 1998, 108, 391 CrossRef CAS.
- J. Pichtel, K. Kuroiwa and H. T. Sawyerr, Environ. Pollut., 2000, 110, 171 CrossRef CAS.
- E. Galán, J. L. Gómez-Ariza, I. González, J. C. Fernández-Caliani, E. Morales and I. Giráldez, Appl. Geochem., 2003, 18(3), 409 CrossRef CAS.
- J. L. Gómez-Ariza, I. Giráldez, D. Sánchez-Rodas and E. Morales, Int. J. Environ. Anal. Chem., 1999, 75(1–2), 3 CAS.
- A. E. González, M. T. Rodríguez, J. C. J. Sánchez, A. J. F. Espinosa and F. R. Barragán, Water, Air Soil Pollut., 2000, 121, 11 CrossRef.
- L. Leleyter and J. L. Probst, Int. J. Environ. Anal. Chem., 1999, 73(2), 109 CAS.
- J. Morillo, J. Usero and I. Gracia, Int. J. Environ. Anal. Chem., 2002, 82(4), 245 CrossRef CAS.
- R. Van Ryssen, M. Leermakers and W. Baeyens, Environ. Sci. Policy, 1999, 2, 75 CrossRef CAS.
- E. A. Álvarez, M. C. Mochón, J. C. J. Sánchez and M. T. Rodríguez, Chemosphere, 2002, 47(7), 765 CrossRef CAS.
- I. Lavilla, B. Pérez-Cid and C. Bendicho, Anal. Chim. Acta, 1999, 381, 297 CrossRef CAS.
- J. Scanzar, R. Milacic, M. Strazar and O. Burica, Sci. Total Environ., 2000, 250, 9 CrossRef CAS.
- I. Walter and G. Cuevas, Sci. Total Environ., 1999, 226, 113 CrossRef CAS.
- A. A. Zorpas, T. Constantinides, A. G. Vlyssides, I. Haralambous and M. Loizidou, Bioresour. Technol., 2000, 72, 113 CrossRef CAS.
- Ph. Quevauviller, G. Rauret, H. Muntau, A. M. Ure, R. Rubio, J. F. López-Sánchez, H. D. Fiedler and B. Gierpink, Fresenius’ J. Anal. Chem., 1994, 349, 808 CrossRef CAS.
- Ph. Quevauviller, G. Rauret, J. F. López-Sánchez, R. Rubio, A. Ure and H. Muntau, Sci. Total Environ., 1997, 205, 223 CrossRef CAS.
- A. M. Ure, Ph. Quevauviller, H. Mutuau and B. Griepink, Int. J. Environ. Anal. Chem., 1993, 51, 135 CrossRef CAS.
- C. Kheboian and C. F. Bauer, Anal. Chem., 1987, 59, 1417 CrossRef.
- E. Tipping, N. B. Hetherington, J. Hilton, D. W. Thompson, E. Bowles and J. H. Taylor, Anal. Chem., 1985, 57, 1944 CrossRef CAS.
- S. Xiao-Quan and C. Bin, Anal. Chem., 1993, 65, 802 CrossRef.
- C. Whalley and A. Grant, Anal. Chim. Acta, 1994, 291, 287 CrossRef CAS.
- G. Bird, P. A. Brewer, M. G. Macklin, D. Balteanu, B. Driga, M. Serban and S. Zaharia, Appl. Geochem., 2003, 18, 1583 CrossRef CAS.
- Departament de Medi Ambient, Junta de Residus, Generalitat de Catalunya, Pautes d’analisis, Servei de Publicacions, Barcelona, Spain, 1998 Search PubMed.
- A. Sahuquillo, J. F. López-Sánchez, R. Rubio, G. Rauret, R. P. Thomas, C. M. Davidson and A. M. Ure, Anal. Chim. Acta, 1999, 382, 317 CrossRef CAS.
- J. W. Einax, H. W. Zwanziger and S. Geiβ, Chemometrics in Environmental Analysis, Wiley-VCH Verlag, Berlin, 1997, ch. 5 Search PubMed.
- H. J. M. Bowen, Environmental Chemistry of the Elements, Academic Press, New York, 1979 Search PubMed.
- D. C. Adriano, Trace Elements in the Terrestrial Environment. Biogeochemistry, Bioavailability and Risk of Metals, Springer Verlag, New York, 2001, ch. 6 Search PubMed.
- D. Darmendrail, The French approach to contaminated-land management, BRGM/RP-51098-FR, Bureau de Recherches Géologique et Minières, Orléans, France, 2001 Search PubMed.
- D. S. Manta, M. Angelone, A. Bellanca, R. Neri and M. Sprovieri, Sci. Total Environ., 2002, 300(1–3), 229 CrossRef CAS.
- C. S. Zhang and S. Zhang, GeoJournal, 1996, 40(1–2), 209.
- C. S. Zhang, O. Selinus and J. Schedin, Sci. Total Environ., 1998, 212, 217 CrossRef CAS.
- T. U. Aualiita and W. F. Pickering, Talanta, 1988, 35(7), 559 CrossRef.
- R. A. Sutherland, Environ. Forensics, 2001,(2), 265 CAS.
- Y. Yin, C. A. Impellitteri, S. J. You and H. E. Allen, Sci. Total Environ., 2002, 287, 107 CrossRef CAS.
- W. F. Pickering, Ore Geol. Rev., 1986, 1, 83 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: complete HMs fractionation dataset. See http://www.rsc.org/suppdata/em/b4/b411316k/ |
| This journal is © The Royal Society of Chemistry 2005 |