Fluorine in crystal engineering—“the little atom that could”
Katharina
Reichenbächer
,
Heike I.
Süss
and
Jürg
Hulliger
*
Department of Chemistry and Biochemistry, University of Berne, Freiestrasse 3, 3012 Berne, Switzerland. E-mail: juerg.hulliger@iac.unibe.ch; Fax: +41 31 631 3993; Tel: +41 31 631 4241
First published on 1st December 2004
Abstract
In the last decade interactions of fluorine substituents in a variety of organic compounds have gained interest in life science and solid state materials. This review provides knowledge on fluorine interactions classified into phenyl–perfluorophenyl-, C–F⋯H, F⋯F and C–F⋯πF interactions. Except for phenyl–perfluorophenyl stacking featuring a stabilising energy of about 30 kJ·mol−1, interactions involving fluorine are generally weak. Although, there is still no concise understanding of fluorine interactions, there are numerous examples showing the influence of weak synthons on chemical, physical and biological properties.
 Katharina Reichenbächer | Katharina Reichenbächer was born in 1979 in Räckelwitz, Germany. After working in the group of Professor E. Weber at the Technical University Bergakademie Freiberg, Germany, she received her Diploma in Chemistry in 2002 (Diploma thesis: perfluorophenoxy substituted triazines). She is currently a PhD student in chemistry at the University of Berne working on fluorinated inclusion compounds. |
 Heike I. Süss | Heike I. Süss studied chemistry at the Technical University Bergakademie Freiberg, Germany, attending an intermediate year at the University of Birmingham, Great Britain. Current research interests include trigonal host molecules and property directed crystallisation of channel forming host compounds, which is also the topic of her PhD thesis at the University of Berne. |
 Jürg Hulliger | Jürg Hulliger studied chemistry at ETH and graduated in physical chemistry. In 1984 he received a PhD from the University of Zurich. After a post-doctoral stay at the Pennsylvania State University he moved to the Institute of Solid State Physics at ETH to conduct research in crystal growth. In 1993 he took up his position as a full Professor of Chemistry at the University of Berne (Switzerland). His research fields are: the chemistry and physics of materials; crystal growth. In 2004 he became a Fellow of the Royal Society of Chemistry. |
1 Introduction
In the last decade fluoro compounds gained an increasing significance in the life sciences. Today, fluorinated chemicals enter the daily life of million of people in different ways: Drinking water, dental products, pharmaceutical products or crop protection agents. A few of these applications are summarised in a recent issue of Chimia,1 enclosing a collection of papers presented at the International Symposium of Bürgenstock “Fluorine in the Life Science” (2003).It is known that the replacement of hydrogen atoms by fluorine can heavily alter the physical and chemical properties of compounds, caused by its electronegativity, low polarisability and bond strength, even the electron density distribution of a perfluorinated phenyl ring is the inverse to that of the hydrogen analogues. Therefore, fluorine can have a great influence on inter- and intramolecular forces.
Fluorinated compounds tend to have similar biological activity to the hydrogen analogues, but are more resistant to metabolic degradation. For example, the organic host molecule cyclodextrin is known to increase the bioavailability of many pharmaceuticals by amphiphilic properties obtained through substitutions by perfluorinated groups.2
The present review provides an overview on fluorine interactions, mainly in the solid state, along with reports on some interesting examples of fluorinated compounds that have appeared in the last 10 years. Concerning different types of interactions we will classify the phenyl–perfluorophenyl-, C–F⋯H, F⋯F and C–F⋯πF interactions. Their influence on crystal structures will be discussed by a selected number of examples. Particularly, the differences in the structures between hydrogen components and corresponding fluorine analogues are highlighted.
2 Fluorine in general
Among all elements fluorine features the highest electronegativity value (EN) of 4. As a result of a strong polarisation, the C–F bond strength is the highest (∼480 kJ mol−1) in comparison to other carbon–halogen bonds.Moreover, the negative inductive effect (σI = 0.51) 3 and the positive mesomeric effect (σR = −0.34)3 are influential factors not only on the reactivity but also on intra- and intermolecular interactions of organic fluoro compounds. One impressive example is the electron density distribution of aromatics which shows the influence of the replacement of H by F on the reactivity on phenyl systems. A non-fluorinated aromatic shows a typical π-cloud, whereas a perfluorinated one features an inversed electron density distribution. Thus, the centre of the ring has a positive charge which is illustrated by calculations for stilbene and decafluorostilbene (Fig. 1).
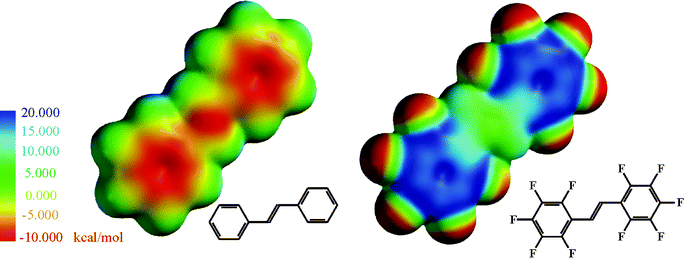 | ||
| Fig. 1 Electron density distribution of stilbene and decafluorostilbene calculated with PC Spartan. | ||
3 Phenyl–perfluorophenyl interactions
The first 1 ∶ 1 mixture of benzene/hexafluorobenzene (B/HFB) was reported by Patrick and Prosser4 in 1960. In contrast to benzene and hexafluorobenzene which crystallise in an edge-to-face (T-shape) structure (Fig. 2a and 2b), the co-crystals show a stacked structure with an alternate sequence of molecules (Fig. 2c). This kind of a stacking motif was observed for many other phenyl–perfluorophenyl complexes.5 Here, the crystal packing is dominated by columns with an alternate arrangement of the molecules, which show a centre to centre separation of 3.4–3.8 Å.6,7 Such stacking is supported by C–F⋯H-interactions between neighbouring rings.Noteworthy, phenyl–perfluorophenyl complexes show a higher melting point in comparison to single component compounds, due to the higher lattice energy of the co-crystals. This energy is a sum of coulombic, polarisation, dispersion and repulsion terms.8 The majority of calculations in the past were based on electrostatic interactions, as phenyl and perfluorophenyl groups show a large, permanent quadrupole moment with similar magnitude but of opposite sign.9 Recently, these methods were regarded as simplifications of the system. Latest calculations by the PIXEL method revealed that dispersion can have a much higher impact on phenyl–perfluorophenyl stacking than electrostatic interactions.8
The energy of the stacking interaction of the B/HFB dimer was estimated both experimentally and theoretically by different methods. The results are summarised in Table 1 showing significant deviations compared to experimental data. However, all theoretical predictions show an interaction energy of the hetero dimer that is 1.5 to 3 times larger than that of a homo dimer.10 In some cases the stabilising energy of the B/HFB dimer was compared with that of the 1,3,5-trifluorobenzene dimer, which shows a small quadrupole moment. The calculations emphasise a significant electrostatic interaction in the B/HFB complex. However, as an essential result of theory, measurements and X-ray analyses, dispersion interactions are as large as—sometimes even larger than (75–80%)—the electrostatic part.10
Cozzi et al. have analysed this kind of an interaction in fluorinated 1,8-diphenylnaphthalines.13 Here, the barrier of rotation for phenyl groups was determined in dependence of an increasing number of fluoro substituents. The repulsion between two stacked phenyl rings resulted in a low barrier of rotation. However, on increasing the fluorination on one phenyl ring the barrier of rotation increased by 3 kJ·mol−1 per fluorine atom independent of the position of the substituent. These results suggest stronger interactions for the stacked phenyl- and perfluorophenyl rings. These facts were confirmed by crystal structures of fluorinated phenylalanines:14 Two crystallographic independent molecules are linked by N–H⋯O interactions, whereby the phenyl groups interact by edge-to-face and displaced face-to-face contacts. However, the fluorination of phenyl substituents lead to an increase of the displaced face-to-face contacts and a declining of the edge-to-face interactions. This arrangement is supported by C–H⋯F interactions between neighbouring fluorinated phenyl rings.
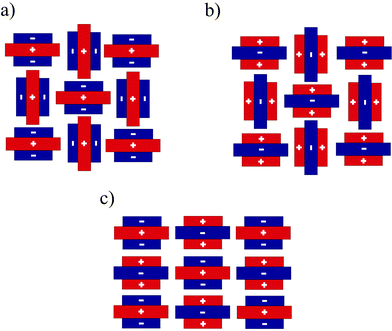 | ||
| Fig. 2 Interaction in the crystal of a) benzene; b) hexafluorobenzene and c) stacking interactions in the crystal of benzene/hexafluorobenzene (B/HFB). | ||
Many examples assume that phenyl–perfluorophenyl stacking interactions are the basis for a number of biological and chemical processes: For illustration, the photocycloaddition and photopolymerisation promoted by stacking interactions may be considered.5 An important condition for photochemical [2+2]cycloaddition reactions is the arrangement of the olefins in a parallel fashion with a centre to centre distances of about 3.5–4.2 Å. Phenyl–perfluorophenyl stacking interactions with centre to centre separations of 3.4–3.8 Å provide appropriate conditions for a cycloaddition. A first example was the reaction between trans-stilbene and trans-decafluorostilbene (Fig. 3a). Here, the structure of the co-crystals resulted in an alternative stacking of molecules with an olefin to olefin separation of about 3.7 Å. The photodimerisation of these molecules showed a yield larger than 98%. A further case is trans,trans-1,4-bis(2-phenylethenyl)-2,3,5,6-tetrafluorobenzene, crystallising also in a stacked structure (olefin distance of 3.8 Å, Fig. 3b). Here, the polymerisation yielded only 15% of the product. One reason for this may be the distance between the reactive centre and the next monomer unit, which becomes larger during the polymerisation.
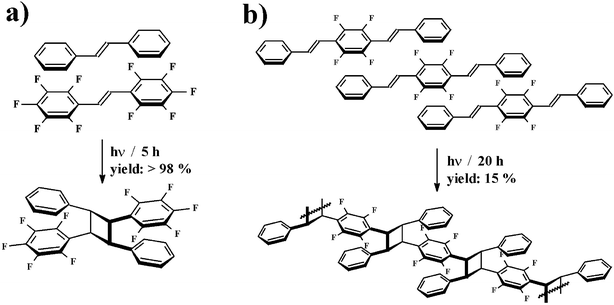 | ||
| Fig. 3 Photocycloaddition: a) stilbene-decafluorostilbene; b) trans,trans-1,4-bis(2-phenylethenyl)-2,3,5,6-tetrafluorobenzene.5 | ||
4 F⋯H interactions
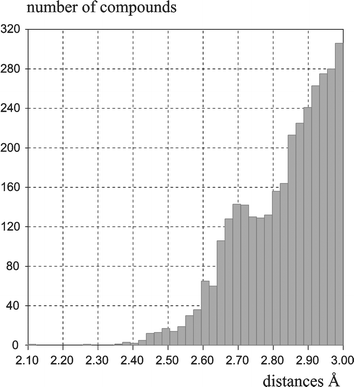
Hydrogen bonds are defined as electrostatic interactions between a hydrogen atom H and an electronegative atom A. According to Pauling's principle15 the strength of the hydrogen bond should increase with the increase of the EN value of the acceptor atom. In this sense, fluorine is predestined to form strong hydrogen interactions. In fact, F− is one of the best H-bond acceptors confirmed experimentally and theoretically.16 In contrast, the C–F group (“organic fluorine”) “… hardly ever accepts hydrogen bonds”17 forming only weak interactions compared to typical H-bond acceptors such as oxygen or nitrogen. Dunitz and Taylor17 suggested that a reason for the structural behaviour is found in the different energy of the competing orbitals that can be influenced by the electron delocalisation of the molecules.Database studies17–19 provided details for F⋯H interactions by applying different geometrical criteria. Here, we classify these interactions into F⋯H–C, F⋯H–N and F⋯H–O types. The sum of the van der Waals radii of fluorine and hydrogen is about 2.67 Å. Consequently, we consider distances up to 2.9 Å as F⋯H interactions. Distances (intra- and intermolecular) and angles (X–H⋯F) found for F⋯H contacts in the CSD are summarised in Fig. 4. However, these contacts are caused not only by hydrogen interactions but also by crystal packing.
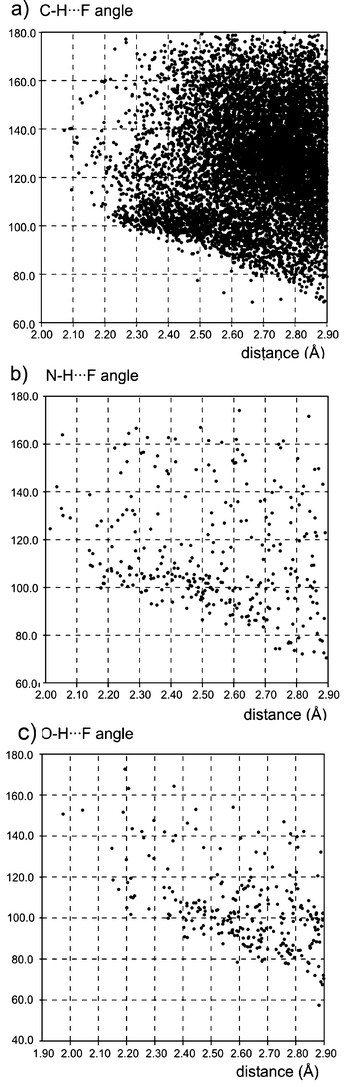 | ||
| Fig. 4 Distances (F⋯H) and angles (X–H⋯F): a) C–F⋯H–C (1475 compounds); b) C–F⋯H–N (151 compounds); c) C–F⋯H–O interactions (134 compounds) found in the CSD (Version: 5.25 (November 2003), using of Vista to create the diagrams; criteria: distances ⩽ 2.9 Å, R < 0.05, not disordered, no ions, only organics, inter- and intramolecular (min. 4 bonds between atoms) contacts). | ||
As expected, most F⋯H contacts were found for the C–H group as a donor. The number of compounds providing donors O–H and N–H is significantly lower, because oxygen and nitrogen act rather as competing acceptors than as hydrogen donors. C–H⋯F angles range from 70° to 180°. Such a wide range suggests weak interactions. Nevertheless, the sum of such weak interactions may have an influence on the structure and properties of organic fluorine compounds. Thalladi et al.20 have analysed the structure of different fluoro substituted aromatics showing supramolecular motifs (Fig. 5) which were similar to the well-known C–H⋯O and C–H⋯N synthons. Furthermore, he could demonstrate that “… H⋯F distances decrease systematically with increasing C–H-group acidity”. In the case of 1,3,5-trifluorobenzene an interaction energy of E = −4.5 kJ·mol−1 (d(H⋯F) = 2.6 Å)8 for H⋯F was calculated by the PIXEL method being essentially smaller than the energy of a O⋯H hydrogen bond (20–40 kJ·mol−1. Therefore, the possibility of a replacement of oxygen by fluorine in a hydrogen bond such as in an enzyme–substrate complex is rare. This was emphasised by a CSD-search by Howard et al.19 showing only a few examples where such a replacement was successful.
A comparison of calculated energies of F⋯H interactions involving aliphatic and aryl bound fluorine indicated that the aliphatic bound fluorine is a better donor. The reason for this behaviour was seen in the conjugation of the fluorine lone pairs with the π-orbitals.19 Thus, the ability to participate in hydrogen bonding decreases.
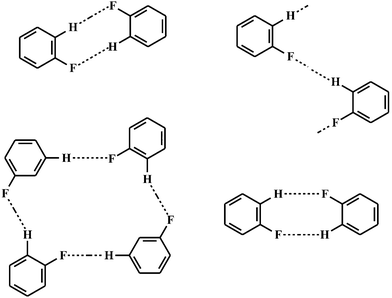 | ||
| Fig. 5 Synthons observed in different fluorobenzenes.20 | ||
F⋯H interactions are seen for example in the fluorinated carborane 9,12-bis(3,5-difluorophenyl)-o-carborane.21 Here, molecules are arranged in chains whereby each molecule is connected by four weak C–H⋯F interactions with two neighbouring molecules. These interactions were identified by IR showing two different C–H stretching frequencies.
The co-crystal of (E)-1,2-bis(4-pyridyl)ethylene and 1,4-diiodotetrafluorobenzene22 features also short F⋯H contacts. Here, molecules are arranged in linear chains due to I⋯N interactions (2.78 Å), whereby adjacent chains are connected by F⋯H hydrogen bonds featuring distances between 2.44 Å and 2.90 Å (Fig. 6). The N⋯I synthon in this lattice is found to be stronger than a typical N⋯H interaction, so that here the nitrogen does not compete for a hydrogen acceptor.
![Structure of the co-crystal (E)-1,2-bis(4-pyridyl)ethylene, 1,4-diiodotetrafluorobenzene (F⋯H interactions are emphasised by dashed lines [F1⋯H3 2.44 Å, F1⋯H5 2.90 Å, F2⋯H1 2.49 Å, F2⋯H6 2.74 Å]).17](/image/article/2005/CS/b406892k/b406892k-f6.gif) | ||
| Fig. 6 Structure of the co-crystal (E)-1,2-bis(4-pyridyl)ethylene, 1,4-diiodotetrafluorobenzene (F⋯H interactions are emphasised by dashed lines [F1⋯H3 2.44 Å, F1⋯H5 2.90 Å, F2⋯H1 2.49 Å, F2⋯H6 2.74 Å]).17 | ||
5 F⋯F interactions
The nature of F⋯F interactions is controversial. According to Pauling's principle15 fluorine has a low polarisability so that the attractive interatomic dispersion forces are rather low. In that sense, F⋯F interactions would be rarely observed and if present they would be weak.In 1986, Ramasubbu et al.23 mentioned that the type II of X⋯X interactions are formed through polarisation of halogen atoms, whereas type I is caused by close packing and does not form stabilising interactions (Fig. 7). This has received confirmation by many other reports comprising halogen–halogen interactions.24 For fluorine, 788 compounds could be found in the CSD with F⋯F distances up to 3.0 Å (Fig. 8) of which only 13 show F⋯F contacts of type II. However, not all of these 13 compounds build up stabilising interactions. Two of them, namely 4,5-(2,2′-difluoropropylenedithio)-1,3-dithiol-2-one25 and 2,3,5,6-tetrafluoro-trans-1,4-diethynylcylohexan-2,5-diene-1,4-diol,26 form short F⋯F contacts of type II with distances smaller than the van der Waals radii (2.94 Å). In both cases the close contacts appear to be driven by the general packing and not by dispersion forces between F atoms.
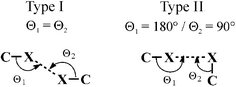
However, there are few compounds showing F⋯F interactions. These interactions could be identified by a combination of NMR and AIM.27 It was observed that experimentally obtained strong F–F coupling constants correlated with the calculated electron density of the bond critical point (ρbcp). The small values of ρbcp and its positive Laplacian suggest close-shell interactions.
One interesting example, the silver complex Ag3(ntb)2(CF3SO3)3, should be mentioned here.28 This complex revealed a three dimensional network {2[Ag3(ntb)2]3+(CF3SO3−)6}, stabilised by N–H⋯O and Ag⋯O interactions (Fig. 9a). A supramolecular (CF3SO3−)6 cluster is held together by twelve F⋯F interactions (Fig. 9b): Feq⋯Feq (2.788 Å) and Fax⋯Feq (2.822 Å).
 | ||
| Fig. 8 Distances of F⋯F interactions found in the CSD (sum of van der Waals radii is 2.94 Å) (CSD-search: Version: 5.25 (November 2003), using Vista to create the diagrams; criteria: distances ⩽ 3 Å, R < 0.05, not disordered, no ions, only organics, inter- and intramolecular (min. 4 bonds between atoms) contacts). | ||
 | ||
| Fig. 9 a) Crystal structure of the complex Ag3(ntb)2(CF3SO3)3; b) structure of the supramolecular (CF3SO3−)6 cluster hold together by twelve F⋯F interactions. (Reproduced with permission from ref. 28.) | ||
Summarising, we can emphasise that in contrast to the heavier halogens, fluorine is more involved in X⋯H than in X⋯X interactions.
6 C–F⋯πF interaction
In 2000, Prasanna and Guru Row29 published a database study showing the influence of C–F⋯π interactions on conformation and crystal packing. A number of compounds were discussed outlining the influence on crystal packing by perfluorinated molecules. However, in these cases there is no common π-cloud any more, because the electron density distribution of a perfluorinated ring is inversed as compared to that of a hydrogen-aromatic system. Therefore, these compounds form a C–F⋯πF interaction built up by a contact between the electronegative fluorine and the electropositive centre of a perfluorinated ring, which was also observed in hexafluorobenzene (Fig. 2b) and other stacked perfluorinated aromatics (section 7.1). Furthermore, the electrostatic repulsion between fluorine and the π-cloud of a non-fluorinated phenyl system leads to a destabilisation of the lattice, confirmed by the inclusion complexes of triphenylmethanol derivatives with methanol.30 Here, it was demonstrated that the temperature of the desorption of guests decreased with an increasing number of fluorine atoms on the host molecule caused by the electrostatic repulsion between fluorine and phenyl group and a loss of C–H⋯π interactions.7 Selected compounds
7.1 Changing the electron density distribution by replacing H by F and its influence on crystal structures
As mentioned previously, the electron density distribution of perfluorinated aromatics is inversed to that of the corresponding hydrogen containing ring. Therefore, a replacement of H by F can lead to changes in crystal structures and also in properties of compounds, which is described in the following for the case of dithiadiazolyl radicals and disubstituted benzenes.Most dithiadiazolyl radicals form a dimer in the solid state through out of plane spin-paired interactions between sulfur atoms.31 The dimerisation energy is about 35 kJ·mol−1. Fluorination of the aromatic ring can inhibit such sulfur–sulfur interactions. For example, the 4-(4-cyano-2,3,5,6-tetrafluorphenyl)-1,2-dithia-3,5-diazolyl radical31 supports the break up of the dimeric structure in the solid state. Two polymorphs of the compound were characterised (α- and β-phase) whereby only the β-phase showed a spontaneous magnetisation at 36 K. For the α- as well as the β-phase short sulfur⋯cyano contacts and some weak CN⋯F and S⋯F interactions, the shortest being 3.252 Å, were reported (Fig. 10).
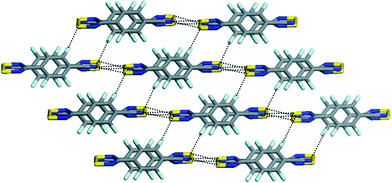 | ||
| Fig. 10 Stacking of the 4-(4-cyano-2,3,5,6-tetrafluorphenyl)-1,2-dithia-3,5-diazolyl radical (β-phase). Dashed lines show S⋯N interactions within sheets and S⋯F interactions between sheets.31 | ||
Further examples stem from disubstituted benzenes. Most known A–C6H4–D compounds (A: acceptor, D: donor) crystallise in a centric space group, because of lateral interactions favouring antiparallel alignment of dipoles. However, it was recently found for A–C6F4–D (A = CN; D = Cl, Br, I) that fluoro analogues preferably crystallise in acentric space groups.32 In contrast to the hydrogen derivates these structures are dominated by interlocking chains, whereby no significant directional interactions between adjacent chains could be found.
7.2 Fluorinated inclusion compounds
Up to now the number of perfluorinated host molecules is low. A few examples from different fields of chemistry will be mentioned here.The first fluorinated phthalcyanine, 29H,31H,1,4,8,11,15,18,22,25-octafluoro-2,3,9,10,16,17,23,24-octaperfluoro(isopropyl)phthalocyanine, F64PcH2 (Fig. 11a), revealed channels making up ∼39% of the unit cell volume (channel dimensions: 13.3 x 10.2 Å2).33 The channels are partly filled with acetone molecules. The crystal structure showed a large dome-like distortion of about 20° (angle between the opposite isoindole units), which might be favored by intermolecular interlocks of perfluoroisopropyl groups.
2,4,6-Tris(pentafluorophenoxy)-1,3,5-triazine (PFPOT)34 forms inclusions with p-xylene and p-chorotoluene with a host–guest ratio of 2 ∶ 1. Guest molecules are arranged in rectangular channels showing dimensions of about 7.6 × 3 Å2 (Fig. 11b). Perfluorophenyl stacking is the dominating supramolecular synthon in the crystal structure. As the channel wall is mainly made up by stacked perfluorophenyl moieties, the resulting host–guest interactions are phenyl–perfluorophenyl, H⋯O, F⋯H and C–H⋯πF contacts. The channel structure of the inclusion PFPOT·(p-xylene)0.5 collapsed under desorption of solvent molecules. However, channels are re-established by sorption from the gas phase.
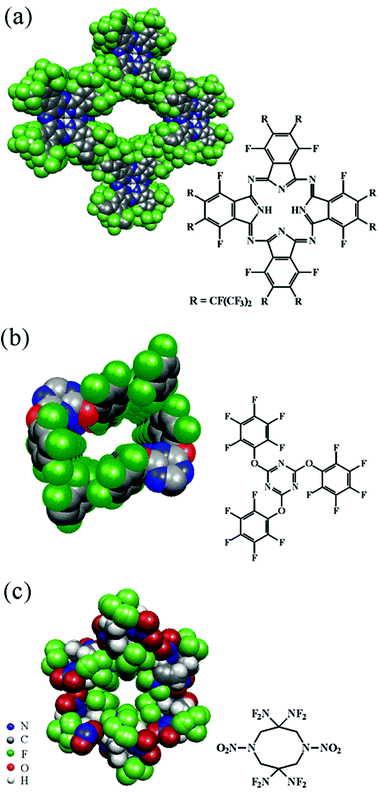 | ||
| Fig. 11 Channel structure of a) F64PcH2, b) PFPOT, c) HNFX. (Fig. 11b Reproduced with permission from ref. 34.) | ||
Another channel-type inclusion compound is 3,3,7,7-tetrakis(difluoramino)octahydro-1,5-dinitro-1,5-diazocine (HNFX) (Fig. 11c).35 Here, 4.6 Å wide channels are completely surrounded by fluorine atoms. There is evidence that solvent molecules in cavities are disordered. Desorption of guests by heating and vacuum led to empty channels. Although this material was not investigated further, it is probably an interesting case of an organic zeolite type material.
Some smaller fluorinated tunnels with dimensions of about 3–5 Å were constructed by double head trifluorolactates.36 These molecules form two dimensional waving sheets caused by a hydrogen bonding network. Here, the sheets seem to be connected by van der Waals interactions between carboxy groups.
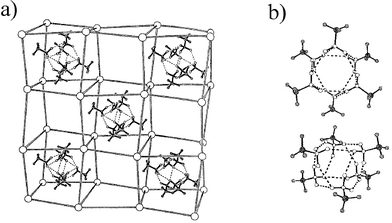
In the field of coordination chemistry, a number of attempts to prepare flexible networks which show no interpenetration have been undertaken so that a number of different guests can be included. In this respect perfluorinated molecules were synthesised, because of the weak intermolecular interactions between fluoro compounds. One example is the p-dipyridylmethyl substituted tetrafluorobenzene coordinated by cadmium, which show a chain, sheet or a diamond structure depending on the structure of included guests.37
7.3 Substituted isoquinolines
Choudhury et al. have discussed the crystal structures of some fluoro substituted isoquinolines.38 The influence of fluorine interactions, namely C–H⋯F, F⋯F and C–F⋯π, on packing are accessible to be studied as no other significant short interactions were recognised in these structures.The changing of the position of fluorine atoms leads to significant changes in the crystal structure. The authors stated that, provided there are no other strong intermolecular forces, the fluorine interactions allow the build up of supramolecular synthons. Occurrence of such interactions depends on the H/F ratio in e.g. crystalline aromatic azines, as shown by comparison of fluoro interactions in mono- and difluoro substituted isoquinolines. Furthermore, a comparison with bromine and chlorine substituted isoquinolines makes clear that the corresponding fluorine compounds form “… well-defined fluorine based interactions … ”.39
7.4 C–F⋯metal interactions
The carbon–fluorine covalent bond can act as a donor towards cations, giving rise to a number of cage complexes, studied by Takemura et al.40 With fluorine having a larger ionisation potential than oxygen or nitrogen this interaction may be characterised as a dipole interacting with a cation. Such complexes feature short C–F⋯cation distances and an elongation of the C–F bonds. Complexes with Li+, Na+, K+, Rb+, Cs+ and NH4+ were investigated. The shortest interatomic distances were found for potassium with F⋯K+ distances of 2.755 and 2.727 Å. Compared to the metal free cage compound (1.356 Å) the C–F bonds are slightly longer in the K+ (1.382 Å) and Cs+ complexes (1.369 Å) (Fig. 12).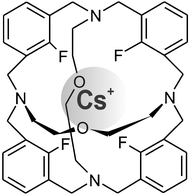 | ||
| Fig. 12 Tetrafluoro cage complex with Cs, F⋯Cs+ distances of 2.944 and 2.954 Å. | ||
To verify the ability of fluoro atoms to act as donor, a cage compound containing no oxygens providing a preorganised cavity of 6 inward looking C–F units was investigated.41 Here, K⋯F distances of 2.56 to 2.92 Å are among the shortest so far reported. A short C–F distance promotes a linear arrangement of the C–F⋯K group. Further evidence stems from the 19F NMR showing a characteristic high field shift.41
7.5 Macrocycles
A new class of planar, electron-deficient ligands was found in perfluorinated porphyrins.42 Here, the electron withdrawing effect of the fluoro atoms leads to a change in pKa. The nonbonding C⋯F distances of the pentafluoroporphyrin lie within the short range. Average contacts of the β-fluorine and the aryl ring carbon are 2.77 Å, whereas the sum of the van der Waals radi is 3.24 Å.For fluorinated calix[n]pyrroles an enhanced affinity to bind to anions was reported.43 The introduction of fluorine in the β-position of the pyrrole leads to a drastic increase of the receptor binding abilities compared to their non-fluorinated analogues. X-ray structure investigations revealed the presence of different conformations for the macrocycle showing close C–F⋯H–N interactions.
Furthermore, perfluoro crown ethers were found among macrocyles. The most interesting difference to their hydrogen analogues is the low melting point and an affinity to coordinate anions such as O2− or F−.44 It was found that the perfluoro crown ethers are non-toxic and biologically inert. These properties make medical applications conceivable, e.g. as oxygen carrying fluids. Perfluoro-15-crown-5 shows only one fluorine resonance and has potential as a 19F NMR imaging agent.44
7.6 Fluorinated fullerenes
The fluorine atom with its small size and high reactivity permits a variety of fluorine-substituted fullerenes to be prepared, reaching from C60F2 to even C60F60 in the gas phase.45 Because of a strong electron withdrawing effect, substitution by fluorine causes drastic changes in the physical and chemical properties of fullerene molecules. There is an increase in electron affinity by 0.05 eV per added fluorine, which gives fluorofullerenes a potential for enhanced acceptors.46In the case of C60F18, fluorination changed the shape of the fullerene ball considerably. The 18 fluorines are bound to one hemisphere of C60 and flatten its shape (Fig. 13).47 C60F18 molecules are highly ordered in the solid state (toluene solvate) due to attractive electrostatic interactions between polarised spheres. This also accounts for short intermolecular C–F⋯C contacts of 2.75 or 2.92 Å. For C60F48 single crystal complexes with different solvent molecules were investigated.48 Here, the repulsion of the fluorine atoms results in a deformation of the cage to generate concave surfaces. The neighbouring F⋯F distances are only 4.2 Å, shielding the double bonds. In fact, C60F48 is the highest fluorinated fullerene which could be isolated. Further fluorination led to a break up of the cage.
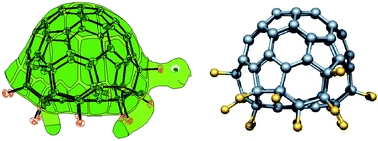 | ||
| Fig. 13 Tortoise-shaped C60F18. (Reproduced from ref. 47 by permission of Wiley-VCH.) | ||
8 Summary and conclusions
The interest in the field of crystal engineering, the prediction of crystal structures and the design of organic compounds with specific properties, has increased significantly in the last years. Especially the field of fluorine compounds is investigated more and more. Here, four main interactions are reviewed: phenyl–perfluorophenyl, C–F⋯H, F⋯F and C–F⋯πF. To date a great number of fluorine compounds have been synthesised to investigate these kind of interactions. Even so, the role of fluorine in crystal engineering is not yet clear in detail. This review provides the knowledge of fluorine interactions through a broad selection of examples from the literature. In spite of phenyl–perfluorophenyl stacking featuring a stabilising energy of about 30 kJ·mol−1, other observed contacts are generally weak. Therefore, the number of characterised fluorine interactions is relatively small as shown by the CSD data. Nevertheless, the sum of these weak interactions can have a significant influence on crystal structures and properties of organic fluoro compounds.The replacement of H by F (“the little atom that could”) has a great influence not only on crystal structures but also on chemical and physical properties. A few examples have illustrated the change of properties in regard to magnetism, inclusion properties or polarity formation.
In view of all developments and advances mentioned, there is still much work needed to understand structural, chemical, physical and biological properties of fluorine compounds for further applications in the life sciences and materials domain.
Acknowledgements
This work was supported by the Swiss NFP 47 program “Functional Supramolecular Materials” (project no. 4047-057476).References
- Chimia, 2004, vol. 58,p. 92 Search PubMed.
- S. Peroche and H. Parrot-Lopez, Tetrahedron Lett., 2003, 44, 241 CrossRef CAS.
- P. Politzer and J. W. Timberlake, J. Org. Chem., 1972, 37, 3557 CrossRef CAS.
- C. R. Patrick and G. S. Prosser, Nature, 1960, 187, 1021 CAS.
- G. W. Coates, A. R. Dunn, L. M. Henling, J. W. Ziller, E. B. Lobkovsky and R. H. Grubbs, J. Am. Chem. Soc., 1998, 120, 3641 CrossRef CAS.
- D. G. Naae, Acta Crystallogr., Sect. B, 1979, 35, 2765 CrossRef CAS.
- C. Dai, P. Nguyen, T. B. Marder, A. J. Scott, W. Clegg and C. Viney, Chem. Commun., 1999, 2493 RSC.
- J. D. Dunitz, A. Gavezzotti and W. B. Schweizer, Helv. Chem. Acta, 2003, 86, 4073 CrossRef CAS.
- P. West, J. R. S. Mecozzi and D. A. Dougherty, J. Phys. Org. Chem., 1997, 10, 347 CrossRef.
- A. Meyer, R. K. Castellano and F. Diederich, Angew. Chem. Int. Ed., 2003, 42, 4120 CrossRef.
- R. Lozman, R. J. Bushby and J. G. Vinter, J. Chem. Soc., Perkin Trans. 2, 2001, 1446 RSC.
- J. Hernández-Trujillo, F. Colmenares, G. Cuevas and M. Costas, Chem. Phys. Lett., 1997, 265, 503 CrossRef CAS.
- F. Cozzi, F. Ponzini, R. Annunziata, M. Cinquini and J. S. Siegel, Angew. Chem., Int. Ed. Engl., 1995, 34, 1019 CrossRef CAS.
- Y. In, S. Kishima, K. Minoura, T. Nose, Y. Shimohigashi and T. Ishiola, Chem. Pharm. Bull., 2003, 51, 1258 CrossRef CAS.
- L. C. Pauling, The Nature of the Chemical Bond, Cornell University, NY, 1960 Search PubMed.
- E. Espinosa, I. Alkorta, J. Elguero and E. Molius, J. Chem. Phys., 2002, 117, 5529 CrossRef CAS.
- D. Dunitz and R. Taylor, Chem. Eur. J., 1997, 3, 89 CrossRef CAS.
- L. Shimoni and J. P. Glusker, Struct. Chem., 1994, 5, 383 CAS.
- J. A. K. Howard, V. J. Hoy, D. O’Hagen and G. T. Smith, Tetrahedron, 1996, 52, 12613 CrossRef CAS.
- V. R. Thalladi, H.-C. Weiss, D. Bläser, R. Boese, A. Nangia and G. R. Desiraju, J. Am. Chem. Soc., 1998, 120, 8702 CrossRef CAS.
- H. Lee, C. B. Knobler and M. F. Hawthorne, Chem. Commun., 2000, 2485 RSC.
- R. Bianchi, A. Forni and T. Pilati, Chem. Eur. J., 2003, 9, 1631 CrossRef CAS.
- N. Ramasubbu, R. Parthasarathy and P. Murray-Rust, J. Am. Chem. Soc., 1986, 108, 4308 CrossRef CAS.
- V. R. Pedireddi, D. Shekhar Reddy, B. Satish Goud, D. C. Craig, A. D. Rae and G. R. Desiraju, J. Chem. Soc., Perkin Trans. 2, 1994, 2353 RSC.
- O. J. Dautel and M. Fourmijué, J. Org. Chem., 2000, 65, 6479 CrossRef CAS.
- N. N. L. Madjaci, G. R. Desiraju, C. Bilton, J. A. K. Howard and F. H. Allen, Acta Crystallogr., 2000, B56, 1063.
- I. Alkorta and J. Elguero, Struct. Chem., 2004, 15, 117 CrossRef CAS.
- C.-Y. Su, B.-S. Kang, Q.-G. Wang and T. C. W. Mak, J. Chem. Soc., Dalton Trans., 2000, 1831 RSC.
- M. D. Prasanna and T. N. Guru Row, Cryst. Eng., 2000, 3, 135 CrossRef CAS.
- N. Hayashi, T. Mori and K. Matsumoto, Chem. Commun., 1998, 1905 RSC.
- A. J. Banister, N. Bricklebank, I. Lavender, J. M. Rawson, C. I. Gregory, B. K. Tanner, W. Clegg, M. R. J. Elsegood and F. Palacio, Angew. Chem., Int. Ed. Engl., 1996, 35, 2533 CrossRef CAS.
- D. Bond, J. Griffiths, J. M. Rawson and J. Hulliger, Chem. Commun., 2001, 2488 RSC.
- H.-J. Lee, W. W. Brennessel, J. A. Lessing, W. W. Brucker, V. G. Young, Jr. and S. M. Gorun, Chem. Commun., 2003, 1576 RSC.
- K. Reichenbächer, H. I. Süss, H. Stöckli-Evans, S. Bracco, P. Sozzani, E. Weber and J. Hulliger, New J. Chem., 2004, 28, 393 RSC.
- R. D. Chapman, R. D. Gilardi, M. F. Welker and C. B. Kreuzberger, J. Org. Chem., 1999, 64, 960 CrossRef CAS.
- T. Katagiri, M. Duan, M. Mukae and K. Uneyama, J. Fluorine Chem., 2003, 120, 165 CrossRef CAS.
- K. Kasai, M. Aoyagi and M. Fujia, J. Am. Chem. Soc., 2000, 122, 2140 CrossRef CAS.
- R. Choudhury and T. N. Guru Row, Cryst. Growth Des., 2004, 4, 47 CrossRef.
- R. Choudhury, K. Nagarajan and T. N. Guru Row, Cryst. Eng., 2004, 6, 43 CrossRef.
- H. Takemura, N. Kon, M. Kotoku, S. Nakashima, K. Otsuka, M. Yasutake, T. Shinmyozu and T. Inazu, J. Org. Chem., 2001, 66, 2778 CrossRef CAS.
- H. Takemura, H. Kariyazono, M. Yasutake, N. Kon, K. Tani, K. Sako, T. Shinmyozu and T. Inazu, Eur. J. Org. Chem., 2000, 141 CrossRef CAS.
- E. K. Woller and S. G. DiMagno, J. Org. Chem., 1997, 62, 1588 CrossRef CAS.
- P. Anzenbacher, Jr., A. C. Try, H. Miyaji, K. Jursikova, V. M. Lynch, M. Marquez and J. L. Sessler, J. Am. Chem. Soc., 2000, 122, 10268 CrossRef.
- T.-Y. Lin, W.-H. Lin, W. D. Clark, R. J. Lagow, S. B. Larson, S. H. Simonsen, V. M. Lynch, J. S. Brodbelt, S. D. Maleknia and C.-C. Liou, J. Am. Chem. Soc., 1994, 116, 5172 CrossRef CAS.
- V. Boltalina, J. Fluorine Chem., 2000, 101, 273 CrossRef.
- R. Taylor, Chem. Eur. J., 2001, 7, 4075 CrossRef CAS.
- S. Neretin, K. A. Lyssenko, M. Y. Antipin, Y. L. Slovokhotov, O. V. Boltalina, P. A. Troshin, A. Y. Lukonin, L. N. Sidorov and R. Taylor, Angew. Chem. Int. Ed., 2000, 39, 3273 CrossRef CAS.
- S. I. Troyanov, P. A. Troshin, O. V. Boltalina, I. N. Ioffe, L. N. Sidorov and E. Kemnitz, Angew. Chem. Int. Ed., 2001, 40, 2285 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |