Materials for organic solar cells: the C60/π-conjugated oligomer approach
José L.
Segura
*a,
Nazario
Martín
*a and
Dirk M.
Guldi
*b
aDepartamento de Química Orgánica, Facultad de Química, Universidad Complutense, E-28040, Madrid, Spain. E-mail: segura@quim.ucm.es; nazmar@quim.ucm.es; Fax: +34 913944103; Tel: +34 913944227
bUniversität Erlangen, Institute for Physical Chemistry, 91058 Erlangen, Germany. E-mail: dirk.guldi@chemie.uni-erlangen.de
First published on 6th December 2004
Abstract
This tutorial review surveys recent advances in the field of C60/π-conjugated oligomer donor–acceptor ensembles. In particular, different synthetic strategies are discussed that were developed to link π-conjugated oligomers, as versatile photoexcited state electron donors, to C60. We highlight relationships between the nature/structural aspects of π-conjugated donor systems and a variety of physico-chemical features. Modifications of the oligomeric components are discussed under aspects of tailoring (i) the absorption cross-section of the chromophore in the visible region, (ii) the oxidation potential of the oligomeric donor moiety, (iii) the size, shape, or chemical makeup of the oligomer, and (iv) the stabilization of the charge-separated radical ion pairs. In the final section, the applicability of selected materials for the fabrication of photovoltaic devices is analyzed.
 José L. Segura José L. Segura | José L. Segura obtained his PhD in organic chemistry in 1994 at the Universidad Complutense de Madrid. After a short stay in the group of Professor W. Daily (University of Pennsylvania) working in the area of strained-ring organic syntheses, he was appointed as a postdoctorate in the group of Professor M. Hanack (University of Tübingen) working on the syntheses of electroluminescent conjugated polymers and in the group of Professor F. Wudl (University of California at Santa Barbara) working on the functionalization of fullerene C60. In 1995 Dr Segura joined the faculty at the Department of Organic Chemistry at the Universidad Complutense de Madrid where he is currently Associate Professor. His current interests involve the synthesis and investigation of the optoelectronic properties of π-conjugated materials. |
 Nazario Martín Nazario Martín | Nazario Martín studied chemistry at the Universidad Complutense de Madrid (UCM), where he obtained his doctorate in 1984. He worked as postdoctoral fellow (1987–1988) at the Institut für Organische Chemie der Universität Tübingen on electrically conducting organic materials. In 1994, he was a visiting Professor at the Institute for Polymers and Organic Solids (IPOS) at the University of California, Santa Barbara (UCSB) working on fullerenes. He is currently full professor of Organic Chemistry at the UCM. His research interests are electroactive molecules with emphasis on the chemistry of fullerenes, electron donor tetrathiafulvalenes (TTFs) and π-conjugated systems in the context of electron transfer processes and photovoltaic applications. He is currently a member of the International Advisory Editorial Board of The Journal of Materials Chemistry, Regional Editor for Europe for the journal Fullerenes and Carbon Nanostructures, and General Editor of the Spanish Royal Society of Chemistry. |
 Dirk M. Guldi Dirk M. Guldi | Dirk M. Guldi graduated from the University of Cologne (Germany) in 1988, where he received his PhD in 1990. In 1992, after a postdoctoral appointment at the National Institute of Standards and Technology, he took a research position at the Hahn-Meitner-Institute Berlin. After a brief stay as a Feodor-Lynen Stipend (Alexander von Humboldt Foundation) at Syracuse University, in 1995 he joined the faculty of the Notre Dame Radiation Laboratory where he was promoted to Associate Scientist in 1996. In 1999 he completed his Habilitation at the University of Leipzig (Germany). He was awarded the Heisenberg-Prize (1999; Deutsche Forschungsgemeinschaft), Grammaticakis-Neumann-Prize (2000; Swiss Society for Photochemistry and Photophysics) and JPP-Award (2004; Society of Porphyrins and Phthalocyanines). His primary research interests are in the areas of new multifunctional carbon-based nanostructures within the context of light-induced charge separation and solar-energy conversion |
Introduction
Since the early discovery of the photovoltaic effect by Becquerel, silicon-based solar cells represent the most efficient photovoltaic devices that help to transform solar light into electrical energy. More recently, a wide range of novel inorganic semiconducting materials, including GaAs and CdTe, have been tested as photo- and electroactive building blocks.1 The development of organic photovoltaic materials as potent alternatives to inorganics has, however, drawn much less attention.Shortly after the first practical applications were found for conjugated polymers, the basic structural and electronic properties of well-structured, monodisperse π-conjugated oligomers were surveyed to mimic and to understand the features of the related, but more complex and polydisperse, polymer analogs. In recent years, the study of monodisperse π-conjugated oligomers has advanced into its own, strong field. This advance is, for the most part, driven by the discovery of a remarkable optoelectronic phenomenon. At the forefront of current initiatives are the developments of new synthetic strategies that permit the synthesis of π-conjugated oligomeric materials with precise length and constitution.2
The advent of fullerenes and their production in multigram quantities at the beginning of the nineties launched a major effort to explore their outstanding electron acceptor features. In this light, blends of semiconducting π-conjugated organic polymers, including p-phenylenevinylenes (PPVs), polythiophenes (PTs), and C60, afford new types of plastic solar cells with comparatively moderate (i.e., relative to silicon-based solar cells), but nevertheless promising, energy transformation efficiencies.3 As far as the activity aspects of these composite materials are concerned, it is crucial to realize that electron transfer events between the electron donating polymers and the electron accepting fullerenes evolve with ultrafast dynamics and high quantum efficiencies. However, the tendency of C60 to phase separate from the polymer and to crystallize is surely one of the prime concerns: it imposes unrealistic measures for its solubility in conjugated polymer matrices.4
Thanks to the pioneering protocols regarding the general chemical functionalization of C60, virtually any functional group can be covalently linked to the highly reactive carbon framework en route to synthetic models of light harvesting arrays and reaction centers. To this end, we and others, we have designed, synthesized, and tested several donor–acceptor ensembles with a monodisperse π-conjugated oligomeric system as integrative photoexcited state electron donors. The major rationales for performing this work were: firstly, the investigation and characterization of the electron and energy transfer processes between photoexcited π-conjugated donor materials and the electron accepting C60. Secondly, the fine-tuning and optimization of the electron transfer efficiencies. Thirdly, the attainment of bicontinuous networks of covalently linked donor–acceptor systems. Finally, the incorporation of these materials into solar cells, but avoiding uncontrolled macrophasic separation processes.
In this review article we will present (i) design principles en route to molecular dyads and triads containing monodisperse π-conjugated chromophores covalently linked to or supramolecularly associated with C60, (ii) a systematic study of the relationship between the structure of the π-conjugated oligomeric system and some physico-chemical properties of the resulting donor–acceptor structures, and (iii) the application of some of these systems for fabricating photovoltaic devices.
Polymer/fullerene blends
For decades oligomers have been considered as model systems for acquiring a comprehensive understanding of optoelectronic properties in polymeric materials.2 Hereby, intense efforts were directed towards the development of solid-state photovoltaic cells that are based on composite films.5,6 Utilizing electron accepting C60 in combination with π-conjugated systems, as photoexcited state electron donors, offers several attractive features. In particular, C60, due to its low reorganization energy in electron transfer reactions, accelerates forward and decelerates back electron transfer, compared to planar electron acceptors. This, as a whole, is beneficial for achieving substantially stabilized charge-separated states in C60-based materials, the ultimate motivation in artificial electron transfer systems.Shortly after reporting the ultrafast photoinduced electron transfer in blends of conjugated polymers and C60, different medium-sized and well-defined π-conjugated oligomers (1–5, Fig. 1) were used in blends with C60. One of the central objectives was to assess the role of the π-conjugation length of the donor.7 In addition, the effects that the carbon- or heterocyclic nature of the monomers has on the electron transfer processes and the morphology of the blends were studied.8
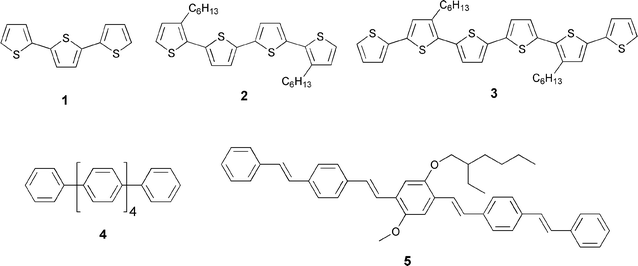 | ||
| Fig. 1 Some selected examples of conjugated oligomers used in blends with C60. | ||
The performance of such photovoltaic devices is based on intermolecular electron transfer events from the photoexcited polymer, as a sacrificial electron donor, to the electron accepting fullerene. A major shortcoming of this kind of device is the tendency, especially of pristine C60, to phase separate and subsequently to crystallize. This imposes important consequences on the solubility of C60 within a conjugated polymer matrix.
Molecular architectures containing monodisperse π-conjugated oligomeric donors linked to C60
Since uniformity and high quality of π-conjugated polymer/C60 thin films are essential requisites for optoelectronic devices, different synthetic strategies have been refined to restrain phase separation. Firstly, the design of soluble functionalized C60 derivatives was pursued with an objective to secure control over the morphology and the phase separation of the interpenetrating networks. In particular, homogeneous and stable blends, containing more than 80% weight of fullerenes, were fabricated by using functionalized fullerene derivatives rather than pristine fullerene materials. Secondly, conjugated polymer structures, bearing covalently linked fullerene moieties (i.e., in-chain versus on-chain) of a different complexity, emerged as viable alternatives for fabricating stable bicontinuous networks. More recently, the benefits of monodisperse π-conjugated oligomers, eminently easier to handle and to characterize, led to the development of oligomer-based donor–acceptor arrays.Among the suitable procedures for the functionalization of C60, the 1,3-dipolar cycloaddition reaction of azomethine ylides is one of the most versatile and widely applied methodologies.9 To date, a large number of fulleropyrrolidine derivatives bearing a variety of electro- and/or photoactive functionalities has been synthesized following this basic concept.10,11 This approach was applied simultaneously and independently by Janssen, Nierengarten, and our group to obtain triad 21b,12a dyad 12a,13 and triad 1814 respectively. Following these 1,3-dipolar cycloaddition conditions, that is, the reaction between sarcosine, C60, and the corresponding π-conjugated systems functionalized with one or two aldehyde groups, led to a variety of full-fledged dyads (6–9,1510,1611,1712,13,1813,1914,2015–16,21Scheme 1) and triads (17–20,14,2221,1222–24,2325,20Scheme 2).
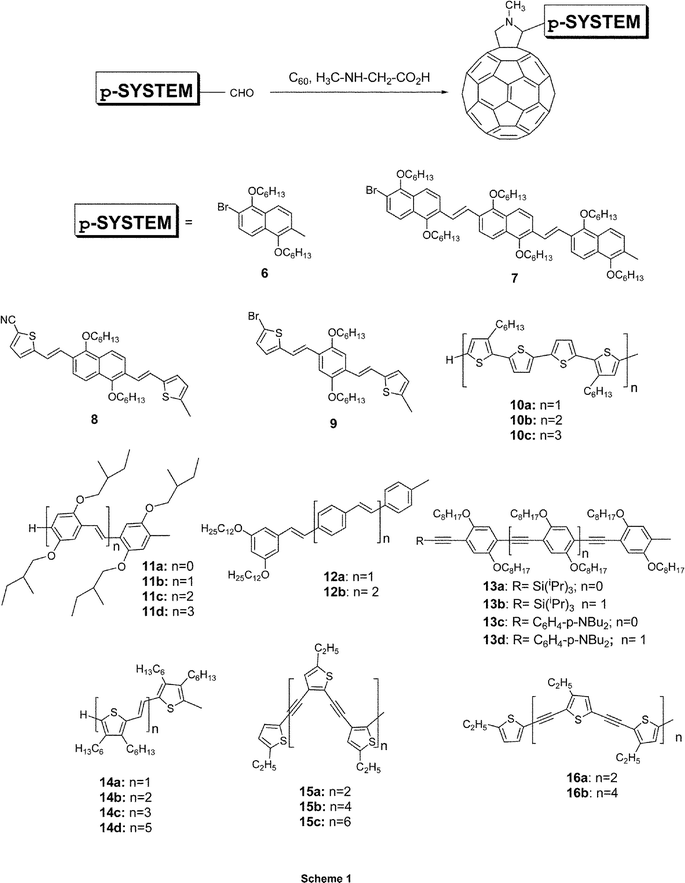 | ||
| Scheme 1 Structure of representative π-conjugated oligomer–C60 dyad materials. | ||
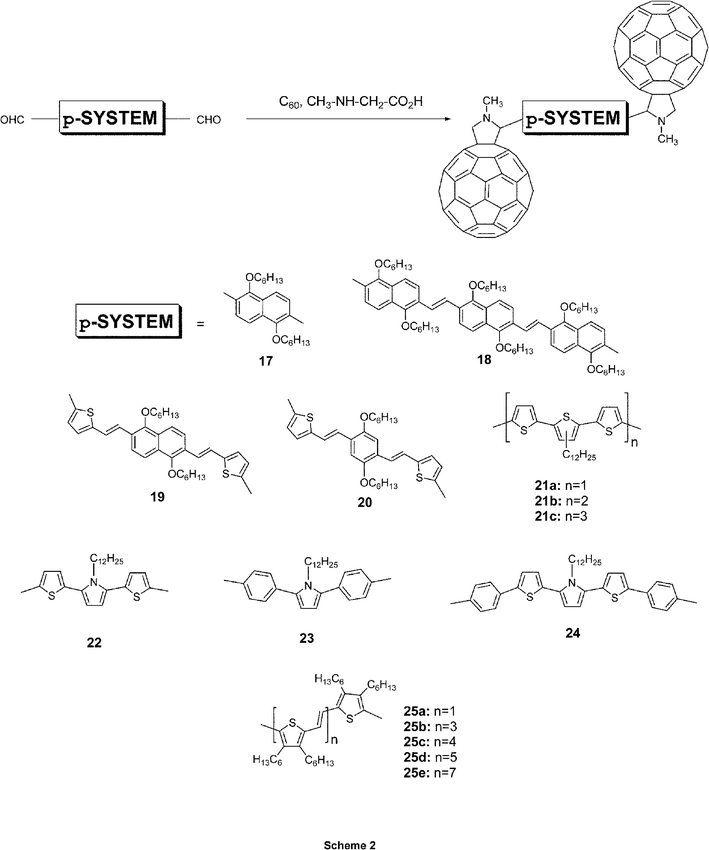 | ||
| Scheme 2 Structure of representative π-conjugated oligomer–C60 triad materials. | ||
Alternative pathways towards C60-based π-conjugated oligomer ensembles involve (i) [4 + 2] cycloaddition reaction of C60 with the heteroanalogous o-quinodimethanes of oligothiophenes 27, generated in situ from sulfone 26, to yield dyad 2824 (Scheme 3a), (ii) 1,3-dipolar cycloaddition reaction of oligomers carrying hydrazone functionalities (29) to C60, which affords dyad 3025 (Scheme 3b), or (iii) nucleophilic addition of oligomers endowed with a lithium acetylide functionality (Scheme 3c).26
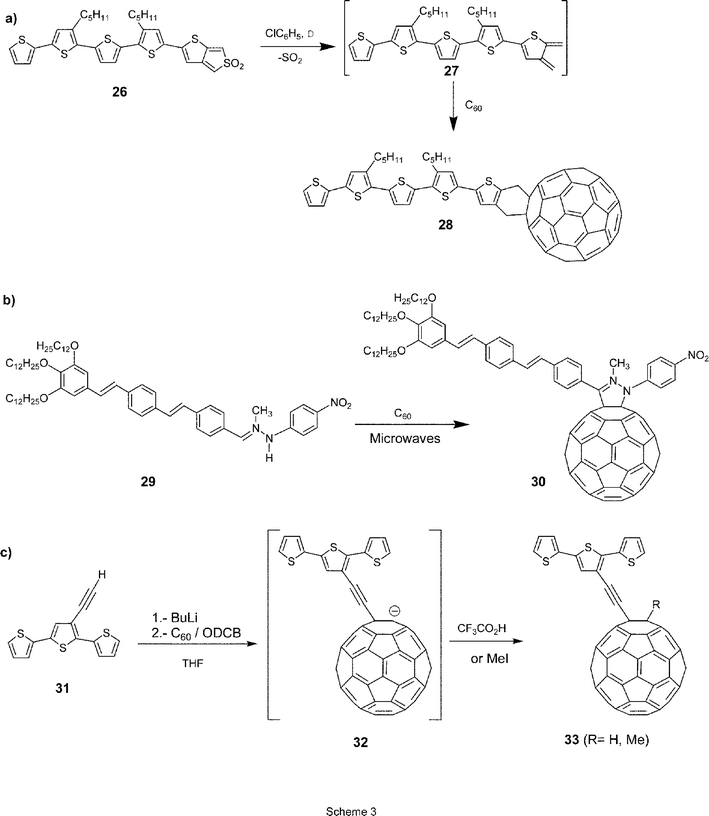 | ||
| Scheme 3 Synthetic approaches towards π-conjugated oligomer–C60 ensembles. | ||
To explore the influences of donor–acceptor orientations on photophysical behavior, Janssen, Roncali and coworkers developed a set of two complementary dyads. They connected C60 in a single (36) or double fashion (37) to the inner β-position of the terminal thiophene rings of a quaterthiophene moiety.27 These new systems (see Scheme 4) were obtained from the corresponding quaterthiophene derivatives endowed with one (34) or two (35) malonate functionalities. Compounds 34 and 35 were subjected to a Bingel reaction28 in the presence of iodine and 1,6-diazabicyclo[5.4.0]undec-7-ene (DBU) to give target dyads 36 and 37 in 42 and 35% yields, respectively. However, due to the lack of regioselectivity, which occurred during the second Bingel reaction, 37 was only isolated as a mixture containing several regioisomers.
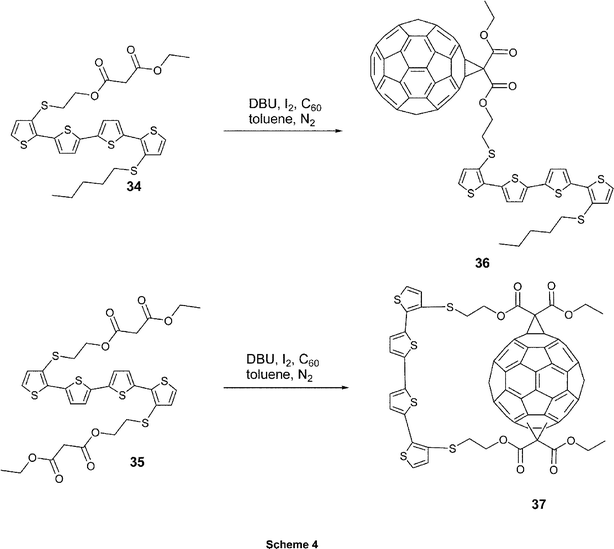 | ||
| Scheme 4 Synthetic approaches to obtain C60 connected in a single (36) or double fashion (37) to the inner β-position of the terminal thiophene rings of a quaterthiophene moiety. | ||
Ever since the discovery of C60, its aesthetically pleasing structure, with perfect icosahedral symmetry, has held a deep fascination for chemists to construct architectures of higher complexity (i.e., core-shell structures, dendrimers, etc.). In this light, the possibility of linking more than a single oligomeric unit to a C60 molecule (38) was explored29 as well as of incorporating several oligomer–C60 dyads into a dendritic type ensemble; see 39,20Fig. 2. Both systems were prepared via the 1,3-dipolar cycloaddition reactions of azomethine ylides, as generated in situ from the appropriate oligomer monoaldehyde and trisaldehyde precursors, with C60.
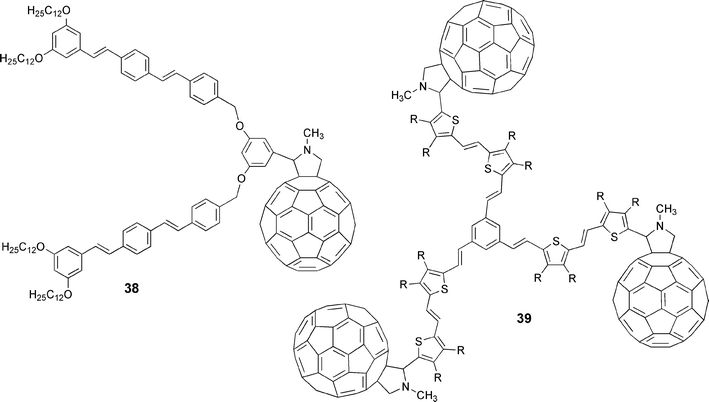 | ||
| Fig. 2 Conjugated oligomer–C60 ensembles containing more than a single oligomer or C60 moiety. | ||
To gain control over the film morphology in molecular devices, Deschenaux and Nierengarten describe an important approach. They synthesized liquid crystalline C60-oligophenylenevinylene conjugates (40), as shown in Fig. 3. Spontaneously, such materials form ordered assemblies, which might be, in the next step, oriented as high-performance thin films.30 Otsubo, Imahori, Sakata and coworkers, on the other hand, have synthesized C60-linked quarterthiophenes and octathiophenes that carry different anchoring groups (41, 42) (see Fig. 3). Anchoring groups, such as disulfides or a rigid triphenylene core with three mercaptomethyl arms, allow the fabrication of photovoltaic cells when grafting the octathiophene ensemble to a gold electrode.31
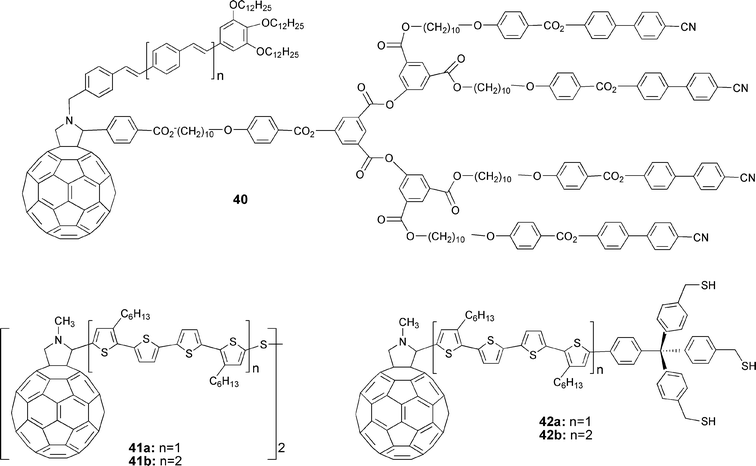 | ||
| Fig. 3 Liquid crystalline conjugated oligomer–C60 ensemble (40) and dyads with anchoring groups (41, 42) to form a self-assembled monolayer. | ||
In an independent and elegant work, quadruple hydrogen-bonding networks have been examined as a supramolecular approach to construct C60/π-conjugated oligomer donor–acceptor architectures.32 A self-complementary network of quadruple hydrogen-bonding, as represented in Fig. 4, leads to heterodimer 43 that holds together two OPV (oligophenylenevinylene) derivatives and one C60 moiety. In particular, the OPV derivative consists of two OPV ureidopyrimidinone subunits connected through a C12 spacer at the 6-position of each isocytosine ring.33 The fullerene derivative, on the other hand, bears two ureidopyrimidinone units.34
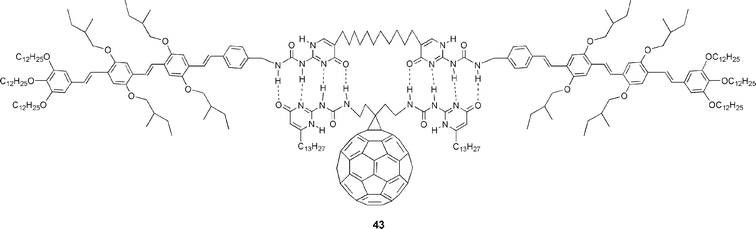 | ||
| Fig. 4 Structure of a hydrogen-bonding supramolecular heterodimer containing two OPV units and a C60 derivative. | ||
Physico-chemical properties of monodisperse π-conjugated oligomer/C60 molecular architectures
Absorption characteristics
A general feature of π-conjugated oligomers is that they exhibit intense and broad absorption bands in the visible region. These chromophoric features are assigned to electronic π–π* transitions and vary with the nature of monomer units and/or with the number of repeat units in the oligomer. Of importance is that the absorptions of π-conjugated oligomers shift to lower energies with increasing repeat units. As an example, when comparing the ground-state absorption of the 1,5-dihexyloxynaphthalene monomer, present in triad 17, with its trimeric analogue, present in triad 18, the maxima red-shift (λmax = 328 nm → λmax = 413 nm). Shifts like these reflect the effects that stem from a more delocalized and extended π-system present in the trimeric structure.The absorption spectra of virtually all the known dyad and triad ensembles (i.e., 10a–c,1611a–d,1713a–d,1914a–d,2015a–c,2116a–b,2121a–c,1122–24,2325a–d,2028,2437,2738,293920) are best described as superimposing features of the individual oligomeric and C60 building blocks. As illustrated in Fig. 5, the distinct and intense absorptions of the π-conjugated systems are typically located in the 250–500 nm range with extinction coefficients as high as 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1.18 Typical fingerprints of C60 monoadducts, on the other hand, show much weaker absorption cross-sections with maxima at 430 and 700 nm and extinction coefficients of around 500 M−1 cm−1. One of the important conclusions of this assay is that, despite linking C60 to any of the π-conjugated chromophores, the separate building blocks retain their electronic properties.
000 M−1 cm−1.18 Typical fingerprints of C60 monoadducts, on the other hand, show much weaker absorption cross-sections with maxima at 430 and 700 nm and extinction coefficients of around 500 M−1 cm−1. One of the important conclusions of this assay is that, despite linking C60 to any of the π-conjugated chromophores, the separate building blocks retain their electronic properties.
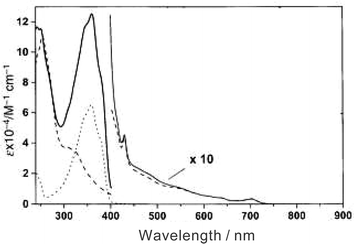 | ||
| Fig. 5 Absorption spectra of 38 (full line), N-methylfulleropyrrolidine (dashed line) and an unsubstituted π-conjugated system (dotted line) in dichloromethane at 298 K. For displaying the 400–875 nm region, a multiplying factor of 10 is used. (Adapted with permission from ref. 29.) | ||
Appreciable deviations are noted in Fig. 7, in which the absorption spectra of dyads 6–915 and triads 17–2022 are compared with the linear superimpositions of the components making up 6–9 and 17–20 (i.e., fulleropyrrolidine 44 and the conjugated π-systems 45–48; see Fig. 6). The differences transpire in non-polar, medium polar and even polar solvents. Representative examples are toluene, THF, dichloromethane, and benzonitrile. It is reasonable to assume that electronic perturbations within the monodisperse π-conjugated donors stem from connecting the redox- and photoactive blocks to each other.
Some degrees of interaction were also registered for 12a,b. A plain superimposition is only certain in the red region, that is, above 500 nm.18 Notable differences evolve in the 400–500 nm range, where the experimental spectra are significantly broader. In addition, in the ultraviolet region, the spectra are 20% less (12a) and 25% more intense (12b) than the sums of their components. A possible rationalization for such deviations implies a rotameric equilibrium of the oligophenylenevinylene moieties in 12a,b.
Contrary to the trend seen in fulleropyrrolidine dyad 49 (Fig. 8), fulleropyrazoline dyad 30 ( Scheme 3) differs from the corresponding component spectra.25 Most importantly, the absorption of the oligophenylenevinylene moiety is found to be red shifted. The differences are due to additional double bonds in the pyrazoline rings, which extend the π-conjugation of the oligomer moiety.
 | ||
| Fig. 6 Component chromophores of dyads 6–9 and triads 17–20. | ||
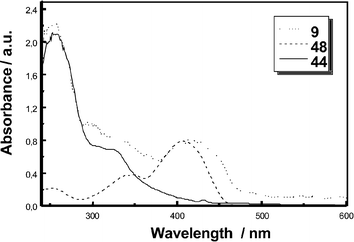 | ||
| Fig. 7 Absorption spectra of dyad 9 together with that of reference compounds fulleropyrrolidine (44) and the unsubstituted conjugated π-system 48. | ||
 | ||
| Fig. 8 Fulleropyrrolidine-based dyad 49. | ||
Electrochemistry
A typical method to investigate the room temperature redox features of monodisperse π-conjugated donors linked to C60 is cyclic voltammetry. However, different experimental conditions render a direct comparison between members of different series an impracticable task in most cases. Considering this, we will limit our summary to general trends.As a general feature, all donor and acceptor components preserve their individual electroactive identities in the donor–acceptor systems. The first oxidation step clearly involves the one-electron oxidation of the π-conjugated systems, while the lowest reduction potential corresponds to the one-electron reduction of C60 (Fig. 9).
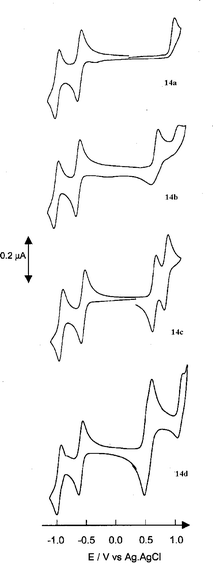 | ||
| Fig. 9 CVs of 14a–d (ca. 10−4 M) in 0.20 M Bu4NPF6–CH2Cl2, scan rate 100 mV s−1. Reprinted with permission from (ref. 20). Copyright (2002) Wiley-VCH. | ||
Saturating one of the double bonds of the C60 core, as happens during the cycloaddition reactions, shifts the C60 centered reduction potentials to more negative values relative to that noted for pristine C60. Even the nature of the cycloaddition reaction, employed to link the oligomeric moiety to the fullerene core, seems to influence the redox feature of the final product. As an example we would like to point out the first reduction potential of fulleropyrrolidine derivative 44 (E1Red = −1.09 V), which is cathodically shifted in comparison with fulleropyrazoline derivative 30 (E1Red = −0.97 V)25 and pristine C60 (E1Red = −0.98 V). Despite these general shifts, the first reduction potentials found for members of the same series are very similar: E1Red (6) = −0.62 V; E1Red (7) = −0.65 V; E1Red (8) = −0.65 V; E1Red (9) = −0.71 V; in dichloromethane vs. SCE.15
As far as the oxidation potentials of the π-conjugated systems are concerned, they depend on the number of monomer units in the oligomer and/or different carbon- or heterocyclic monomers. The impact of these modifications is manifested in the following examples. An increase in the donor strength is observed when going from dihexyloxynaphthalene (6, E1Ox = 1.41 V) to the trimeric analogue (7, E1Ox = 1.31 V) and also upon replacing the peripheral naphthalene units by the π-exceedent thiophene (8, E1Ox = 1.20 V). The lowest oxidation potential is seen for the π-conjugated system, in which the central naphthalene moiety is replaced by a phenylene unit (9, E1Ox = 1.01 V).15
A similar behavior is concluded in 10a–c16 (10a, E1Ox = 1.04 V; 10b, E1Ox = 0.76 V; 10c, E1Ox = 0.67 V; in benzonitrile vs. AgCl/Ag), 11a–d17 (11a, E1Ox = 1.19 V; 11b, E1Ox = 0.97 V; 11c, E1Ox = 0.85 V; 11d, E1Ox = 0.77 V; in dichloromethane vs. SCE), 12a–b18 (12a, E1Ox = 1.31 V; 12b, E1Ox = 1.16 V; in o-dichlorobenzene vs. AgCl/Ag) and 14a–d20 (14a, E1Ox = 0.99 V; 14b, E1Ox = 0.69 V; 14c, E1Ox = 0.59 V; 14d, E1Ox = 0.49 V; in dichloromethane vs. AgCl/Ag). Overall, the donor ability of the resulting oligomer moieties steadily increases as a function of monomer units in the oligomer backbone within all of the different series.
On the contrary, the choice of connecting either a fulleropyrrolidine or a fulleropyrazoline has no impact on the oxidation potentials. In 30 and 49, the potentials are indistinguishably around 0.60 V in dichloromethane vs. Fc/Fc+.25
It is also worth pointing out that the redox features in the dyad and triad analogues are comparable: when comparing, for instance, dyads 6–915 with triads 17–2022 almost identical redox potentials are observed, despite the presence of either one C60 or two C60 moieties.
Photophysics—building blocks
The wide variety of π-conjugated oligomers, which are linked through a carefully chosen design to C60, provides a rich platform for detailed studies of their photophysical properties. The study of the photophysical behavior of monodisperse π-conjugated/C60 molecular architectures necessitates, in a first step, the characterization of the separate building blocks (i.e., π-conjugated oligomers and C60).Most π-conjugated oligomers are extremely strong chromophores, with singlet excited state fluorescence that approaches unity quantum yields. Their singlet excited state energies are typically around 3.5–2.5 eV. These features render them convenient probes for photophysical measurements, especially regarding C60-containing materials. As a general trend, the fluorescence spectra are in excellent agreement with their mirror imaged absorption features, see for example 45,46,47 and 48. Parallel to the high fluorescence yields, the investigated models show short-lived fluorescence lifetimes (i.e., ∼1–2 ns).
Transient absorption changes of the singlet excited states (i.e., 45–48) involve characteristic singlet–singlet transitions in the far-visible–near-infrared spectra. For example, following picosecond laser pulses, photoexcitation of 46 leads to the instantaneous formation of a strong maximum in the 800–900 nm region, see Fig. 10a. These attributes are ascribed to the singlet excited state absorption features of the conjugated π-systems in photoexcited 45–48. At longer times the singlet excited states deactivate via mono-exponential rate laws and a rate of ∼109 s−1 to produce the long-lived triplet excited state. The triplet absorbances in the 500–700 nm region generally decayed via dose-independent first-order kinetics and resulted in a complete restoration of the ground state.
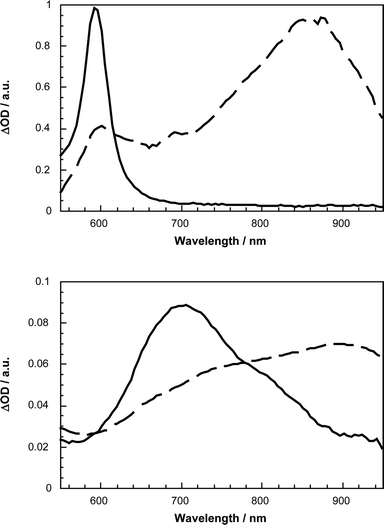 | ||
| Fig. 10 Transient absorption spectrum visible–near-infrared part recorded 20 ps (dashed line) and 5000 ps (line line) upon flash photolysis of 46 (upper spectrum) and 44 (lower spectrum) at 355 nm in deoxygenated toluene, indicating the oligomer/C60 singlet–singlet and triplet–triplet features, respectively. | ||
A model C60 derivative, namely N-methylfulleropyrrolidine (44)9 reveals nearly solvent-independent fluorescence in the near-infrared (i.e., 1.76 eV), much lower than the highly energetic singlet excited states of most π-conjugated oligomers (i.e., 3.5–2.5 eV). Also the fluorescence quantum yields are much lower, with a value of 6.0 × 10−4.35 The fluorescence lifetime of ∼1.5 ns, on the other hand, is comparable to that of π-conjugated oligomers.
In transient absorption measurements, pumping of the C60 ground state with ultraviolet or visible light leads to the population of the singlet excited state. The lifetime of this intermediate state is relatively short, as C60 and most of its derivatives intersystem cross rapidly (5 × 108 s−1) to the much longer lived triplet excited state with nearly unit yield. The spectral characteristics of these main excited states are shown in Fig. 10b. Both the singlet and triplet excited states give rise to transient absorptions in the near-infrared with maxima around 900 and 700 nm. Perturbation of the fullerene π-system, which stems from the removal of a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond upon functionalization, brings about noticeable blue shifts of the excited state relative to those of C60.36
C double bond upon functionalization, brings about noticeable blue shifts of the excited state relative to those of C60.36
Photophysics—steady state fluorescence with donor–acceptor ensembles
Due to the strong absorption cross-section of π-conjugated materials in the solar spectrum, visible light excitation reaches quantitatively the oligomer chromophore, while it fails to excite C60. By and large, in most of the monodisperse π-conjugated/C60 molecular architectures, the characteristic fluorescence of the oligomers is nearly quantitatively quenched. Leading examples are 6–915 and 17–20,22 where the strong emission of the reference π-conjugated systems, with quantum yields that reach up to 72%, is substantially quenched and reveals quantum yields between 0.03 and 0.05% (Fig. 11). It is important to note that, despite the weak fluorescence, the emission patterns of the conjugated π-systems remain unchanged and are not impacted noticeably by the presence of C60.15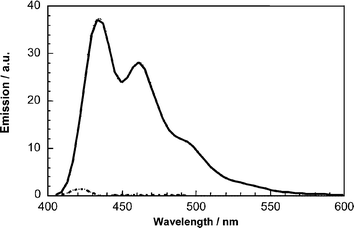 | ||
| Fig. 11 Fluorescence spectra of triad 18 (dashed line) in toluene solution together with that of the corresponding unsubstituted π-conjugated system 46 (solid line). | ||
Similarly, in 11b–d,1712a,b,1821a–c12 and 30, the fluorescence quenching of the π-conjugated moieties reaches more than three orders of magnitude.25 Quenching of between two and three orders of magnitude was observed for 15a–c21 and 16a,b21, while the quenching in 38 is essentially quantitative.29
Oligothiophenes are likewise affected. Despite the generally strong fluorescence of oligothiophenes, quantitative emission quenching of the oligothiophene moiety was observed in 10a–c. When comparing the flexible and singly bonded derivative 36 with the doubly linked dyad 37, the residual oligothiophene fluorescence in toluene and o-dichlorobenzene is about ten times larger for 36.27
As far as a mechanistic interpretation of the fluorescence quenching is concerned, steady-state fluorescence spectroscopy is a lot more powerful than just measuring the deactivation of the photoexcited oligomers. Upon inspecting the fluorescence data in detail, the fluorescence of C60 is also discernible, see Fig. 12. Specifically, the familiar C60 fluorescence spectrum appears in the near-infrared with a *0–0 emission at around 715 nm, despite exclusive excitation of the π-conjugated moiety at 400 nm. This behavior emerges as a general trend in 6–9,1510a–c,1611a–d,1712a–b,1815a–c,2116a–b,2117–20,2221a–c,1222–24,2330,2536,273727 and 38.29
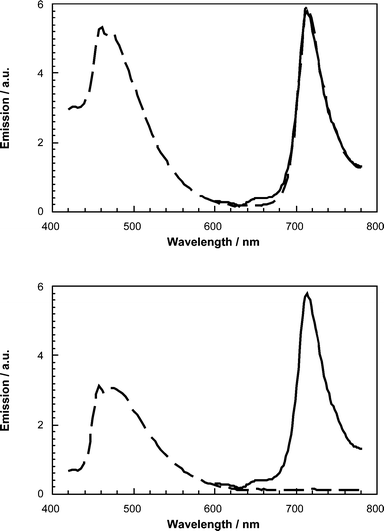 | ||
| Fig. 12 Fluorescence spectra of 44 (solid line) and 9 (dashed line) in toluene (upper spectrum) and THF (lower spectrum). | ||
Additional excitation spectra of the 715 nm emission, which coincide with the absorption spectra of the π-conjugated oligomer, corroborate the transduction of singlet excited state energy from the initially excited oligomer to C60. This conclusion is an excellent reflection of the singlet excited state energy levels of π-conjugated oligomers (3.5–2.5 eV) and of C60 (1.76 eV).
Yet a second pathway takes place and competes with the intramolecular energy transfer in the overall deactivation of the photoexcited chromophore (i.e., π-conjugated systems) in monodisperse π-conjugated/C60 molecular architectures. This notion derives from fluorescence measurements in solvents of different polarity, as illustrated in Fig. 12. At this point it is worth pointing out that the π-conjugated systems reveal no characteristic changes, when different solvent polarities are tested. The fullerene fluorescence quantum yields, on the other hand, deviate substantially from this observation. Looking at dyads 6–9 helps to exemplify this trend. In 6–9, the quantum yields measured in THF solutions diverge from those measured in toluene.15 At first glance, a highly exothermic electron transfer scenario, direct electron transfer, is considered. Another plausible alternative implies sequential energy and electron transfer—indirect electron transfer—events. Fig. 13 summarizes the photophysical processes that take place in monodisperse π-conjugated/C60 molecular architectures after photoexcitation.
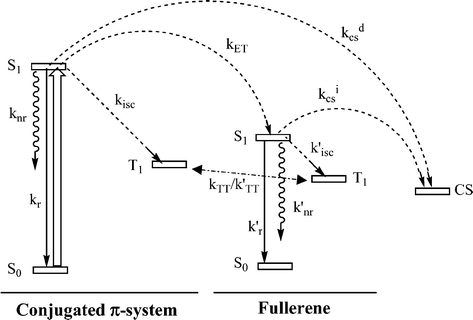 | ||
| Fig. 13 Schematic diagram describing most of the possible photophysical processes for dyads of the π-conjugated system–C60 together with the energy levels of the singlet (S0 and S1), triplet (T1) and charge-separated (CS) states. The energy transfer (kET) and the indirect (kCSi) and direct (kCSd) charge separation processes are indicated with curved dashed arrows. The solid arrow describes the initial excitation of the π-conjugated system. kr and k′r represent the radiative rate constants, knr and k′nr the non-radiative decay constants and kisc and k′isc the intersystem crossing rate constants. | ||
Photophysics—time-resolved fluorescence and transient absorption measurements with donor–acceptor ensembles
Up to this point, we have summarized the steady-state fluorescence behavior of monodisperse π-conjugated/C60 molecular architectures. The synopsis of the fluorescence studies along with the thermodynamic calculations is that an intramolecular singlet–singlet energy transfer dominates the deactivation of the photoexcited π-conjugated systems. However, indications that suggest excited state deactivation via electron transfer are also gathered. To find decisive evidence for the energy/electron transfer scenarios necessitates investigations by time-resolved photoinduced absorption spectroscopy, light-induced electron spin resonance and solid-state measurements. Considering the complexity that such investigations bring about, we will discuss in the next section of this review article the different series of compounds separately.Photophysics—dyads 6–915 and triads 17–2022
In toluene, transient absorption changes, recorded upon the 18 ps laser excitation of 6–9 and 17–20, are all superimposable with those observed for the reference fulleropyrrolidine 44, despite the exclusive photoexcitation of the π-conjugated system. Time profiles illustrate this trend: they show that the singlet excited state of C60 is formed instantaneously (i.e., > 5.0 1010 s−1) and in a single step. Hereafter, intersystem crossing is the predominant deactivation of the fullerene singlet excited state (k′ISC in Fig. 13), which affords the C60 triplet excited state in quantitative yields.A different reactivity is observed in a strongly polar solvent like benzonitrile. In particular, dihexyloxybenzene–thiophene systems 9 and 20 give rise to the immediate occurrence of a 690 nm band (Fig. 14), which is assigned to the π-radical cation of the π-conjugated system. The fast growth of the π-radical cation band (≤ 18 ps) verifies a direct electron transfer mechanism (kCSd in Fig. 13) to form C60˙−–oligomer˙+.
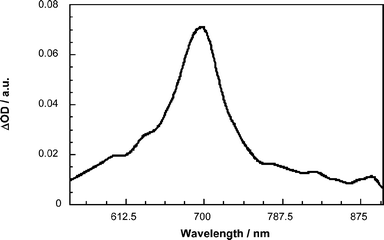 | ||
| Fig. 14 Transient absorption spectrum (i.e., visible–near-infrared part) recorded 20 ps upon flash photolysis of 20 (2.0 × 10−5 M) at 355 nm in deoxygenated benzonitrile, indicating the oligomer π-radical cation features (λmax ∼ 700 nm). | ||
Raising the oxidation potential of the donor, for example, in the dihexyloxynaphthalene–thiophene containing systems 8 and 19 leads to the detection of two photoproducts. Charge separation, namely, generation of C60˙−–oligomer˙+, is inferred on the basis of the instantaneously produced 700 nm maximum, while the presence of a 900 nm absorption, an intersystem crossing rate of 7.8 108 s−1, and high C60 triplet quantum yields are clear attributes connected with the energy transfer product.
In dialkoxynaphthalene systems 6, 7, 17 and 18, the picosecond changes are overshadowed by the dominance of the fullerene photophysics, confirming an all energy transfer scenario.
The different behavior in 6–9 and 17–20 is explained by reference to the relative energy levels of the charge-separated state and of the fullerene singlet excited state. For the stronger donor materials (9,20) the energy of the charge-separated state (1.49 eV) is below that of the fullerene singlet-excited state (1.76 eV). Reducing the donor strength (8,19) brings the energy of the charge-separated state (1.74 eV) close to that of the fullerene singlet excited state. Further lowering the donor ability in these dyads (6,7) and triads (17,18), the charge-separated state (1.90 eV) even passes that of the fullerene singlet state.
Particular benefits are noted when attaching two fullerene cores (i.e. a triad) to the oligomeric structure. In benzonitrile, for example, the triad system 20 shows a 67% enhanced quantum yield and a longer lifetime (1.34 times) of charge separation relative to the dyad system 9. These observations clearly indicate that cooperative effects evolve between the two fullerene moieties.
Photophysics—dyads 11a–d17, 1420b triads 21a–c,1222,23,2423 and 2520b
Janssen and coworkers have performed photoinduced absorption spectroscopy measurements (PIA) to probe 11a–d (Fig. 15a).17 In o-dichlorobenzene, PIA measurements reveal the fingerprints of charge separation (i.e., C60˙−–oligomer˙+). Thus, an electron transfer process is inferred for 11c–d, which follows the initial energy transfer event. For 11b, although the fullerene emission is partly quenched, the C60 triplet excited state is still discernible in the PIA spectrum. This observation indicates a combination of energy transfer and intersystem crossing. In 11a, the PIA spectrum in o-dichlorobenzene shows only the absorption of the fullerene singlet excited state. This behavior corresponds well with the better donor ability of the oligomers, which increases with the number of monomer units.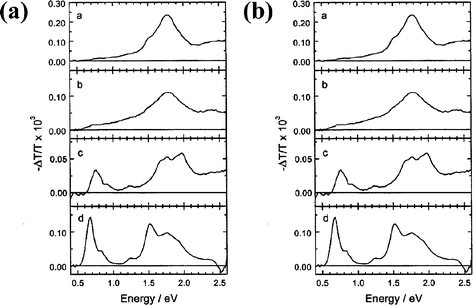 | ||
| Fig. 15 (a) PIA spectra of 11a–d in ODCB (4 × 10-4 M) at 295 K. (b) PIA spectra of 11a–d thin films on quartz. Reprinted with permission from (ref. 17). Copyright (2000) American Chemical Society. | ||
In films (Fig. 15b), solid-state PIA measurements exclude photoinduced electron transfer for 11a–b. More precisely, the behavior matches that of fulleropyrrolidine reference 44. Direct spectral evidence for C60˙−–oligomer˙+ is found only for 11c and 11d. These activities track the donor ability of the oligomeric moieties: 11a, E1Ox = 1.19 V; 11b, E1Ox = 0.97 V; 11c, E1Ox = 0.85 V; 11d, E1Ox = 0.77 V; dichloromethane vs. SCE.
The average charge-separated state lifetime (11c,11d) in the film is of the order of 0.5–1.5 ms, much larger than that observed in molecular ensembles, whose lifetimes usually do not extend into the microsecond regime.10,11 This finding suggests that, in films, the charge-separated state becomes a material property rather than remaining that of a molecule. Migration of holes and/or electrons to neighboring molecules in the films is responsible for stabilizing the charge separation product.
Photophysical investigations have also been carried out with oligothiophene containing triads (21a–c).12 In toluene solutions, the PIA spectra of 21a and 21b are identical to that of fulleropyrrolidine 44, while that of 21c corresponds to a superimposition of the triplet absorption of the fulleropyrrolidine and the triplet absorption of the oligomer. A relay of energy transfer reactions is responsible for this reactivity: photoexcitation of the oligomer is followed by singlet–singlet energy transfer (kET in Fig. 13) to the fullerene, intersystem crossing to afford the fullerene triplet excited state (k′isc in Fig. 13), and, finally, triplet energy transfer back to the oligothiophene (k′TT in Fig. 13).
In a more polar solvent, such as o-dichlorobenzene or benzonitrile, no PIA signals were detected at all for 21a–c. Intermolecular electron transfer processes are, however, established in solutions containing fulleropyrrolidine and reference oligomers. Thus, the lack of PIA signals in 21b and 21c prompts the short-lived nature of C60˙−–oligomer˙+, which decays faster to the ground state than the near steady-state conditions of the PIA time scale allow to detect. Interestingly, solid-state measurements of 21b and 21c thin films show the characteristic absorptions of the fulleropyrrolidine radical anion and oligomer radical cation.
For dyads 14a,b and 25c containing oligothienylenevinylenes covalently linked to one or two C60 units, different photophysical behavior is observed depending on the relative energy positions of the singlet excited state of the fulleropyrrolidine and the charge separated states, which can be controlled by the polarity of the solvent.20b Thus, an intramolecular charge separated state is formed in ODCB in a two-step mechanism involving a fast singlet energy transfer followed by electron transfer. In the less polar toluene, competition between two photophysical processes are observed: (a) electron transfer to yield the intramolecularly generated charge-separated state and (b) fluorescence and intersystem crossing followed by triplet energy transfer.
Photophysics—dyads 3627 and 3727
Orientation effects on the photophysical properties are probed in quaterthiophene-C6036 and 37.27 The different connections provide a completely free orientation for the singly attached system 36 and a face-to-face orientation for the doubly attached dyad 37. In toluene, submillisecond charge separation lifetimes prevent the detection of PIA charge-separated absorptions. In o-dichlorobenzene, the PIA spectrum of 36 shows charge separation that undergoes bimolecular decay and lifetimes exceeding 10 ms. Similar measurements for 37 show only spectra that resemble those of the C60 triplet state.When testing a mixture of the reference compounds in o-dichlorobenzene solutions, the PIA spectra reveal fingerprints of the C60 triplet state as well as the quaterthiophene radical cation. Changing the modulation frequency in the PIA measurements discloses that the C60 triplet absorption band contains contributions of a short-lived and a long-lived component and that the intensity of the band is quenched in comparison to the triplet absorption of the fullerene reference. As the authors point out, all these features support a rapid charge recombination for C60˙−–oligomer˙+, which is reflected by the low intensity of the triplet fullerene absorption.
Rate constants of deactivation in the geometrically constrained dyad 37 are one order of magnitude larger than those for flexible 36. This suggests that the constraint imposed on the orientation of the chromophores in 37 lead to much stronger interactions between the excited states of the oligothiophene and C60. One of the major conclusions of this fascinating work is that ultrafast photoinduced electron transfer processes, as observed in blends containing conjugated polymers and fullerenes, are due to face-to-face orientations between the two moieties formed spontaneously in the film.
Photophysics—dyads 10a–c16,37,38
Oligothiophenes of different length covalently linked to C60 were recently studied by Otsubo, Ito and coworkers. In a first study they tested the charge separation and charge recombination processes in a tetrathiophene-based dyad (10a).37Measurements carried out in toluene indicate a fast singlet excited state energy migration from the oligothiophene to C60. On the other hand, photolysis in benzonitrile features the radical cation band of oligothiophene at 1140 nm, while the fingerprint of the one-electron reduced C60 is masked by the much stronger oligomer radical cation band. Kinetic investigations show an increase in the quantum yield of the charge separation process when increasing the solvent polarity. Estimated values are as high as 0.97, for example, in THF. Additional photolysis measurements indicate that the charges recombine to yield mainly the triplet excited state. An unusually long lifetime of the charge-separated state (6.3 μs) was rationalized to stem from equilibrium conditions between the charge-separated state and the triplet excited state.
Fine particles of 10a–c, with granular-type diameters of about 100 nm, provide important information on the photophysical activity in a crystalline phase.38 These fine particles show extensive fluorescence quenching. In subpicosecond laser photolysis measurements, the radical cation absorption of the oligomer moiety develops. It is, however, worth mentioning that the absorption band due to the charge-separated state is small when compared to that seen in benzonitrile (Fig. 16b). The reduced yields in the cluster are attributed to the existence of competitive deactivation pathways in the singlet excited cluster samples. One of the competition pathways includes exciton migration.
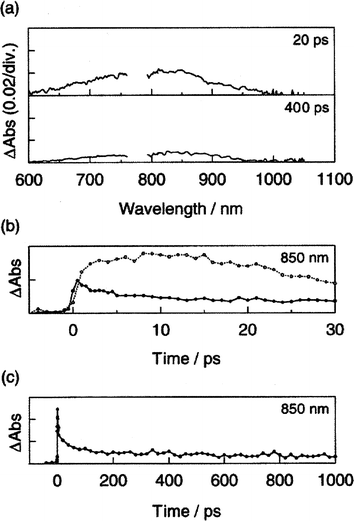 | ||
| Fig. 16 (a) Transient absorption spectra of the 10b water dispersion observed by 388 nm laser excitation. (b) Absorption–time profiles at 850 nm of 14b fine particles (solid line) and 14b in benzonitrile (dot line) after a laser pulse. (c) Absorption–time profile at 850 nm of 10b fine particle after a laser pulse. Reprinted with permission from (ref. 38). Copyright (2001) American Chemical Society. | ||
Concerning the deactivation of C60˙−–oligomer˙+, a two-step decay mechanism is postulated. Firstly, a fast decaying process occurs, which corresponds to an intramolecular charge recombination within the dyad molecule. The second contribution is a slow, intermolecular decay. It is plausible that the generated radical cations and/or radical anions migrate to adjacent molecules in the clusters.39 As a result of this migration the average distance between the radical ions increases and a fast charge recombination is prevented. This process is further augmented by the hole transport ability of oligothiophenes.
Photophysics—dyads 12a,b,183025 and triad 3829
Armaroli, Nierengarten and coworkers have also carried out photophysical investigations with phenylenevinylene-based systems. In toluene solutions, 12a,b show, following the initial photoexcitation, no trace of the oligomer fluorescence, despite 60% light absorption by the oligomer fragment. Instead, the characteristic fullerene fluorescence is observed. Similar characteristics are concluded in polar benzonitrile. Therefore, under these conditions, photoinduced energy transfer is the only intracomponent process. This picture did not change for the analogue (38) that bears two oligomer moieties per C60.Photovoltaic devices based on monodisperse π-conjugated oligomer/C60 molecular architectures
The most efficient polymer solar cells fabricated today are based on the concept of bulk heterojunctions formed by p-type conjugated polymers mixed with an acceptor moiety, as n-type material.6 The different solubilities of the two components in organic solvents, used, for example, for the film preparation, and their limited miscibilities result in severe phase segregation. The tendency of phase segregation needs to be controlled to handle and optimize charge generation and charge transport. An intimate mixing of the donor and acceptor emerges as a central prerequisite for optimal charge generation conditions, since the charges are preferentially formed at the donor–acceptor interface. It is also necessary to attain an efficient transport of holes through the donor phase and of electrons through the acceptor phase. For these reasons the donor and acceptor components have to be organized in the form of nanoscopic phase segregated bicontinuous networks. In this light, the most significant improvements of device efficiencies have been attributed to favorable morphologies.40One approach to attain an intimate mixing of the donor–acceptor components and to control the film morphology implies the concept of “double cables”. These kinds of materials consist of a hole conducting conjugated polymer chain carrying pendant electron conducting moieties. In this way, electron-acceptor and donor moieties are forced to form a p- and n-conducting network with well-defined links. We and others have recently used similar structures consisting of a conducting polymer covalently connected to electron accepting units as photovoltaic devices.41
An optimized electron transfer is another prerequisite for devising a high performance photovoltaic device. Since the donor–acceptor ensembles presented above are low molecular weight analogues of the double-cable materials, some of them have been sandwiched between asymmetric electrodes to test their photovoltaic behavior. The dyad 9, which in solution shows all electron transfer behavior, is incorporated into a device by spin-coating a 1.2% toluene–o-chlorobenzene (3 : 2) solution to reach a film thickness of approximately 60 nm on top of an ITO/PEDOT/PSS (indium tin oxide/poly(3,4-ethylenedioxythiophene)/poly(styrenesulfonate)) substrate. A LiF/Al (6 Å/100 nm) layer is then thermally deposited as a top electrode (Fig. 17a).15 Typical current to voltage characteristics of 9, under illumination and also in the dark, are given in Fig. 17b. The dark I–V curve is symmetric in the region between +0.5 and −0.5 V, which is, in part, due to a small shunt resistivity of the diode leading to an ohmic contribution. A diode turn on behavior is observed at voltages higher than 0.7 V, with rectification ratios of more than two orders of magnitude. Under irradiation with AM 1.5 solar simulated light with 80 mW cm−2, the open circuit voltage (Voc) is 0.67 V and the short circuit current (Isc) is 0.75 mA cm−2. The fill factor (FF), defined as (Imax × Vmax)/(Isc × Voc) with Imax and Vmax corresponding to the point of maximum power output, is < 0.3. Taking all these parameters into account, an overall white light conversion efficiency of ≤0.2% is calculated. This is one of the highest values reported for a solution processed single component solar cell. It is also worth mentioning that the monochromatic efficiency of 10% peaks at 440 nm. Between 500 and 700 nm, this value drops below 4%. Spectrally resolved photocurrent measurements reveal contributions from both the oligomer and the fullerene moiety to the photocurrent generation.
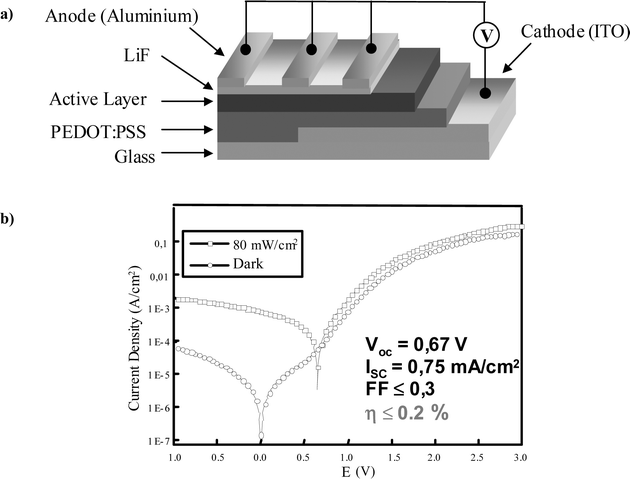 | ||
| Fig. 17 (a) Set up of the ITO/PEDOT/9/LiF/Al diode; (b) I–V characteristics of the diode under 80 mW cm−2 white light (open squares) from a solar simulator and in the dark (open circles). | ||
When photovoltaic devices are fabricated with 11d as an active layer, the I–V curves are completely reversible and the device shows diode behavior with a rectification ratio between −2 V and +2 V of approximately 100.17 Under ∼65 mW cm−2 white light illumination, the following parameters are determined: an open circuit voltage of 650 mV, a short circuit current of 235 μA cm−2, and a fill factor of 0.25 V.
To incorporate 12a and 12b into photovoltaic devices, chloroform solutions (i.e., 4% weight) are spin-coated onto ITO/glass substrates, which leads to a film thickness of between 100 and 140 nm.18 The devices performance for 12a in the dark and under illumination (i.e., λ = 400 nm with an intensity of 12 mW cm−2) includes an open circuit voltage of 0.46 V and a short circuit current of 10 μA cm−2. A fill factor of 0.3 is calculated and the monochromatic conversion efficiency is about 0.01%. An analogous device fabricated with 12b as the active layer shows, under similar conditions, a 0.03% monochromatic power conversion efficiency
Gross, Hadziioannou, Nierengarten and coworkers have investigated the photovoltaic properties of devices with 13a–d.42 All dyads are sandwiched between (PEDOT-PSS) covered indium tin oxide and aluminium electrodes. The monochromatic efficiencies under illumination (i.e., 400 nm, 1W cm−2) are 0.0012, 0.0026, 0.020 and 0.018 % for 13a, 13b, 13c and 13d, respectively. The performances of the devices prepared from the N,N-dialkylaniline terminated derivatives 13c,d are significantly improved, when compared to 13a or 13b. This is in agreement with the better donor ability of the N,N-dialkylaniline donors (i.e., 13a, E1Ox = +0.80 V; 13b, E1Ox = +0.72 V; 13c, E1Ox = +0.32 V; 13d, E1Ox = +0.33 V) in comparison with the trimethylsilyl substituted analogues. In the dark, the current in reverse bias is higher by two orders of magnitude for the device fabricated with 13a in comparison with that fabricated with 13b. A similar trend is observed when comparing devices made with 13c and 13d. Apparently, derivatives with shorter oligomer chains exhibit better charge transport properties, because of a higher intrinsic C60/oligomer mass ratio.
Aluminum and gold electrodes in combination with 10a–c were explored by Aso, Otsubo and Harima.43 The short-circuit currents are highly dependent on the oligothiophene chain lengths. An increase of the photocurrent is observed when testing the long chain oligothiophenes. This trend agrees well with the fact that electron transfer occurs more efficiently in the dyads bearing longer oligothiophene chains. For the device fabricated with the dyad covalently linked to the hexadecamer, an open circuit voltage of 0.64 V, a short-circuit density of 190 nA cm−2 and a fill factor of 0.34 are derived. On the basis of these data, the monochromatic power conversion efficiency of this device is 0.4%.
Obviously, the performances of these devices are somewhat inferior relative to solar cells fabricated with blends of conjugated polymer and fullerene derivatives. Among the many factors that limit the intrinsic efficiency of these devices, the following should be considered. Firstly, the absorption spectrum of the oligomers fails to cover the wavelength range of the parent polymers, due to the reduced conjugation length. Secondly, the intrinsic C60/oligomer ratio in the dyads and triads differs from the optimized C60/polymer weight ratio of 4, which is currently used in the most efficient solar cells. Nevertheless, the above results confirm that charge generation as well as current rectification is realized within these kinds of donor–acceptor ensembles, thereby combining essential features for photovoltaics within a single molecule.
Conclusions and outlook
The photophysical behavior of π-conjugated oligomer and C60 based donor–acceptor ensembles has been fine-tuned by (i) using different carbon- and heterocycles as oligomeric component units, (ii) increasing the number of repeat units within the oligomeric moiety, (iii) using more than one fullerene unit per oligomer moiety, which leads to cooperative effects evolving between both fullerenes, (iv) the use of different links between the electroactive moieties, (v) investigation of orientational effects, and (vi) photophysical measurements carried out in solutions, in fine particles, and in the solid state.Variations in the number of monomer units within a family of oligomers and of the arylene units forming the π-conjugated oligomers—benzene, naphthalene, thiophene—result in control over the absorption wavelength and the oxidation potentials of the oligomeric donor moiety. Depending on the donor ability of the oligomeric fragment, no interaction or only slight interactions are observed between both electroactive moieties in the ground state.
A general trend observed is that the characteristic fluorescence of the oligomeric moiety is nearly quantitatively quenched in the donor–acceptor ensembles. Photophysical processes following the photoexcitation of the oligomeric moiety involve (i) singlet–singlet energy transfer to generate the fullerene singlet excited state followed by intersystem crossing to the fullerene triplet excited state, (ii) singlet–singlet energy transfer to generate the fullerene singlet excited state followed by charge separation, and (iii) charge separation.
Together with the intramolecular processes observed in solution, intermolecular processes are also seen both in the solid-state and in investigations carried out with fine particles. Generally, charge-separated state lifetimes tend to be shorter in solutions than in thin films. Two different kinetics are observed for the decay of the charge generated states in clusters: the fast decaying process indicates charge recombination in the dyad while the slow decay is due to intermolecular processes. Taking all of this into account, we conclude that in the solid state, the lifetime of a charge-separated state is a material rather than a molecular property.
However, an optimized electron transfer is an imperative prerequisite for devising a high performance photovoltaic device. Considering that the photophysical properties of conjugated oligomers are similar to those of the corresponding polymers, the applicability of selected conjugated oligomer–fullerene ensembles in the fabrication of photovoltaic devices has been explored. Charge generation as well as current rectification is realized within these types of materials.
In summary, the C60/π-conjugated oligomer approach constitutes a recent and powerful field that deserves further attention. Future work, which should be motivated by overcoming some of obstacles that remain in front of these materials before becoming integrative components in fabricating efficient solar cells, will involve the use of fullerene–oligomer-donor triads with even stronger donors to produce even longer-lived charge-separated states. The morphology of these materials in thin films should be further explored as this evolves as a key parameter to achieve good transport properties. Materials that absorb light in a wider range of the visible spectra in combination with the above requirements remain a challenge to achieve good monochromatic efficiencies and also better overall light conversion efficiencies.
Acknowledgements
This work has been partially supported by the MCyT of Spain (Project BQU2002-00855), by the European Commission (Contract JOR3CT980206), by the Comunidad de Madrid (project 07N/0004/2002) and the Office of Basic Energy Sciences of the U.S. Department of Energy (contribution No. NDRL-4582 from the Notre Dame Radiation Laboratory). We also thank all those colleagues who have participated in the development of this emerging field of organic photovoltaics and, particularly, to our partners engaged in the former European projects “Molecular Plastic Solar Cells” and “EUROMAP”.References
- For a comprehensive summary of photovoltaics (solar cells), see: Clean Electricity from Photovoltaics, ed. M. D. Archer and R. Hill, Imperial College Press, 2001 Search PubMed.
- (a) Electronic Materials: The Oligomer Approach, ed. K. Müllen and G. Wegner, Wiley-VCH, Weinheim, 1998 Search PubMed; (b) For a recent review, see: J. L. Segura and N. Martín, J. Mater. Chem., 2000, 10, 2403 Search PubMed.
- M. T. Rispens, J. C. Hummelen, in Fullerenes: From Synthesis to Optoelectronic Properties, ed. D. M. Guldi and N. Martín, Kluwer Academic Publishers, Dordrecht, 2002 Search PubMed.
- (a) G. Yu, Y. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789 CrossRef CAS; (b) K. Matsumoto, M. Fujitsuka, T. Sato, S. Onodera and O. Ito, J. Phys. Chem. B, 2000, 104, 11632 CrossRef CAS.
- N. S. Sariciftci and A. J. Heeger, in Handbook of Organic Conductive Molecules and Polymers, ed. H. S. Nalwa, Wiley, New York, 1996 Search PubMed.
- C. J. Brabec, N. S. Sariciftci and J. C. Hummelen, Adv. Funct. Mater., 2001, 1, 15 CrossRef CAS.
- R. A. J. Janssen, M. P. T. Christiaans, K. Pakbaz, D. Moses, J. C. Hummelen and N. S. Sariciftci, J. Chem. Phys., 1995, 102, 2628 CrossRef CAS.
- (a) L. Ouali, V. V. Krasnikov, U. Stalmach and G. Hadziioannou, Adv. Mater., 1999, 11, 1515 CrossRef CAS; (b) S. C. Veenstra, G. C. Malliaras, H. J. Brouwer, F. J. Esselink, V. V. Krasnikov, P. F. van Hutten, J. Wildeman, H. T. Jonkman, G. A. Sawatzky and G. Hadziioannou, Synth. Met., 1997, 84, 971 CrossRef CAS; (c) L. Pasimeni, A. L. Maniero, M. Ruzzi, M. Prato, T. Da Ros, G. Barbarella and M. Zambianchi, Chem. Commun., 1999, 429 RSC; (d) G. Hadziioannou, Polym. Prepr., 2000, 41, 797 CAS; (e) A. Y. Andreev, G. Matt, H. Sitter, C. J. Brabec, C. Badt, H. Neugebauer and N. S. Sariciftci, Synth. Met., 2001, 116, 235 CrossRef CAS.
- M. Prato and M. Maggini, Acc. Chem. Res., 1998, 31, 519 CrossRef CAS.
- (a) N. Martín, L. Sánchez, B. Illescas and I. Pérez, Chem. Rev., 1998, 98, 2527 CrossRef CAS; (b) D. M. Guldi and N. Martin, J. Mater. Chem., 2002, 12, 1978 RSC; (c) D. M. Guldi, Chem. Soc. Rev., 2002, 31, 22 RSC.
- (a) H. Imahori and Y. Sakata, Adv. Mater., 1997, 9, 537 CAS; (b) H. Y. Imahori, Eur. J. Org. Chem., 1999, 2445 CrossRef CAS.
- (a) R. A. J. Janssen, P. A. van Hal, J. Knol, J. C. Hummelen, Communication presented to the European Conference on Organic Solar Cells, ECOS, Cadarache (France), 1998 Search PubMed; (b) P. A. van Hal, J. Knol, B. H. W. Lamgeveld-Voss, S. C. J. Meskers, J. C. Hummelen and R. A. J. Janssen, J. Phys. Chem. A, 2000, 104, 5974 CrossRef CAS.
- J.-F. Nierengarten, J.-F. Eckert, J.-F. Nicoud, L. Ouali, V. Krasnikov and G. Hadziioannou, Chem. Commun., 1999, 617 RSC.
- J. L. Segura and N. Martín, Tetrahedron Lett., 1999, 40, 3239 CrossRef CAS.
- D. M. Guldi, C. Luo, A. Swartz, R. Gómez, J. L. Segura, N. Martín, C. Brabec and N. S. Sariciftci, J. Org. Chem., 2002, 67, 1141 CrossRef CAS.
- T. Yamashiro, Y. Aso, T. Otsubo, H. Tang, Y. Harima and K. Yamashita, Chem. Lett., 1999, 443 CrossRef CAS.
- E. Peeters, P. A. van Hal, J. Knol, C. J. Brabec, N. S. Sariciftci, J. C. Hummelen and R. A. J. Janssen, J. Phys. Chem. B, 2000, 104, 10174 CrossRef CAS.
- J.-F. Eckert, J.-F. Nicoud, J.-F. Nierengarten, S.-G. Liu, L. Echegoyen, F. Barigelletti, F. N. Armaroli, L. Ouali, V. Krasnikov and G. Hadziioannou, J. Am. Chem. Soc., 2000, 122, 7467 CrossRef CAS.
- T. Gu and J.-F. Nierengarten, Tetrahedron Lett., 2001, 42, 3175 CrossRef CAS.
- (a) C. Martineau, P. Blanchard, D. Rondeau, J. Delanuy and J. Roncali, Adv. Mater., 2002, 14, 283 CrossRef CAS; (b) J. J. Aperloo, C. Martineau, P. A. van Hal, J. Roncali and R. A. J. Janssen, J. Phys. Chem. A, 2002, 106, 21 CrossRef.
- Y. Okara, K. Takimiya, Y. Aso and T. Otsubo, Tetrahedron Lett., 2001, 42, 6877 CrossRef CAS.
- D. M. Guldi, C. Luo, A. Swartz, R. Gomez, J. L. Segura and N. Martin, J. Phys. Chem. A, 2004, 108, 455 CrossRef CAS.
- E. H. A. Beckers, P. A. van Hal, A. Dhanabalan, S. C. J. Meskers, J. Knol, J. C. Hummelen and R. A. J. Janssen, J. Phys. Chem. A, 2003, 107, 6218 CrossRef CAS.
- (a) F. Effenberger and G. Grube, Synthesis, 1998, 1373; (b) S. Knorr, M. Mehring, G. Grube and F. Effenberger, J. Chem. Phys., 1999, 110, 3502 CrossRef CAS.
- N. Armaroli, G. Accorsi, J.-P. Gisselbrecht, M. Gross, M. V. Krasnikov, D. Tsamouras, G. Hadziioannou, M. J. Gómez-Escalonilla, F. Langa, J.-F. Eckert and J.-F. Nierengarten, J. Mater. Chem., 2002, 12, 2077 RSC.
- Y. Murata, M. Suzuki and K. Komatsu, Org. Biomol. Chem., 2003, 1, 2624 RSC.
- P. Van Hal, E. H. A. Beckers, S. C. J. Meskers, R. A. J. Janssen, B. Jonsselme, P. Blanchard and J. Roncali, Chem.–Eur. J., 2002, 8, 5415 CrossRef CAS.
- C. Bingel, Chem. Ber., 1993, 126, 1957 CrossRef CAS.
- (a) N. Armaroli, F. Barigeletti, P. Ceroni, P. J.-F. Eckert, J.-F. Nicoud and J.-F. Nierengarten, Chem. Commun., 2000, 599 RSC; (b) J.-F. Eckert, N. Armaroli, F. Barigelletti, P. Ceroni, J.-F. Nicoud and J.-F. Nierengarten, Electrochem. Soc., Proc., 2000, 10, 256.
- S. Campidelli, R. Deschenaux, J.-F. Eckert, D. Guillon and J.-F. Nierengarten, Chem. Commun., 2002, 656 RSC.
- D. Hirayama, T. Yamashiro, K. Takimiya, Y. Aso, T. Otsubo, H. Norieda, H. Imahori and Y. Sakata, Chem. Lett., 2000, 570 CrossRef CAS.
- E. H. A. Beckers, A. P. H. Schenning, P. A. van Hal, A. El-ghayoury, L. Sánchez, J. C. Hummelen, E. W. Meijer and R. A. J. Janssen, Chem. Commun., 2002, 2888 RSC.
- A. El-ghayoury, A. P. H. Schenning, P. A. van Hal, J. K. J. van Duren, R. A. J. Janssen and E. W. Meijer, Angew. Chem., Int. Ed., 2001, 40, 3660 CrossRef CAS.
- L. Sánchez, M. T. Rispens and J. C. Hummelen, Angew. Chem., Int. Ed., 2002, 41, 838 CrossRef CAS.
- D. M. Guldi and M. Prato, Acc. Chem. Res., 2000, 33, 695 CrossRef CAS.
- (a) D. M. Guldi and K. D. Asmus, J. Phys. Chem. A, 1997, 101, 1472 CrossRef CAS; (b) D. M. Guldi and M. Maggini, Gazz. Chim. Ital., 1997, 127, 779 CAS.
- (a) M. Fujitsuka, O. Ito, Y. Yamahiro, Y. Aso and T. Otsubo, J. Phys. Chem. A, 2000, 104, 4876 CrossRef CAS; (b) M. Fujitsuka, K. Matsumoto, O. Ito, T. Yamashiro, Y. Aso and T. Otsubo, Res. Chem. Intermed., 2001, 27, 9930 CrossRef CAS.
- M. Fujitsuka, H. Masahura, H. Kasai, H. Oikawa, H. Nakanishi, O. Ito, T. Yamashiro, Y. Aso and T. Otsubo, J. Phys. Chem. B, 2001, 105, 9930 CrossRef CAS.
- K. G. Thomas, V. Biju, D. M. Guldi, P. V. Kamat and M. V. George, J. Phys. Chem. B, 1999, 103, 8864 CrossRef CAS.
- S. E. Shaheen, C. J. Brabec, N. S. Sariciftci, F. Padinger, T. Fromherz and J. C. Hummelen, Appl. Phys. Lett., 2001, 78, 841 CrossRef CAS.
- For a review on the “double cable” approach, see: A. Cravino and A. N. S. Sariciftci, J. Mater. Chem., 2002, 12, 1931 Search PubMed.
- T. Gu, D. Tsamouras, C. Melzer, V. Krasnikov, J.-P. Gisselbrecht, M. Gross, G. Hadziioannou and J.-F. Nierengarten, ChemPhysChem, 2002, 1, 124 CrossRef.
- N. Negishi, K. Yamada, K. Takimiya, Y. Aso, T. Otsubo and Y. Harima, Chem. Lett., 2003, 32, 404 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2005 |