Nucleobases as supramolecular motifs
Sona
Sivakova
and
Stuart J.
Rowan
*
Department of Macromolecular Science and Engineering, Case Western Reserve University, 2100 Adelbert Road, Cleveland, OH, USA. E-mail: stuart.rowan@case.edu; Fax: +1 216 368 4202; Tel: +1 216 368 4242
First published on 6th December 2004
Abstract
The five main natural nucleobases adenine, cytosine, guanine, thymine and uracil are involved in the self-assembly of one of nature's most interesting and intriguing class of biopolymers, namely the nucleic acids DNA and RNA. As such, these nucleobases have held a fascination to researchers in a diverse range of fields. With the growth in the field of supramolecular chemistry and consequently a better understanding of how molecules interact with each other, more and more information is emerging on the complex supramolecular behaviour of these nucleobase. This tutorial review tries to bring together some of the basic concepts of how nucleobases can interact not only with each other, but also with other small organic molecules as well as metals and then looks at how such an understanding is starting to influence the development of new materials and polymers.
 Sona Sivakova Sona Sivakova | Sona Sivakova was born in Presov, Slovakia. She came to the United States in 1989 and obtained her BS at Case Western Reserve University in Biomedical Engineering in 2000. She returned to Case to pursue her PhD in the Macromolecular Science and Engineering department under the supervision of Stuart Rowan. Her work focuses on utilizing DNA nucleobases in the construction of supramolecular polymeric systems. |
 Stuart J. Rowan Stuart J. Rowan | Stuart J. Rowan received his BSc and PhD from the University of Glasgow, where he worked with Dr David D. MacNicol on Supramolecular Crystal Engineering. In 1994 he moved to Cambridge to work with Professor Jeremy K. M. Sanders and continued his postdoctoral studies with Professor J. Fraser Stoddart at UCLA. In 1999 he was appointed as an Assistant Professor to the Department of Macromolecular Science at Case Western Reserve University. His research interests focus on the potential of dynamic chemistry (covalent and non-covalent) in the construction and properties of polymeric materials and developing new synthetic methods for the construction of complex polymeric architectures. |
Introduction
The non-covalent bond is a powerful synthetic tool in the preparation of complex molecular architectures. This combined with the ability to utilize such dynamic bonds to impart tuneable, responsive properties on the resulting aggregates has led to a dramatic growth in activity and subsequent understanding of this area over the last 30 years. The nature of such self-assembled products has a strong dependence not only on the molecular components they contain but also on the type of interactions which hold them together. What helps to make this field so intriguing is the wealth of supramolecular bonds that molecules use to interact with each other and their subtle interplay. Such non-covalent interactions range from purely electrostatic bonds to hydrophobic effects, from hydrogen bonding to phase segregation and from metal–ligand chemistry to π–π interactions. Each of these interactions offers differences in strength, binding kinetics, directionality and useful media that allow one to pick and choose the appropriate interaction for the desired purpose. For example, nature has excelled at utilizing supramolecular chemistry to store, transmit and replicate information in a challenging environment with a limited number of structural units. DNA and DNA-like materials offer the opportunity of preparing controlled self-assembled architectures. The interaction between two DNA strands is primarily mediated by four nucleobases adenine (A), thymine (T), guanine (G) and cytosine (C).1 The two anti-parallel strands of DNA are held together by A–T and C–G base pairs to form the famous double helix. While the selectivity of these base-pair interactions is controlled mainly by hydrogen bonding, both π–π stacking and hydrophobic effects also play a role in stabilizing the resulting structure. Utilization of the common nucleobases in supramolecular chemistry offers the flexibility of exploiting four different binding units A, C, G, and T (or U), all of which offer different binding characteristics. A limiting factor, however, in the utilization of single base-pairing interactions lies in the fact that in polar solvents, in which functionalized purine (A, G) and pyrimidine (C, T) derived systems are generally most soluble, the energies of the complementary base-pair interactions are small. Biological systems have overcome this problem by utilizing a combination of non-covalent interactions, in addition to multiple base pairs, to contribute toward the formation of the desired complexes. Fig. 1, as an example, shows some of the possible non-covalent interactions of guanine and its corresponding nucleotide deoxyguanosine monophosphate which could be utilized in supramolecular systems. All nucleobases offer a similar plethora of potential non-covalent interactions, and so a multiple site binding approach, where the weak base-pairing interactions are complemented by additional binding forces, adds an extra dimension to the use of nucleobase recognition in supramolecular chemistry. Furthermore, nucleotides, which also contain a charged sugar phosphate, expand on these molecules’ useful repertoire of non-covalent bonds by including electrostatic interactions.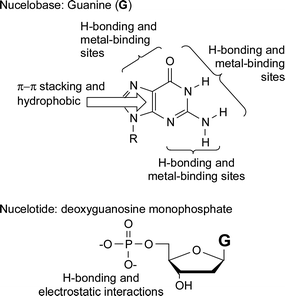 | ||
| Fig. 1 Some of the non-covalent interactions which are present in the nucleobase guanine and the nucleotide deoxyguanosine monophosphate. | ||
This review will mainly focus on the recent use of single nucleobase pairs in supramolecular chemistry concentrating on three different but inter-related areas. The first two sections will examine recent developments which primarily use mono-nucleobases as either hydrogen bonding motifs or as metal-binding ligands, while the final section will look at the recent uses of individual nucleobase interactions in polymeric systems. The idea of this review is to show the flexibility of the natural nucleobases as supramolecular motifs.
Hydrogen bonding of nucleobases
There are two major nucleobase binding motifs present in nucleic acids, adenine–thymine, AT (or adenine–uracil, AU in RNA) and guanine–cytosine, GC. These nucleobase pairs interact via 2 or 3 hydrogen bonds, respectively (Fig. 2a). However, while this Watson–Crick base pairing is dominant within nucleic acids, it is important to note that the nucleobases are not exclusive in their binding behaviour. There are 28 possible base-pairing motifs that involve at least two hydrogen bonds which can be formed between the four common nucleobases.1 These include reverse Watson–Crick (Fig. 2a), Hoogsteen and ‘wobble’ (or mismatched) base pairs (Fig. 2b). These, and other nucleobase binding modes, can play important roles in any nucleobase self-assembly process which is controlled by its hydrogen bonding, especially but not exclusively, when the interaction is not constrained by the geometry of the double helix. Furthermore, the purines, A and G, also contain two major hydrogen bonding sites, the Watson–Crick and the Hoogsteen sites. Thus, in addition to being able to form 1 : 1 complexes through either of these two sites, both of these nucleobases can form 2 : 1 aggregates (or base-triplets) with an appropriate partner (Fig. 2c). Such complexes are involved in the formation of triple helix DNA.2 In the case of the C+ : G : C base-triplet, the N3 of one of the cytosines needs to be protonated in order to allow the donation of a hydrogen bond to the N7 of guanine. Furthermore, in addition to interacting with other nucleobases, each of the nucleobases can also homodimerize. In the case of purines, A and G, larger aggregates are also possible on account of the two binding sites. Guanine in particular is known to self aggregate3 into tapes and macrocycles aided by its relatively high association constant for dimerization (KGGca. 102–104 M−1 in CDCl3, cf. KAAca. <5 M−1 in CDCl3).4,5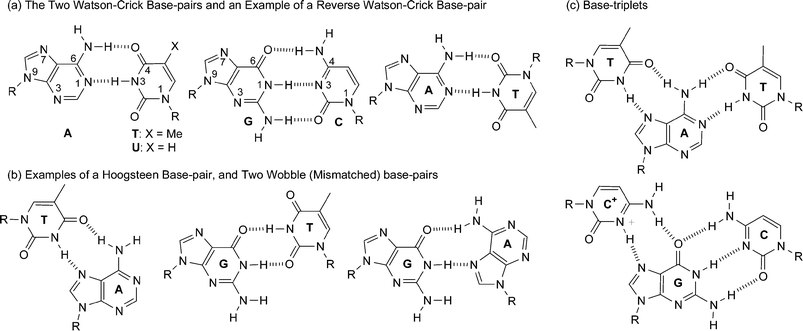 | ||
| Fig. 2 Some different hydrogen-bonding modes of nucleobases. | ||
Adenine and/or thymine
The degree of interaction between adenine and thymine (KATca. 100 M−1 in CDCl3) is significantly lower than between cytosine and guanine (KCGca. 104–105 M−1 in CDCl3).4 Nonetheless, over the years, the A–T interaction has been utilized in a variety of interesting supramolecular systems (e.g. see Fig. 3) by a variety of groups.6–10 Generally, multiple nucleobase binding sites are utilized to obtain significant binding constants. For example, all the systems shown in Fig. 3 utilize hydrogen bonding in conjunction with additional hydrogen bonding sites and/or π–π stacking interactions.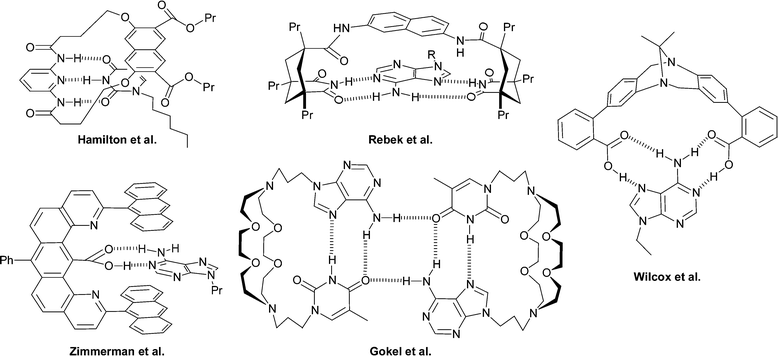
More recently, the Watson–Crick base pairing between A and T has been exploited in such a way that its recognition generates output signals that are in accordance with logic gate behaviour.11 4,6′-Diamidino-2-phenylindole (1) is a DNA-binding dye which utilizes a combination of π–π stacking and electrostatic interactions to favour binding into AT rich DNA regions. Therefore, the nucleotides 2 and 3 were used as the logic gate inputs with the fluorescence response of the system acting as the output. A concurrent blue shift and reduction in intensity of the dye's emission peak is observed only upon binding the 2·3 complex (Fig. 4). It is proposed that the interaction of the 2·3 complex with the dye causes a microenvironmental change and possible changes in the protonation state of the amidino functionalities, resulting in spectral changes in the dye. A single nucleotide is insufficient to produce this particular behaviour which means that this system operates in a similar fashion to a NAND logic gate. Interestingly enough, this system is selective to the nature of the nucleotide used as the input, showing no drop in emission intensity (at 455 nm) with other nucleotide combinations (e.g. deoxycytidine 5′-phosphate and deoxyguanosine 5′-phosphate).
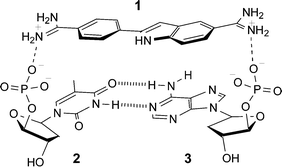 | ||
| Fig. 4 Proposed ternary complex (1·2·3) that results in fluorescence intensity changes. | ||
It is important to note that the substituents attached to the nucleobase can play a role in controlling the nature of the supramolecular assembly that is formed. An example from Ward and Barigelletti is shown in Fig. 5,12 where an adenine is attached to a 2,2′-bipyridine ligand which is then subsequently complexed to RuII moieties. When this ligand is complexed to RuII(2,2′-bipyridine)2 to yield 4 then the resulting supramolecular structure in the solid state is the expected Watson–Crick hydrogen bonded dimer (Fig. 5a). However, if the same adenine derived ligand is bound to the bulkier RuII(5,5′di-tert-butyl-2,2′-bipyridine)2 complex resulting in 5, then a dramatically different supramolecular array is observed (Fig. 5b). In this system, adenine undergoes self-association in the solid state to form a linear ribbon through an unusual set of hydrogen bonds, namely N3⋯H-N6 and N1⋯H–CH. It is proposed that the dominant controlling feature in the formation of this hydrogen-bonded ribbon is the requirement to maximise the separation between the positively charged metal complex units. It was hoped that the formation of an AT base pair between 4 (or 5) and a related thymine containing osmium complex would facilitate photoinduced energy transfer between the metal centres. Unfortunately, the degree of interaction (Kaca. 60 M−1) of this nucleobase pair was not large enough to see any significant effect in the luminescent behaviour in solution. This led the same group to prepare Ru(II) and Os(II) complexes similar to ones shown in Fig. 5 but with cytosine and guanine as the pendant nucleobases.13 The much larger degree of interaction of this nucleobase pair (Ka > 5 × 103 M−1) was such that a Ru → Os photoinduced energy transfer (rate constant, ken = 9.3 × 107 s−1) was observed on account of the formation of the hydrogen bond mediated supramolecular structure.
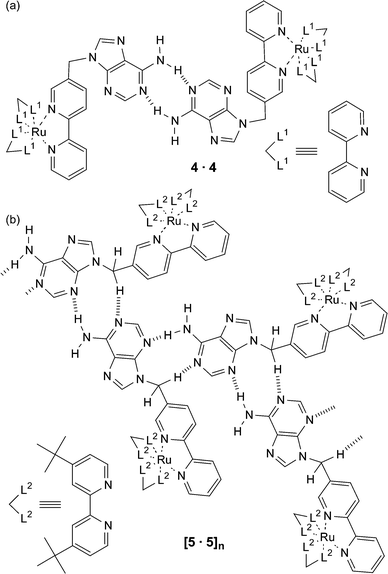 | ||
| Fig. 5 A schematic representation of the different modes of hydrogen bonding that occur in the crystal lattice of two related adenine derivatives. | ||
Cytosine and guanine
As we have just discussed above, the GC interaction is attractive as a supramolecular motif on account of its relatively strong degree of interaction (>104 M−1 in CDCl3). However, the more extensive synthesis required to make derivatives of these nucleobases and the reduced solubility of the guanine derivatives has somewhat limited its use in synthetic systems. Nonetheless, there are some elegant examples where this binding motif has been utilized in supramolecular chemistry. In an early example14 from Sessler and Harriman, cytidine and guanosine bearing porphyrins were used to examine the effect of non-covalent interactions on a photoinduced energy transfer process between donor and acceptor units.Connecting the protected nucleosides cytidine and guanosine moieties together should result in the spontaneous formation of oligomeric structures. Sessler et al. have placed a double bond between these complementary units resulting in a monomer unit which self-assembles into a trimeric species 63 (Fig. 6), which is stable enough to be observed by GPC over a broad range of concentrations (0.001–0.01 M).15
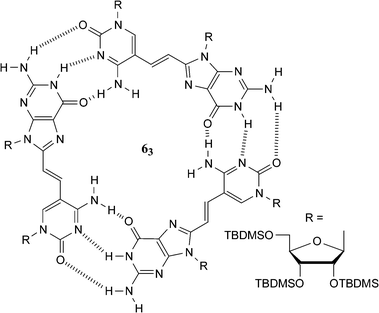 | ||
| Fig. 6 A guanosine–cytidine dinucleoside that self-assembles into a cyclic trimer (63). | ||
Sessler et al. have also utilized electrostatic interactions to complement the interaction of guanine and cytosine derivatives. In this case, a positively charged sapphyrin–cytosine conjugate (7) was utilized to bind the nucleotide, guanosine 5′-monophosphate 8, which contains the charged sugar phosphate moiety.16 The positively charged sapphyrin was designed to interact electrostatically with the phosphate group of the guest nucleotide while the cytosine unit interacts via hydrogen bonding, which not only aids the formation of the complex but also imparts some nucleobase selectivity to the host molecule. The resulting complex (Fig. 7) allows the selective transport of guanosine 5′-monophosphate 8, over other nucleotides, across a membrane. More recently, the same group prepared a similar nucleotide binding system based on calix[4]pyrroles which uses hydrogen bonding to the phosphates rather than electrostatic interactions to enhance the nucleobase binding.17
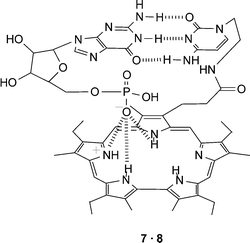 | ||
| Fig. 7 A combination of electrostatic and hydrogen bonding interactions are utilized in the proposed complex formed between sapphyrin–cytosine conjugate 7 and guanosine 5′-monophosphate 8. | ||
The major groove in DNA provides an important binding site for a variety of molecules. The nucleobases provide a range of multiple hydrogen bonding motifs that are placed along the major groove. For example, aided by the formation of Hoogsteen hydrogen bonding interactions (Fig. 2c), an extra nucleic acid strand can be bound in this region resulting in the formation of triple helix DNA. The natural nucleobases generally only recognize the major groove hydrogen bonding sites expressed by either GC or TA. This has led to a number of studies focused at designing receptors which are capable of selectively binding to other DNA base pair major groove sites. For example, within the major groove, the CG base pair expresses a different array of hydrogen bonds than not only TA or AT base pairs but also the GC base pair. So a system designed to hydrogen bond to the CG base pair should show selective binding in the major groove. The urocanamide derivative 9 (Fig. 8) was designed to interact with a CG base pair through two hydrogen bonds with the 4-amino group of C and the 6-carbonyl oxygen of G.18 NMR titrations measured an association constant of 37 M−1 in CD2Cl2. A receptor (10) which is also capable of interacting with the CG base pair has been designed by Zimmerman et al.1910 can interact with the CG base pair through three hydrogen bonds and this extra hydrogen bond results in a much stronger binding interaction (ca. 103 M−1 in CDCl3). The benefit of this extra hydrogen bond is highlighted by a large reduction in the degree of interaction of a similar receptor in which the ureido moiety is replaced by an amide. This modification reduces the possible hydrogen bonding sites from three to two and as a result the binding constant drops to ca. 70 M−1 in CDCl3.
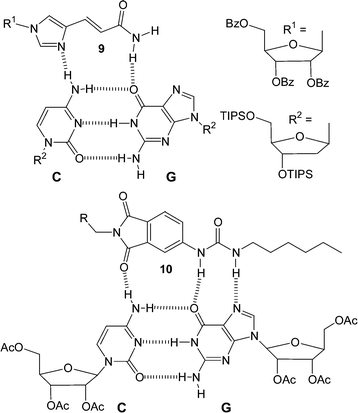 | ||
| Fig. 8 Two examples of molecules designed to bind to the CG base pair through hydrogen bonding. | ||
Guanine–guanine interactions
All nucleobases have the ability to homodimerize. However, of the natural nucleobases, homooligomers of guanine derivatives are perhaps the most interesting. The presence of two complementary hydrogen bonding arrays, a donor/donor site comprising the N1H amide and N2H amine and an acceptor/acceptor site comprising the O6 and N7 functionalities, within the guanine molecule allows it to oligomerize via hydrogen bonding. This supramolecular setup combined with the strength of the interaction (KGGca. 102-104 M−1 in CDCl3), makes it the most studied homonucleobase interaction.3,5,20Guanine derivatives can utilize hydrogen bonding to self-assemble into either linear tapes21 or macrocycles.3Fig. 9a shows an example of a guanine linear tape which self-assembles through Hoogsteen interactions. The other possibility is for the self-assembly to result in macrocycles. In fact, guanine is well known to self-assemble into hydrogen-bonded cyclic tetramers, the so-called G-quartet, in the presence of metal ions (Fig. 9b). In recent times there has been a significant amount of interest in the G-quartet. This, in part, has been fuelled by the observation that this self-assembled arrangement not only occurs in a variety of G-rich nucleic acid structures, but has also been implicated in a variety of biological functions. In fact, there exist such nucleic acid structures where G-quartets form four-stranded column-like superstructures called G-quadruplexes (Fig. 9c). This superstructure is not limited to nucleic acids, for example, work on lipophilic guanosine derivatives, e.g. 11 (Fig. 10), has shown that metal ions, such as K+, template the formation of not only the G-quartet but also the G-quadruplex in solution.22 NMR evidence is consistent with 11 self-associating in organic solvents to form stable dimeric G-quartet complexes in which the metal ion is believed to be sandwiched between the two quartets (G8-M). These octamers can further assemble into higher order structures. In the solid-state, a lipophilic G-quadruplex is formed which consists of 16 units of 11 and four equivalents of a metal picrate. This G-quadruplex consists of four stacked G quartets that are stabilized by a column of K+ cations in a central channel (Fig. 10a). The exterior surface of the G quadruplex has modified sugars, is hydrophobic, and has a belt of four lipophilic picrate ions (not shown) encircling the filled ion channel.
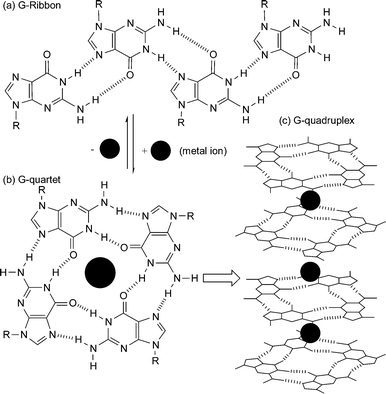 | ||
| Fig. 9 Self-assembly of guanine derivatives into (a) G-ribbon, (b) G-quartet, and (c) G-quadruplex formed by stacking of G-quartets around a column of cations. | ||
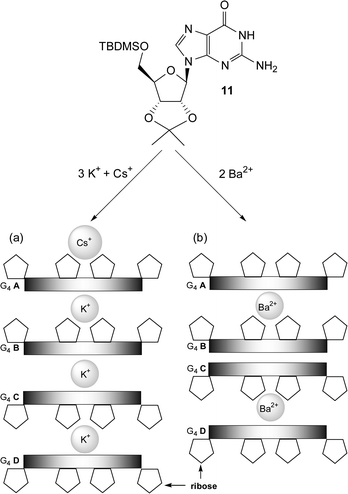 | ||
| Fig. 10 Schematic representations of the crystal structures of the G-quadruplexes formed by four G-quartets (G4A–G4D) of 11 in the presence of (a) K+ picrate and Cs+ picrate and (b) Ba2+ picrate. The picrate anions are not shown for clarity. | ||
Guanosine quartets are capable of binding either mono or divalent cations to form G8-M sandwiches. While both K+ and Ba2+ have similar ionic radii, the nature of these templating ions has a large effect on both the resulting structure and dynamics of G-quadruplexes.23 NMR integration has showed that 11 dissolved in methylene chloride can extract Ba2+ picrate from water to give a hydrogen bonded complex consisting of eight equivalents of nucleoside bound to each Ba2+ picrate. The crystal structure of the Ba2+ complex shows that the Ba2+ ions are only found between individual octamers and no cation is located between the “inner” two G quartets (Fig. 10b). Furthermore, when K+ is used as the templating ion with (D,L)-11, a mixture of heterochiral diastereoisomers are formed. However, Ba2+ directs enantiomeric self-recognition of the quadruplexes, resulting in the formation of the two homochiral species effectively displaying chiral resolution at the supramolecular level (Fig. 11). The Ba2+·G-quadruplex is more thermodynamically and kinetically stable then its corresponding K+ templated G-quadruplex. For example, stirring a dichloromethane solution of the K+ G-quadruplexes with an aqueous solution of Ba2+ picrate establishes an equilibrium favoring the homochiral Ba2+ quadruplexes. It is proposed that the increase in the divalent cations charge density controls the enantiomeric self-recognition process by strengthening the G-quartet hydrogen bonds, G-quadruplex–picrate interactions and cation–dipole interactions. Together, these provide enough enthalpic stabilization to overcome the unfavorable entropy associated with enantiomeric self-assembly.
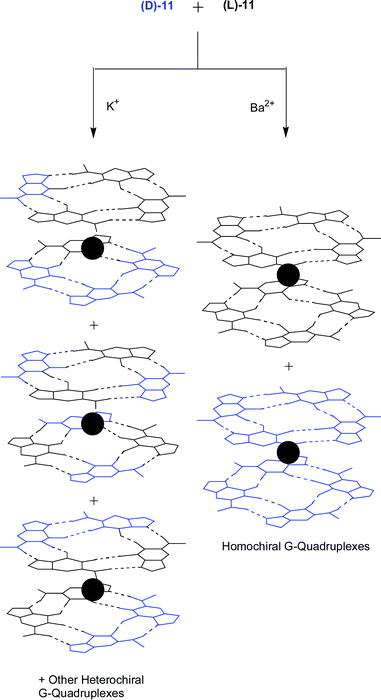 | ||
| Fig. 11 The effect of the templating ion on the formation of G-quadruplexes from (D,L)-11. The Ba2+ ion templates the enantiomeric self-recognition of the quadruplexes, enabling chiral resolution on the supramolecular level. | ||
G-Quartets have further been utilized to design self-assembled nanotubes from calix[4]arene–guanosine conjugates.24 The approach aimed at attaching four G moieties to a calix[4]arene-1,3-alternate scaffold (12) (Fig. 12). The 1,3-alt core in the monomer orients two orthogonal pairs of self-complementary G nucleosides so that they are well positioned for intermolecular hydrogen bonding with neighboring monomers. Electron microscopy images of precipitated material showed micrometre-long strands, visible as either single tubes or as bundles of tubes with a diameter ca. 3 nm. This assembly process was also completely reversible. Both temperature and pH can be used to control the cation-templated aggregation.
![(a) G4-Calixarene (12). (b) Nanotube formation via12 utilizing cation-templated self-assembly. Squares represent the 1,3-alt-calix[4]arene core, ovals represent the guanosine moieties, and solid spheres represent the Na+ cations.](/image/article/2005/CS/b304608g/b304608g-f12.gif) | ||
| Fig. 12 (a) G4-Calixarene (12). (b) Nanotube formation via12 utilizing cation-templated self-assembly. Squares represent the 1,3-alt-calix[4]arene core, ovals represent the guanosine moieties, and solid spheres represent the Na+ cations. | ||
Interestingly enough, Sessler et al. have demonstrated that certain derivatives of guanosine can form G-quartets without the presence of templating cations.25 The guanosine derivative 13, which has a N,N-dimethylaniline substituent on the 8-position of the guanine as well as the ribose unit protected with isobutyryl groups (Fig. 13), forms the G-quartet in both solution and solid state. It is believed that the presence of the aryl group on the 8-position of the guanine and/or hydrogen bonding between the N2-H of the guanine and the C5′ isobutyryl carbonyl forces 13 into a syn rather than the more conventional anti conformation and as a consequence favours the formation of G-quartets rather than linear structures.
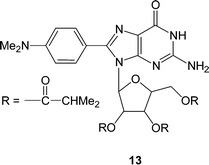 | ||
| Fig. 13 The syn conformation adopted by the 8-(N,N-dimethylaniline) guanosine derivative 13 which forms G-quartets without the presence of the templating cations | ||
Another synthetic modification of guanosine derivatives has allowed Sessler et al. to design a new base-dimerization motif. Conversion of a N2-H into a formamidine moiety (NCHNH2) imparts on the guanine units the ability to dimerize via a four-hydrogen bonded array.26Fig. 14 shows a dimeric complex 14·14 that is formed by the interaction of two sets of these four hydrogen bonded guanosine derivatives. This supramolecular structure, which is held together by a total of eight hydrogen bonds, is believed to be so stable that it persists even in strongly hydrogen bonding competing solvents, such as DMSO.
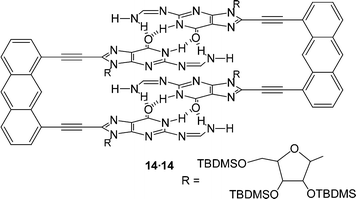 | ||
| Fig. 14 Guanine derivative 14 constrained within a rigid framework held together by a pair of four-point hydrogen bonds. | ||
Metal binding of nucleobases
The presence of accessible nitrogen and oxygen lone pairs in the nucleobases opens up the possibility of these molecules acting as metal ligands. Studies into metal ion–nucleobase interactions are of great interest since metal ions play a crucial role in the structure and function of nucleic acids and genetic information transfer. In addition, supramolecular assemblies containing transition metal ions are of particular interest on account of the functional properties conferred upon them by the presence of the metal centers. The wealth of potential metal binding sites on the heterocyclic bases of DNA makes this an interesting and complex field. Recently, a number of excellent reviews27 have been published detailing this area so rather than focusing on all the different possibilities, this section of the review will outline some basic concepts and focus on some recent examples where the nucleobase–metal interaction is used as a supramolecular motif.In DNA itself, major metal binding sites include those accessible in either the major groove, such as the N7 positions of both purines (A and G), or the minor groove, such as N3 of adenine and the O2 of thymine. For single nucleobases it is generally possible to metallate almost any site of the nucleobase including the carbon atoms. Of course, some metal binding sites are more accessible to metal binding than others. Fig. 15 shows some of the major metal ion binding sites in the four major nucleobases. There are a number of metal sites which are accessed upon deprotonation (or proton shift) of the nucleobase, e.g. exocyclic amines of A, C and G and endocyclic N-Hs in G and T.
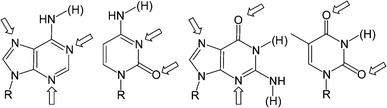 | ||
| Fig. 15 Arrows showing the major metal-binding sites on the four nucleobases. The H atoms in parentheses are attached to additional sites which can be involved in metal binding upon removal of the hydrogen. | ||
Metal binding can have a number of effects on the supramolecular chemistry of the nucleobases. We have already seen an example where metal ions, which can bind to the O6 of guanine, template the formation of the guanine quartet, favouring this macrocyclic structure over linear analogues. The nucleobases can act as simple ligands interacting with each other through the metal ion. Alternatively, the metal can block the hydrogen bonding site to which it is bound. This results in either preventing the nucleobase from participating in hydrogen bonding or changing the mode of hydrogen bonding the nucleobase can participate in. Metal binding can also have an electronic effect on hydrogen bonding altering the strength of this supramolecular interaction or even result in the formation of rare nucleobase tautomers allowing the stabilization of mismatched base pairs.28
Adenine and thymine metal coordination
A number of supramolecular receptors have been designed that utilize the metal–nucleobase interaction as the major mode of binding. For example, a binuclear Zn(II) complex with an oxa-aza macrocyclic ligand has been shown to bind two deprotonated thymine (or uracil) moieties.29 Ligand 15 (Fig. 16) contains two major binding sites, namely two triamine chains which are separated by two weakly coordinating polyoxa chains. Each triamine unit can bind to one Zn(II) ion and present an unsaturated coordinated sphere. The Zn(II) ion can then bind to thymine (or uracil) at neutral or slightly basic pH (pH < 9). In addition to the metal–nucleobase interaction, which occurs through the deprotonated N3 of the thymine, two hydrogen bonds are formed by the thymine oxygens and the ligand’s nitrogen protons. The binuclear Zn(II) complex displays similar binding features towards thymine and uracil forming stable 1 : 1 and 1 : 2 complexes in aqueous solutions (K1ca. 105, K2ca. 105 M−1) indicating an advantage of using metal–ligand interactions in a strong hydrogen bonding environment such as water.2 displaying the intramolecular hydrogen bonds and the hydrogen bond pairing between thymine units of different [Zn2·15·T2] complexes.](/image/article/2005/CS/b304608g/b304608g-f16.gif) | ||
| Fig. 16 Ligand 15 which consists of two triamine chains and two weakly coordinating polyoxa chains and a schematic representation of crystal packing [Zn2·15·T2](ClO4)2 displaying the intramolecular hydrogen bonds and the hydrogen bond pairing between thymine units of different [Zn2·15·T2] complexes. | ||
In addition to the intramolecular hydrogen bonding, the crystal packing revealed the formation of hydrogen-bond pairing between the thymine units of different [Zn2·15·T2]2+ complexes (Fig. 16). Thus, while intramolecular hydrogen bonds aid the stability of the complex, intermolecular hydrogen bonding is involved in the assembly of supramolecular architectures based on infinite pillars of [Zn2·15·T2]2+ units.
Molecular receptors have been designed that employ not only metal–nucleobase coordination and hydrogen bonding but π–π stacking interactions as well.30 Loeb et al. designed receptor 16 which can bind adenine, through simultaneous metal–nucleobase interaction, via sigma-donation from an aromatic N atom to a Pd center, hydrogen bonding between the adenine and polyether O atoms, and π-stacking between the electron poor aromatic rings of the nucleobase and the electron-rich aromatic spacing units in the host. (Fig. 17)
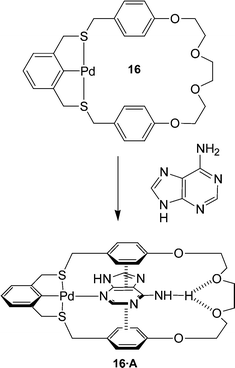 | ||
| Fig. 17 The thiocyclophane metalloreceptor which utilizes a combination of metal coordination, π–π stacking and hydrogen bonding to bind the adenine substrate. | ||
The binding mode observed for the 16·A complex involves the coordination to the N3 of adenine which orients the amino group for hydrogen bonding while simultaneously allowing for π-stacking interactions. Interestingly, adenine–metal binding usually occurs through the more basic N7 site suggesting that the other non-covalent interaction might be influencing the nature of the metal–substrate interaction to maximize their binding efficiency. Receptor 16 has also been shown to bind to guanine although this time, the metal–nucleobase interaction is through the expected N7 site.
Guanine and cytosine with metal coordination
Since the discovery of the antitumor activity of Cisplatin in 1969, studies into platinum–nucleobase interactions have played an important role in the investigation of nucleobases as ligands.27 The kinetically inert platinum–nucleobase interaction can be used to alter the nature of the hydrogen bonding exhibited by the nucleobase. For example, N7 platinated tris(9-methylguanine) exhibits a versatile range of hydrogen bonding interactions which can lead to the formation of 17, a twofold metallated guanine quartet (Fig. 18a). 17 can also interact with cytosine giving rise to interesting supramolecular structures.27,28,31 A centrosymmetric aggregate 18 (Fig. 18b) is produced which consists of two subunits of an unusual [GC]2 quartet. In each quartet the two 1-methylcytosine units are hydrogen bonded, in a Watson–Crick manner, to the trans-positioned 9-methylguanine ligands of the tris(guanine)amine–platinum(II), and interact with each other through a bridging water molecule. These two subunits are connected through an interesting G-G base pair. One of the guanine units is deprotonated allowing it to form a triple hydrogen bonded interaction with the second neutral guanine.![(a) Association of a twofold platinated guanine quartet 17. (b) View of supramolecular cation 18 consisting of two [guanine,cytosine]2 quartets and a gunanine,guanine base pair.](/image/article/2005/CS/b304608g/b304608g-f18.gif) | ||
| Fig. 18 (a) Association of a twofold platinated guanine quartet 17. (b) View of supramolecular cation 18 consisting of two [guanine,cytosine]2 quartets and a gunanine,guanine base pair. | ||
NMR experiments examining hydrogen bonding between N7 platinated guanines and free cytosine derivatives show that the strength of the Watson–Crick interaction between the N7 platinated guanine and cytosine is 2–3 times greater than in the case of 9-ethylguanine and cytosine.32 This increase of the association constant is attributed to the increase in the acidity of the N1H and the N2H2 sites, which is caused by the electron-withdrawing effects of the metal, resulting in both these sites becoming better hydrogen bond donors. It should be noted, however, that the metal binding also decreases the hydrogen bond accepting ability of the O6 site and this can reduce the binding constant of the base pair if too much electron density is withdrawn from the purine.
Nucleobases in polymeric and material systems
The attraction of using nucleobase interactions in material systems is many-fold. For long nucleic acid sequences the predictability of the interaction and their potential to interface efficiently with biological systems is very attractive from the point of view of forming controlled nano-structures and/or new biomaterials.33 However, in the last section of this review we will focus on some recent developments where single nucleobase interactions are used to control the self-assembly of new materials.Chain-end nucleobase modified monomers and polymers
In an effort to utilize the complementary nature of the nucleobase hydrogen bond interactions there have been a number of investigations aimed at examining the effect that placement of such supramolecular motifs on the ends of small molecules or polymer chains has on the organization of the systems and subsequently its material properties.Gelators are low molecular weight molecules that are capable of organizing themselves into higher-ordered structures. The aggregation of these gelators into, for example, rodlike or tubular structures results in the formation of a gel. Cholesterol-derivatives have been shown to be excellent gelators and they frequently exhibit a helical structure in gel-fibers. Shinkai, Reinhoudt and Snip have designed 19, a cholesterol derivative with a pendant uracil unit (Fig. 19) in an attempt to impart recognition properties to the gel.3419 forms stable gels in various non-polar solvents exhibiting high gelation temperatures. Addition of even a small amount of adenine resulted in a weakening of the gels suggesting that the uracil unit is involved in intergelator hydrogen bonds, which aids the stability of the gel. Furthermore, removal of the hydrogen bond donor sites via methylation of 19 results in 20, a material with a dramatically reduced gelation temperature. SEM microscopy micrographs show well developed networks consisting of fibrils having a helical structure.
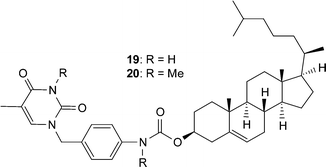 | ||
| Fig. 19 Cholesterol with pendant nucleobase. | ||
The chain-end functionalization of oligomethylene spacers with nucleotides has also resulted in the formation of gels.35 Shimizu and coworkers investigated the self-assembly of such bolaamphiphiles, which contain 3′-phosphorylated thymidine moiety at each end of a chain (Fig. 20), in aqueous solution and found that they form hydrogels. The oligomethylene core requires at least 18 carbons to exhibit excellent gelation ability under both neutral and weak alkaline aqueous conditions, suggesting that intermolecular hydrophobic interactions are the driving force of the hydrogelation. Microscopy studies of 21 confirmed the hydrogels consist of an intertwined network of nanofibers (with diameters ca. 10–30 nm).
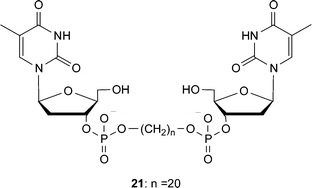 | ||
| Fig. 20 Thymine functionalized bolaamphiphile which can form hydrogels. | ||
The same group have also prepared nucleobase-appended bolaamphiphiles (without the phosphate sugar component) with thymine or adenine residues separated by oligomethylene spacers (Fig. 21a). This time, the molecules self-assemble in water to produce supramolecular nanofibers in a crystalline state without forming a hydrogel.36 The thymine bolaamphiphile 22 produced interesting double-helical ropes in 10% ethanolic/aqueous solutions. These ropes are thought to be initiated by a small amount of macrocyclic dimers [cyclo(22)2] which are formed via thymine photodimerization. The adenine appended 23 did not form fibers by itself however nanofibers were formed from an equimolar mixture of the homoditopic 22 : 23 as well as from the heteroditopic 24. X-Ray studies of these materials containing the complementary nucleobases is consistent with the formation of a multilamellar arrangement in the solid-state with nucleobase interactions extending the chain length and a combination of hydrogen bonding through the amide linkers and nucleobase π–π stacking between the chains (Fig. 21b).
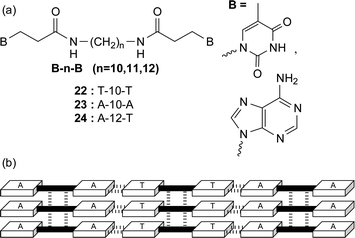 | ||
| Fig. 21 (a) Nucleobase terminated bolaamphiphiles and (b) a possible hydrogen bond scheme for the heteroassembly formed from an equimolar mixture of 22 and 23. | ||
Low molecular weight macromonomers that have nucleobases placed on either one or both of their chains-ends can result in the material behaving like a high molecular weight polymer.37 For example, attachment of nucleobase derivatives on to low molecular weight poly(tetrahydrofuran) has been shown to have a profound effect on the materials’ properties.37a The commercially available bis(3-aminopropyl) terminated poly(tetrahydrofuran) (Mn ca. 1400), is a soft waxy solid with a melting point of around 21 °C. Attachment of a nucleobase derivative—either thymine acetic acid or N6-(4-methoxybenzoyl)-9-(carboxymethyl)adenine (AAnCH2CO2H)—at each chain end resulted in a marked change in the properties of the material with an increase of over 100 °C in the melting point (Fig. 22a). The adenine-derived supramolecular telechelic polymer 26 self-assembles in the solid state to yield materials with enough mechanical stability to be melt processed. Upon heating and cooling the material an optically clear film is formed which shows pronounced thermally sensitive behaviour. Above 130 °C, this material forms a low viscous melt. However, upon cooling below this ceiling temperature, a marked change in material properties is observed and the supramolecular material can be processed into films (Fig. 22b) and fibers. Interestingly, the thymine modified material 25 did not exhibit such properties due to a high crystallization temperature (ca. 145 °C) which prevents any film or fiber-forming ability. It is believed that a number of non-covalent forces allow the formation of these thermoreversible supramolecular materials. The weak dimerization constant of the AAn (< 5 M−1 in CDCl3) suggests that the self-assembly was additionally driven by other factors such as π–π stacking, dipole–dipole interactions, phase segregation between the hard nucleobase components and the soft poly(tetrahydrofuran) chain, as well as the hydrogen bonding of the amide bond, which links the nucleobase to the polymer.
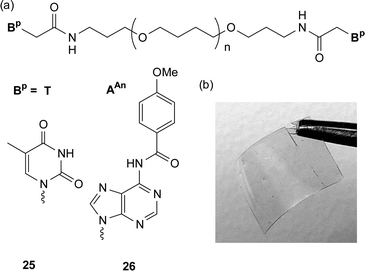 | ||
| Fig. 22 (a) Supramolecular telechelic macromonomer and (b) film from the adenine modified macromonomer 26. | ||
The nucleobase derivatives T and AAn have also been attached to a functional core such as the mesogenic, fluorescent bis(4-alkoxy)-substituted bis(phenylethynyl)benzene core (Fig. 23a).38 The underivatized alkoxy-substituted bis(phenylethynyl)benzene is a known mesogen that exhibits several nematic and smectic liquid crystalline transitions. However, upon attachment of the nucleobases to its terminus, the material showed no propensity for liquid crystalline behaviour and the materials behaved as low molecular weight, high melting solids. Mixing the two complementary nucleobase-terminated monomers results in a dramatic decrease in the melting temperature along with a concurrent formation of a viscous birefringent phase. Fig. 23b shows the polarized optical micrograph of the viscous birefringent phase formed by a 1 : 1 mixture of 27 and 28. In addition to forming these birefringent phases, the mixed materials also displayed polymer-like properties. For example, oriented fibers could be obtained from the LC phase of this system (Fig. 23c). In addition, on account of the fluorescent core, these fibers were also fluorescent. Thus a combination of the functionality of the core unit and the self-assembling ability of the nucleobases results in the formation of a supramolecular, liquid crystalline, fluorescent polymeric material.
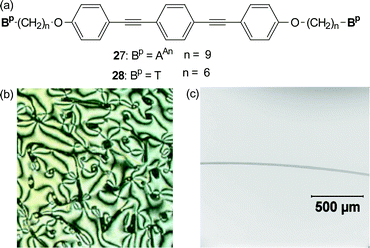 | ||
| Fig. 23 (a) The nucleobase terminated bis(4-alkoxy)-substituted bis(phenylethynyl)benzene derivatives, 27 and 28. (b) Optical micrograph of birefringent phase between. (c) Optical micrograph of a fiber of 27·28. | ||
The three hydrogen bonded thymine–2,6 diaminotriazine supramolecular motif has been utilized to make self-assembled pseudo-block copolymers.39 One problem frequently encountered in polymer blends is that of polymer immiscibility. Poly(isobutylene) (PIB) and poly(etherketone) (PEK) are two immiscible polymers that macrophase separate upon blending. The addition of complementary binding units to the ends of these polymers should provide an enthalpic driving force to aid miscibility. Blending the telechelic PEK with 2,4-diamino-1,3,5-triazine moieties, and PIB with thymine units attached, shows a dramatic increase in their miscibility and electron microscopy shows that a nanophase separated structure with a periodicity of 70 nm is formed. Interestingly enough if PIB with cytosine instead of thymine is blended with the functionalized PEK then the result is a disordered structure with phase separation on the micrometre scale. This can be explained by the nature of the non-covalent interaction within these two blended materials. The thymine and 2,6-diaminotriazine binding constant (890 M−1 in CDCl3) is much larger than the homodimerization of the supramolecular components, thus encouraging the formation of an alternating self assembled system. However this is not the case for the cytosine and 2,6-diaminetriazine where the heterodimer formation is much less than the homodimerization of the cytosine (8 M−1vs. 40 M−1 in CDCl3), which would therefore encourage a more phase separated structure.
Side-chain nucleobase modified polymers
As well as functionalizing chain ends of a polymer, it is of course also possible to place nucleobases on polymer side-chains. Rotello and co-workers have investigated this area utilizing side chain functionalized low molecular weight polystyrene and the three hydrogen bonded thymine/diamidopyridine supramolecular motif. For example, the low molecular weight polystyrene which is derivatized with diamidopyridine units (29) can be non-covalently crosslinked with bis-thymine units (30).40 Upon combination in non-competitive solvents, discrete stable micron-scale spherical aggregates were formed arising from specific three-point polymer-crosslinker hydrogen bonding interactions (Fig. 24a). The diameter of these gel microspheres could be controlled by varying the length of the spacer in the crosslinker. The cross-linking process was fully thermally reversible, with complete dissolution observed at 50 °C and reformation of the aggregates upon return to ambient temperature. This process could be repeated, with lower particle dispersity observed as a result of annealing.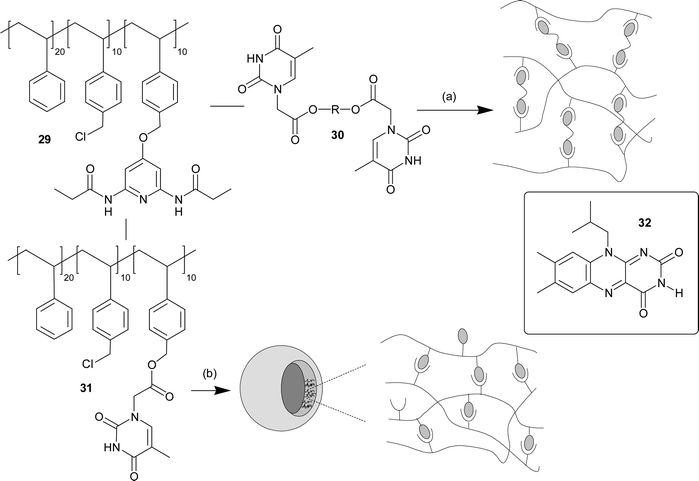 | ||
| Fig. 24 (a) Schematic illustration of non-covalent polymer cross-linking of copolymer 29 using a complementary bifunctional cross-linker 30. (b) Schematic representation of the diacyldiaminopyridine- and thymine-based random copolymers, 29 and 31, as assembled in three dimensional vesicles. | ||
If thymine is itself attached to the side chain of the low molecular weight polystyrene polymer (31), then addition to 29 results in slightly different behaviour with the formation of spontaneously self-assembling giant vesicles (Fig. 24b).41 These recognition induced polymersomes (RIPs) are formed through very specific interactions between the complementary random copolymer chains and thus are sensitive to the presence of units which contain the complementary supramolecular motif. For example, addition of monovalent imide guest 32 results in an initial swelling of the RIPs before the vesicles dissociated due to competitive binding of the small molecule guest resulting in decrosslinking of the polymers. Incorporation of multivalent guests was also investigated by adding gold nanoparticles which have about ca. 10 thymine units on their surface per nanoparticle. Here a contraction is observed in the size of the vesicle as the nanoparticle is incorporated. This behaviour was shown to require the supramolecular interaction as nanoparticles containing a methyl group on the N3 of the thymine units showed no incorporation into the vesicles.
Summary
When confronted with nucleobase interactions most people will automatically think of the Watson–Crick base pair, but over the years research from a variety of different disciplines has shown that nucleobases are much more promiscuous with their non-covalent interactions. What we have tried to do in this review is to bring just a small percentage of this research together from some of these disciplines and highlight the wealth of non-covalent interactions that can be utilized with the five main nucleobases. The ability to further understand how nucleobases interact both with themselves as well as with other molecules will not only allow the construction of systems which can bind more specifically to targeted DNA sequences but will also allow us to gain valuable insights on how we might be able to harness these interesting biological molecules to construct complex nanostructures and materials.Acknowledgements
The authors would like to thank the NIH (NIBIB: EB-001466-01), the NSF (CAREER: CHE-0133164), the US Army Research Office (DAAD19-03-1-0208) and the Case School of Engineering for financial support.References
- See for example: W. Saenger, Principles of Nucleic Acid Structures, Springer, New York, 1984 Search PubMed.
- See for example: V. N. Soyfer, V. N. Potaman, Triple-Helical Nucleic Acids, Springer, New York, 1995 Search PubMed.
- J. T. Davis, Angew. Chem., Int. Ed., 2004, 43, 668 CrossRef CAS and references cited therein.
- J. Sartorius and H.-J. Schneider, Chem. Eur. J., 1996, 2, 1446 CrossRef CAS and references cited therein.
- G. Gottarelli, S. Masiero, E. Mezzina, G. P. Spada, P. Mariani and M. Recanatini, Helv. Chim. Acta, 1998, 81, 2078 CrossRef CAS.
- See for example: A. D. Hamilton and D. Van Engen, J. Am. Chem. Soc., 1987, 109, 5035 Search PubMed.
- See for example: B. Askew, P. Ballester, C. Buhr, K. S. Jeong, S. Jones, K. Parris, K. Williams and J. Rebek, Jr., J. Am. Chem. Soc., 1989, 111, 1082 Search PubMed.
- See for example: S. C. Zimmerman, W. Wu and Z. Zeng, J. Am. Chem. Soc., 1991, 113, 196 Search PubMed.
- See for example: O. F. Schall and G. W. Gokel, J. Am. Chem. Soc., 1994, 116, 6089 Search PubMed.
- J. C. Adrian, Jr. and C. S. Wilcox, J. Am. Chem. Soc., 1989, 111, 8055 CrossRef.
- H. T. Baytekin and E. U. Akkaya, Org. Lett., 2000, 2, 1725 CrossRef CAS.
- C. M. White, M. F. Gonzalez, D. A. Bardwell, L. H. Rees, J. C. Jeffrey, M. D. Ward, N. Armaroli, G. Calogero and F. Barigelletti, J. Chem. Soc., Dalton Trans., 1997, 727 RSC.
- S. Encinas, N. R. M. Simpson, P. Andrews, M. D. Ward, C. M. White, N. Armaroli, F. Barigelletti and A. Houlton, New. J. Chem., 2000, 24, 987 RSC.
- See for example: J. L. Sessler, B. Wang and A. Harriman, J. Am. Chem. Soc., 1995, 117, 704 Search PubMed.
- J. L. Sessler, J. Jayawickramarajah, M. Sathiosatham, C. L. Sherman and J. S. Broabelt, Org. Lett., 2003, 5, 2627–2630 CrossRef CAS.
- V. Král, J. L. Sessler and H. Furuta, J. Am. Chem. Soc., 1992, 114, 8704 CrossRef CAS.
- J. L. Sessler, V. Král, T. V. Shishkanova and P. A. Gale, Proc. Natl. Acad. Sci. USA, 2002, 99, 4848 CrossRef CAS.
- M. G. M. Purwanto, D. Lengeler and K. Weisz, Tetrahedron. Lett., 2002, 43, 61 CrossRef CAS.
- E. Mertz, S. Mattei and S. C. Zimmerman, Org. Lett., 2000, 2, 2931 CrossRef CAS.
- For example, see: W. GuschlBauer, J.-F. Chantot and D. Thiele, J. Biomol. Struct. Dyn., 1990, 8, 491 Search PubMed and references cited therein.
- For a recent example, see: T. Giorgi, F. Grepioni, I. Manet, P. Mariani, S. Masiero, E. Mezzina, S. Pieraccini, L. Saturni, G. P. Spada and G. Gottarelli, Chem. Eur. J., 2002, 8, 2143–2152 Search PubMed and references cited therein.
- S. L. Forman, J. C. Fettinger, S. Pieraccini, G. Gottarelli and J. T. Davis, J. Am. Chem. Soc., 2000, 122, 4060 CrossRef CAS.
- X. Shi, J. C. Fettinger and J. T. Davis, J. Am. Chem. Soc, 2001, 123, 6738 CrossRef CAS.
- V. Sidorov, F. W. Kotch, M. El-kouedi and J. T. Davis, Chem. Commun., 2000, 2369 RSC.
- J. L. Sessler, M. Sathiosatham, K. Doerr, V. Lynch and K. A. Abboud, Angew. Chem., Int. Ed., 2000, 39, 1300–1303 CrossRef CAS.
- J. L. Sessler and R. Wang, Angew. Chem., Int. Ed., 1998, 37, 1726 CrossRef CAS.
- See for example: B. Lippert, Coord. Chem. Rev., 2000, 200–202, 487–516 Search PubMed and references cited therein.
- B. Lippert, J. Chem. Soc., Dalton Trans., 1997, 3971–3976 RSC and references cited therein.
- C. Bazzicalupi, A. Bencini, E. Berni, A. Bianchi, S. Ciattini, C. Giorgi, P. Paoletti and B. Valtancoli, Eur. J. Inorg. Chem., 2001, 629 CrossRef CAS.
- J. E. Kickham, S. J. Loeb and S. L. Murphy, Chem. Eur. J., 1997, 3, 1203 CrossRef CAS.
- E. Freisinger, S. Meier and B. Lippert, J. Chem. Soc., Dalton Trans., 2000, 3274–3280 RSC.
- R. K. O. Sigel, E. Freisinger and B. Lippert, JBIC, J. Biol. Inorg. Chem., 2000, 5, 287 Search PubMed.
- See for example: N. C. Seeman, Chem. Biol., 2003, 10, 1151 Search PubMed; J. J. Storhoff and C. A. Mirkin, Chem. Rev., 1999, 99, 1849 CrossRef CAS and references cited therein.
- E. Snip, S. Shinkai and D. N. Reinhoudt, Tetrahedron Lett., 2001, 42, 2153 CrossRef CAS.
- R. Iwaura, K. Yoshida, M. Masuda, K. Yase and T. Shimizu, Chem. Mater., 2002, 14, 3047 CrossRef CAS.
- T. Shimizu, R. Iwaura, M. Masuda, T. Hanada and K. Yase, J. Am. Chem. Soc., 2001, 123, 5947 CrossRef CAS.
- (a) S. J. Rowan, P. Suwanmala and S. Sivakova, J. Polym. Sci: Part A: Polym. Chem., 2003, 41, 3589 CrossRef CAS; (b) K. Yamauchi, J. R. Lizotte and T. E. Long, Macromolecules, 2002, 35, 8754 CrossRef.
- S. Sivakova and S. J. Rowan, Chem. Commun., 2003, 2428 RSC.
- (a) W. H. Binder, M. J. Kunz and E. Ingolic, J. Polym. Sci: Part A: Polym. Chem., 2004, 42, 162 CrossRef CAS; (b) W. H. Binder, M. J. Kunz, C. Kluger, G. Hayn and R. Saf, Macromolecules, 2004, 37, 1749 CrossRef CAS.
- R. J. Thibault, P. J. Hotchkiss, M. Gray and V. M. Rotello, J. Am. Chem. Soc., 2003, 125, 11249 CrossRef CAS.
- R. J. Thibault, T. H. Galow, E. J. Turnberg, M. Gray, P. J. Hotchkiss and V. M. Rotello, J. Am. Chem. Soc., 2002, 124, 15249 CrossRef.
| This journal is © The Royal Society of Chemistry 2005 |