Photoisomerisation of dibenzobarrelenes—a facile route to polycyclic synthons†
Danaboyina
Ramaiah
*a,
Meledathu C.
Sajimon
a,
Joshy
Joseph
a and
Manapurathu V.
George
*ab
aPhotosciences and Photonics Division, Regional Research Laboratory (CSIR), Trivandrum 695 019, India. E-mail: d_ramaiah@rediffmail.com; Fax: +91- 471 2490186; Tel: +91- 471 2490392
bJawaharlal Nehru Centre for Advanced Scientific Research, Bangalore 560 064, India. E-mail: mvgeorge@rediffmail.com
First published on 10th December 2004
Abstract
Triplet state mediated di-π-methane rearrangements of dibenzobarrelenes give a variety of interesting synthons, formed as primary and secondary photoproducts. These synthons could find use for the synthesis of complex synthetic targets. This tutorial review highlights the photoisomerisation of some bridgehead substituted dibenzobarrelenes and the products derived from them. Selected examples of photoisomerisations proceeding through a tri-π-methane pathway are also included.
 Danaboyina Ramaiah Danaboyina Ramaiah | Danaboyina Ramaiah was born in Karimnagar, Andhra Pradhesh, India in 1958. He received his PhD degree in 1988 from the Indian Institute of Technology, Kanpur. He joined the Regional Research Laboratory, Trivandrum in 1988 and is associated with the Photosciences and Photonics Division. He was an Alexander von Humboldt Fellow with Professor Dr. Waldemar Adam at the University of Würzburg, Germany (1992–1993) and Post-doctoral Fellow at the Georgia Institute of Technology, Atlanta, with Professor Gary B. Schuster (1997–1998). He was a Visiting Scientist at the University of Mainz, Germany with Professor Dr. Bernd Epe (1999–2002) and at the Georgia Institute of Technology, Atlanta, USA with Professor Gary B. Schuster (2002–2003). His current research interests include the photochemistry of multichromophoric organic compounds and design of sensitisers for photodynamic therapeutical applications, photoactivated DNA cleaving agents and probes for nucleosides and nucleotides. |
 Meledathu C. Sajimon Meledathu C. Sajimon | M. C. Sajimon was born in Kerala, India in 1974. He obtained the MSc degree in Chemistry (1996) from the Mahatma Gandhi University, Kottayam, India and PhD degree (2001) from the University of Kerala, Trivandrum, India. He worked with Dr. D. Ramaiah for his PhD degree on some aspects of the phototransformations of dibenzobarrelenes and related compounds, at the Photosciences and Photonics Division of the Regional Research Laboratory, Trivandrum. Subsequently he spent a year at the University of Würzburg, Germany with Professor Waldemar Adam (Alexander von Humboldt Fellow, 2001–2002). Presently he is working as a Postdoctoral Research Fellow at Northwestern University, USA with Professor Frederick D. Lewis. His research interests include organic reactions, photochemistry and laser chemistry, study of new photoreactions and mechanistic investigations. |
 Joshy Joseph Joshy Joseph | Joshy Joseph was born in Kerala, India in 1976. He obtained the MSc degree in Chemistry (1998) from the Mahatma Gandhi University, Kottayam, Kerala and PhD degree (2004) from the University of Kerala, Trivandrum. He worked for his PhD degree on synthesis, photophysical and DNA binding properties of acridine and acridinium derivatives under the supervision of Dr. D. Ramaiah at the Photosciences and Photonics Division of the Regional Research Laboratory (CSIR), Trivandrum. Currently he is working as a Postdoctoral Fellow at Georgia Institute of Technology, Atlanta, USA with Professor Gary B. Schuster. His research interests include organic photochemistry, design of photoactivated DNA cleaving agents and study of electron and energy transport in biological systems. |
 Manapurathu V. George Manapurathu V. George | M. V. George was Professor of Chemistry at the Indian Institute of Technology, Kanpur during 1965 to 1988 and since November 1988 he has been associated with the Photochemistry Research Unit (current name: Photosciences and Photonics Division) of the Regional Research Laboratory (CSIR), Trivandrum, India. Currently he is an Honorary Professor of the Jawaharlal Nehru Centre for Advanced Scientific Research, Bangalore at RRL, Trivandrum. He is a fellow of the Indian Academy of Sciences, Indian National Science Academy and the Third World Academy of Sciences. His research interests include study of organic reactions, new photoreactions and mechanistic investigations. He is the Executive Director of the Foundation for Capacity Building in Science (FCBS) (a foundation for the promotion of science education in India). |
1 Introduction
The di-π-methane rearrangement of 1,4-dienes containing two π-systems separated by a methane unit was first reported by Zimmerman and Grunewald in 1966 in the photoisomerisation of barrelene (1) to semibullvalene (2)1 (Scheme 1). Subsequent studies by various workers have shown that this is a very general reaction which takes place usually with high chemical and quantum yields giving rise to vinylcyclopropanes or related products, which are not readily available through alternative routes.2–4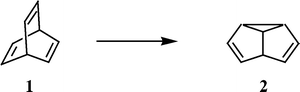 | ||
| Scheme 1 | ||
Extensive studies have been carried out on different aspects of this reaction such as the multiplicity of the excited state involved, the nature and stability of diradical intermediates, substituent effects, regioselectivity and stereoselectivity. In view of the pioneering and extensive contributions of Zimmerman and coworkers to illustrate the generality of di-π-methane rearrangement, this rearrangement is also referred to as the Zimmerman rearrangement. The application of di-π-methane reactions to other systems such as oxa-di-π-methane3,5 and aza-di-π-methane3 has also been illustrated.
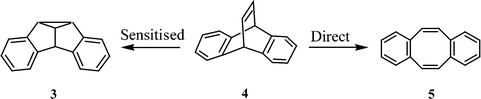
Di-π-methane rearrangement occurs readily when one of the two π-bonds is part of an aromatic ring. An illustrative example is the case of dibenzobarrelene (also referred to as 9,10-ethenoanthracene) (4), which gives dibenzosemibullvalene (3)6 under sensitised irradiation and dibenzocyclooctatetraene (5)7 under direct irradiation (Scheme 2). The photochemistry of several dibenzobarrelene derivatives has been examined in detail since then. A recent review by Scheffer and Yang highlights the results of some of these studies.8
 | ||
| Scheme 2 | ||
From our studies employing 11,12-dibenzoyl-substituted dibenzobarrelenes (or 11,12-dibenzoyl-9,10-dihydro-9,10-substituted-9,10-ethenoanthracenes) and those of others involving different substituted dibenzobarrelenes, it is evident that several interesting and synthetically significant polycyclic systems are derived from the primary photoreaction of these substrates and also through the secondary reactions of the initially formed photoproducts. In the present review, we shall focus on the photoreactions of substituted dibenzobarrelenes and with particular reference to 11,12-dibenzoyl-substituted dibenzobarrelenes and related systems. Although the primary photoproducts can be transformed further to give a variety of secondary photoproducts, we shall be discussing only those secondary photoproducts formed in the one pot reaction and not others derived through separate reactions.‡§
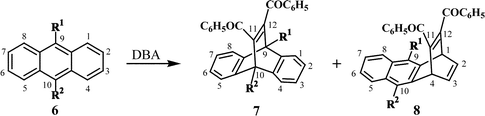
11,12-Dibenzoyl-substituted dibenzobarrelenes (7) can be conveniently prepared through the Diels–Alder addition of appropriately substituted anthracenes (6) with dibenzoylacetylene (DBA), either thermally or in the presence of Lewis acid catalysts such as aluminium chloride. Depending on the substituents and reaction conditions, the reaction yields either 11,12-dibenzoyl-substituted dibenzobarrelene (7) (formed through addition across the 9,10 positions of anthracene) or a mixture of 7 and dibenzoyl-substituted naphthobarrelene (8) (formed through addition across the 1,4 positions of anthracene) (Scheme 3).9
 | ||
| Scheme 3 | ||
It is generally known that dibenzobarrelenes give rise to dibenzosemibullvalenes (or 4b,8b,8c,8e-tetrahydrodibenzo[a,f]cyclopropa[c,d]pentalenes) through triplet state mediated pathways, whereas dibenzocyclooctatetraenes are formed through singlet state mediated pathways.6,7 In the case of 11,12-dibenzoyl-substituted dibenzobarrelenes, there will be a greater propensity for the formation of triplet state mediated products, due to the presence of the two benzoyl groups. In the following sections, we shall examine some of the different types of polycyclic ring systems that are formed in the solution photochemistry of 11,12-dibenzoyl-substituted dibenzobarrelenes.
2 Dibenzosemibullvalenes
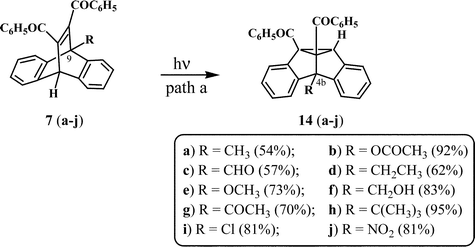
Dibenzosemibullvalenes are the products formed in the triplet state mediated isomerisation of dibenzobarrelenes. The di-π-methane route for the isomerisation of dibenzobarrelenes proceeds through initial “benzo–vinyl” bridging to give diradical intermediates. In the case of symmetrically substituted dibenzobarrelenes having C2v molecular symmetry, all four possible pathways for the “benzo–vinyl” bridging will lead to the same product. The situation however is different in the case of unsymmetrically substituted dibenzobarrelenes. In the case of bridgehead-monosubstituted dibenzobarrelene 7, for example, the di-π-methane pathway leads to the formation of the two regioisomeric dibenzosemibullvalenes 13 and 14, as shown in Scheme 4. Path a involves “benzo–vinyl” bridging (C(12)–C(9a) bridging) leading to the diradical intermediates 9 and 10, which would then be transformed to the 8b-substituted dibenzosemibullvalene 13. Path b, on the other hand (C(11)–C(4a) bridging), involves diradical intermediates 11 and 12, which ultimately lead to the 4b-substituted dibenzosemibullvalene 14. Interaction of the vinyl group and the phenyl ring on the left side (C(12)–C(8a) and C(11)–C(10a) bridging), however, will lead to the corresponding diradical intermediates, which will ultimately give the enantiomers of the dibenzosemibullvalenes 13 and 14.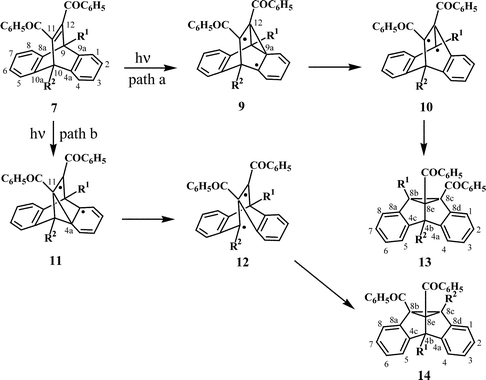 | ||
| Scheme 4 | ||
We have examined the photoisomerisation of several 9-substituted-11,12-dibenzoyldibenzobarrelenes to understand the factors responsible for the preferred regioselectivity leading to either 4b- or 8b-substituted dibenzosemibullvalenes. Scheme 5 summarises the percentage of 4b-isomer formed from several 9-substituted dibenzobarrelenes.
 | ||
| Scheme 5 | ||
In general, dibenzobarrelenes (7a–j) with bridgehead substitution by alkyl groups, nitro groups and halogens exclusively afford the 4b-substituted semibullvalenes (14a–j).10–12 Dibenzobarrelenes with substituents such as hydroxy, cyano and aryl groups give the 8b-substituted dibenzosemibullvalenes as intermediates, which then undergo transformation to other products (see later section). The conjugative stabilisation of the diradical intermediates by these substituents could be a decisive factor that governs the observed regioselectivity.
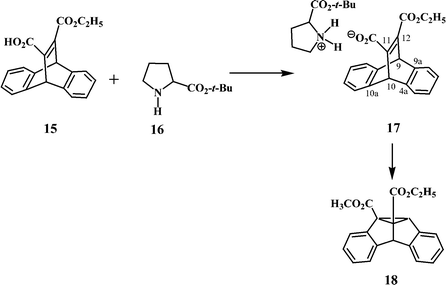
Scheffer and coworkers have investigated the photorearrangements of several dibenzobarrelene derivatives and have shown that the reaction proceeds well, in most cases, in the crystalline phase and in solution.13 They have shown that the solid state reactions of dibenzobarrelenes lead to greater regio-, stereo- and enantioselectivity compared to their solution phase reactions. By irradiating enantiomorphously pure crystals of a dibenzobarrelene derivative, they could isolate the corresponding dibenzosemibullvalene in nearly quantitative enantiomeric excess.14 They were able to chart the absolute steric course of the rearrangement by measuring the absolute configurations of the reactant and its photoproduct by X-ray crystallography.15 Through their studies they have been able to identify the topochemical factors responsible for the observed high enantioselectivity. The enantioselectivity in the solid state photoreaction of several dibenzobarrelenes has been enhanced further by Scheffer and coworkers through the “ionic auxiliary” concept.16 In this approach, the chromophore is linked to a sensitiser or a heavy atom, followed by the solid state irradiation of the salts, resulting in increased yields of the triplet photoproduct. An illustrative example is the irradiation of the salt 17 formed between the dibenzobarrelene ester-acid 15 and the tert-butyl ester of (S)-proline, 16 (Scheme 6). Irradiation of crystals of 17, followed by acidic work-up to remove the ionic chiral auxiliary and the esterification of the resulting carboxylic acid with diazomethane gave exclusively the regioisomer 18.17 It was concluded that the initial “benzo–vinyl” bridging occurs between C(11) and C(4a) to give the observed enantiomer 18. The authors have suggested that one of the contributing factors for the preferential enantioselectivity is an intramolecular steric effect in which the substituents on the vinyl group would like to remain far apart during the “benzo–vinyl” bridging. The steric interference is less in the case of C(11)–C(4a) bridging as compared to C(11)–C(10a) bridging.
 | ||
| Scheme 6 | ||
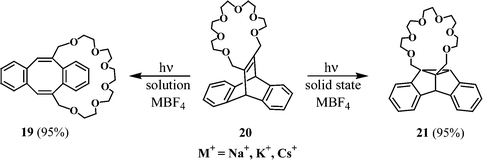
A recent study shows that the photochemical properties of a chromophore are modified by alkali metal ions through crown ether complexation.18 The dibenzobarrelene 20, which is annulated with a crown ether, complexes with alkali metals such as sodium, potassium and caesium, on treatment with NaBF4, KBF4 and CsBF4, respectively. Irradiation of 20 alone, or its alkali metal complexes in acetonitrile or benzene, leads preferentially to the dibenzocyclooctatetraene 19 as the photoproduct. In contrast, the solid state irradiation of the alkali metal complexes of 20 leads exclusively to the dibenzosemibullvalene 21, which is attributed to a cation effect (Scheme 7).
 | ||
| Scheme 7 | ||
3 Dibenzocyclooctatetraenes
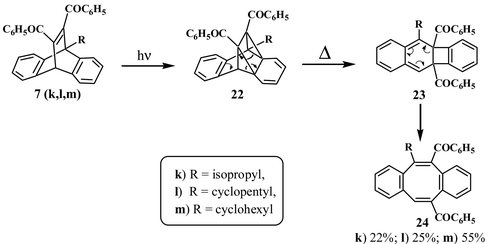
Direct irradiation of dibenzobarrelenes leads to the corresponding dibenzocyclooctatetraenes. On the basis of detailed investigations of the photochemistry of analogous benzo- and naphthobarrelene systems, it has been suggested that the dibenzocyclooctatetraene formation takes place through the singlet excited states of dibenzobarrelenes and involves initial intramolecular [2+2] cycloaddition.8,18Although the triplet state mediated pathway is predominant in most of the 11,12-dibenzoyl-substituted dibenzobarrelenes, we have observed that in certain 9-alkyl-substituted-11,12-dibenzoyldibenzobarrelenes, the corresponding dibenzocyclooctatetraenes are also formed.19 Thus, when the bridgehead positions are substituted by isopropyl, cyclopentyl or cyclohexyl groups (7k, l, m), substantial amounts of dibenzocyclooctatetraenes19 are formed (Scheme 8). A probable route to the formation of dibenzocyclooctatetraenes 24, on the basis of analogy, is shown in Scheme 8. The reaction is assumed to proceed through the initially formed [2+2] adduct 22, which rearranges to 23 and ultimately to the dibenzocyclooctatetraene 24.
 | ||
| Scheme 8 | ||
Certain bridgehead-substituted dibenzobarrelenes undergo photorearrangement through a “tri-π-methane” route leading to 1,4-diradical intermediates, which subsequently lead to dibenzocyclooctatetraenes with C2 symmetry. Irradiation of 11,12-dibenzoyl-9,10-dimethyl-substituted dibenzobarrelene (25) in benzene and under complete absence of oxygen, for example, gave a mixture of the dibenzocyclooctatetraene 28 and the dibenzopentalene derivative 29 (Scheme 9).20
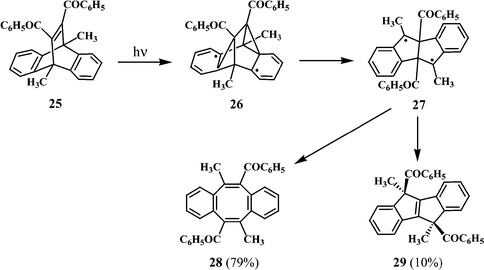 | ||
| Scheme 9 | ||
Excitation of 25 would lead to the diradical intermediate 26, through a tri-π-methane route.21 Further transformation of 26 will result in the benzylic 1,4-diradical 27. A Grob type fragmentation of 27 will lead to the dibenzocyclooctatetraene 28, whereas rearrangements involving benzoyl group migration would give the dibenzopentalene derivative 29. It may be mentioned that in the irradiation of 25, a small amount of a hexacyclic peroxycarbinol is also formed. A probable route to the formation of this product is discussed in a later section.
Scheffer and coworkers were the first to report the tri-π-methane pathway for the photoisomerisation of dibenzobarrelenes.21 They have observed that irradiation of dimethyl 9,10-dimethyl-9,10-dihydro-9,10-ethenoanthracene-11,12-dicarboxylate gives photoproducts, analogous to 28 and 29, arising from the diradical intermediate corresponding to 27. The involvement of a similar diradical intermediate (33) was postulated in the photoisomerisation of dimethyl 9-chloromethyl-9,10-dihydro-9,10-ethenoanthracene-11,12-dicarboxylate (30) (Scheme 10).22 Irradiation of 30 in solvents like benzene, acetonitrile and chloroform gave a mixture of products, 31 and 32, arising through the di-π-methane pathway and 35 and 36, arising through the tri-π-methane pathway and involving the diradical intermediate 33.
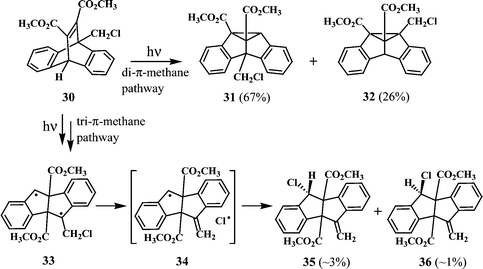 | ||
| Scheme 10 | ||
A similar example of dibenzocyclooctatetraene formation through a tri-π-methane route is found in the phototransformation of dimethyl 9-hydroxymethyl-10-methyl-9,10-dihydro-9,10-ethenoanthracene-11,12-dicarboxylate (37), leading to the dibenzocyclooctatetraene lactone 40 (Scheme 11).23
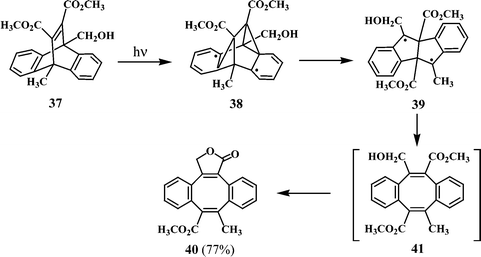 | ||
| Scheme 11 | ||
Dibenzocyclooctatetraenes are also formed from dibenzosemibullvalenes. A recent report24 states that the dibenzocyclooctatetraene 45 is formed as a secondary photoproduct from the dibenzosemibullvalene 42 (Scheme 12). It has been suggested that the reaction proceeds through σ-bond cleavage to give the 1,3-diradical 43, which subsequently undergoes an aryl 1,2-shift to give the 1,4-diradical 44. Further transformation of 44 will give the dibenzocyclooctatetraene 45.
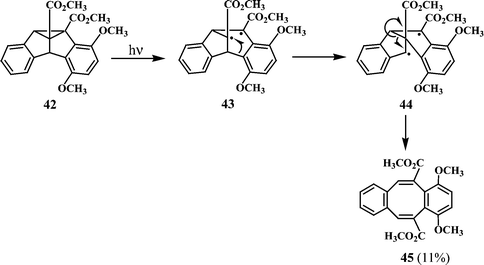 | ||
| Scheme 12 | ||
4 Dibenzopentalenodihydrofurans
Several dibenzosemibullvalenes undergo facile thermal isomerisation to give exclusively the corresponding dibenzopentalenodihydrofurans (Scheme 13).12,25–27 The ease of isomerisation is found to depend on the nature and position of the substituents. For example, the 8b-aryl-substituted 11,12-dibenzoyl-dibenzosemibullvalenes (13k, l, m, n, p, q), formed from the corresponding 9-aryl-substituted dibenzobarrelenes (7k, l, m, n, p, q) could be detected only at low temperatures. At ambient conditions, only the dibenzopentalenodihydrofurans (46k, l, m, n, p, q) could be isolated as the photoproducts (Scheme 13).25 However some of the 4b-substituted (14b, h, j) and 4b,8b-disubstituted (47q, r, s) dibenzosemibullvalenes were found to be quite stable under ambient experimental conditions. Thermal isomerisation of these dibenzosemibullvalenes to the corresponding dibenzopentalenodihydrofurans (48b, h, j and 48q, r, s) could be achieved at elevated temperatures (100–200 °C) (Scheme 14).12, 26,27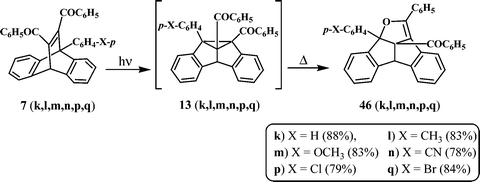 | ||
| Scheme 13 | ||
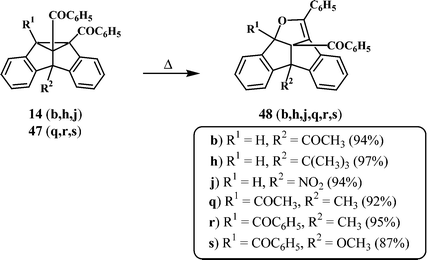 | ||
| Scheme 14 | ||
The thermal isomerisations of the dibenzosemibullvalenes to the corresponding dibenzopentalenodihydrofurans (13 to 46 and 47 to 48) are analogous to the rearrangement of vinylcyclopropanes to cyclopentenes.28 These isomerisations could proceed through a concerted pathway, involving a [1,3] sigmatropic shift or through a 1,3-diradical intermediate.29 The thermodynamic parameters of these isomerisations are in support of a concerted sigmatropic rearrangement. The thermal isomerisation of dibenzosemibullvalenes can proceed through an adiabatic pathway involving the ground state potential energy surface and similar thermally reversible photoisomerisations are reported in the literature.30
It has been reported that the dibenzopentalenodihydrofurans (48) are thermally stable, but photochemically isomerisable to the corresponding dibenzosemibullvalenes (13 and 47). A probable pathway is shown in Scheme 15, which involves the cleavage of the C–O bond to give diradical intermediates. Thus the dibenzopentalenodihydrofuran 46m, for example, will give on excitation the triplet diradical 49m, through a likely triplet mediated pathway (Scheme 15). Further transformation (intersystem crossing) of the triplet diradical 49m to the corresponding carbon centered singlet 1,3-diradical would lead to the corresponding dibenzosemibullvalene, 13m. The involvement of the diradical intermediate 49m was confirmed through oxygen quenching and isolation of the endo-peroxide 50m.31
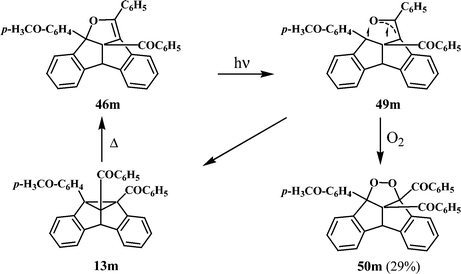 | ||
| Scheme 15 | ||
5 Dibenzopentalenes
Direct photolysis of dibenzosemibullvalenes does not lead to dibenzopentalenes. However, the σ-bond in the cyclopropane ring can be conveniently cleaved by hydrogenolysis to give the corresponding dibenzopentalene. Thus, hydrogenation of the dibenzosemibullvalene 14 using 5% Pd on charcoal gave the corresponding dibenzopentalene 51a (Scheme 16).10 Similar dibenzopentalenes 51b and 51c11 were obtained from the corresponding dibenzosemibullvalenes 14a and 13t, respectively.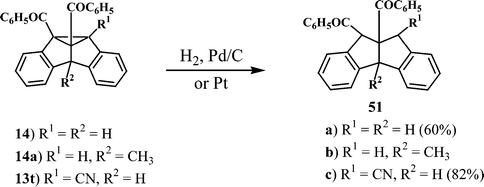 | ||
| Scheme 16 | ||
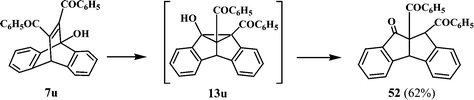
11,12-Dibenzoyl-9-hydroxydibenzobarrelene (7u), on photolysis in acetone is reported to give the dibenzoyldibenzopentalenone (52) (Scheme 17). The formation of 52 in the photolysis of 7u suggests that the 8b-hydroxydibenzosemibullvalene 13u is initially formed, which undergoes cleavage of the cyclopropane ring to give 52 (Scheme 17).11 A similar reaction has been reported earlier in the case of dimethyl 9-hydroxydibenzobarrelene-11,12-dicarboxylate.32 The preferred regioselectivity for the formation of the 8b-isomer has been attributed to a decrease in the radical stabilising ability of the ester group due to hydrogen bonding. This would favour radical formation next to the non-hydrogen bonded CO2CH3 group, which leads ultimately to the 8b-hydroxydibenzosemibullvalene.
 | ||
| Scheme 17 | ||
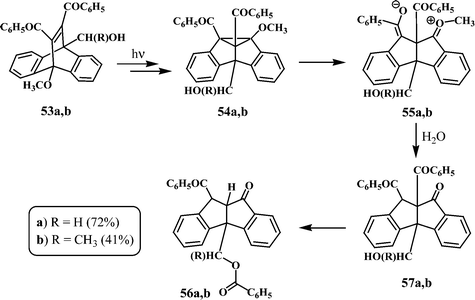
An interesting dibenzopentalenone (56a) was observed in the photolysis of 11,12-dibenzoyl-9-hydroxymethyl-10-methoxydibenzobarrelene (53a) (Scheme 18).33 It has been suggested that 53a, on irradiation, gives 4b-hydroxymethyl-8b-methoxydibenzosemibullvalene (54a), which gives the dipolar intermediate 55a, through an ionic ring opening of the cyclopropyl functionality in 54a. The demethylated, dibenzopentalenone 57a would be formed through the hydrolysis of 55a under the reaction conditions. The bridgehead hydroxymethyl and benzoyl groups in 57a are close to each other and the formation of the rearranged dibenzopentalenone 56a could proceed through an aldol intermediate (Scheme 18).
 | ||
| Scheme 18 | ||
A similar dibenzopentalenone (56b) has been reported in the photolysis of 11,12-dibenzoyl-9-(1-hydroxyethyl)-10-methoxydibenzobarrelene (53b) in benzene.33 In addition to 56b, the dibenzopentalenopyran, 60 is also formed in the photolysis of 53b. The formation of 56b from the dibenzobarrelene 53b could follow a similar path as in the case of 56a (Scheme 18).
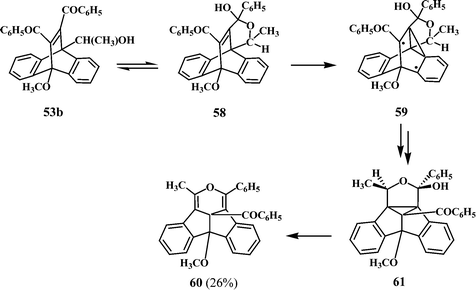
The formation of the dibenzopentalenopyran 60, however follows a different route as shown in Scheme 19. It is known33 that the starting 11,12-dibenzoyl-9-(1-hydroxyethyl),10-methoxydibenzobarrelene 53b exists in equilibrium with its cyclic carbinol 58. This may be due to steric interaction exerted by the methyl group in 53b, favoring the interaction between the hydroxyethyl substituent at the 9-position with the benzoyl group at the 12-position leading to the carbinol 58. The formation of the dibenzopentalenopyran 60 could be through the diradical intermediate 59 and the hexacyclic dibenzosemibullvalene intermediate 61. Elimination of water from 61 under the reaction conditions could lead to 60 (Scheme 19).
 | ||
| Scheme 19 | ||
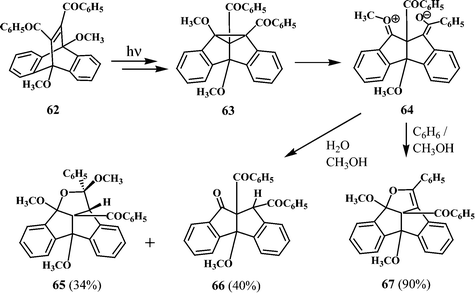
Dibenzopentalenones are also formed by the irradiation of 11,12-dibenzoyl-9,10-dimethoxydibenzobarrelene (62).33 The irradiation of 62 in a mixture of methanol and benzene (4 ∶ 1) gave exclusively the dibenzopentalenodihydrofuran (67). However, when the irradiation of 62 was carried out in a mixture of benzene and aqueous methanol, a mixture of products consisting of the pentacyclic methanol adduct (65) and the dibenzopentalenone (66) is formed (Scheme 20). The formation of the different products (65, 66 and 67) from 62 can be understood in terms of the initial formation of the dibenzosemibullvalene 63 and its further transformations as shown in Scheme 20.
 | ||
| Scheme 20 | ||
6 Oxygen containing heterocycles
In an earlier section of this report, it was mentioned that the irradiation of a bridgehead disubstituted dibenzobarrelene such as 11,12-dibenzoyl-9,10-dimethyl-substituted dibenzobarrelene (25) in benzene gives a mixture of the dibenzocyclooctatetraene (28) and a dibenzopentalene derivative 29 (Scheme 9), along with a small amount of a hexacyclic peroxy carbinol. The structure of the peroxy carbinol has been confirmed as 70 on the basis of X-ray crystallographic analysis.20 The formation of the hexacyclic peroxy carbinol 70 from the dibenzobarrelene 25 is somewhat intriguing. It appears that 25 undergoes transformation through a di-π-methane pathway to give the dibenzosemibullvalene 68. The fact that none of the dibenzosemibullvalene 68 was isolated in the reaction of 25 would suggest that it is undergoing further transformations. One of the possible modes is through cleavage of the cyclopropane ring to give a new 1,3-diradical 69, which can combine with oxygen to give the endo-peroxide 72. Further transformations of 72 as shown in Scheme 21 would lead to the hexacyclic peroxy carbinol 70. It may be mentioned in this connection that Scheffer and coworkers have isolated a peroxide analogous to 72, from the photorearrangement of dimethyl 9,10-dichlorodibenzobarrelene-11,12-dicarboxylate.34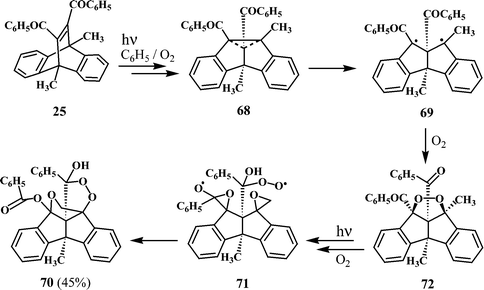 | ||
| Scheme 21 | ||
An interesting example of a heptacyclic peroxy compound formation is observed in the phototransformation of 11,12-dibenzoyl-9-ethyldibenzobarrelene (73).35 Thus, irradiation of 73 in acetone gave a mixture of dibenzosemibullvalene (74), a heptacyclic peroxy compound (77) and a lactone product (76) (Scheme 22).
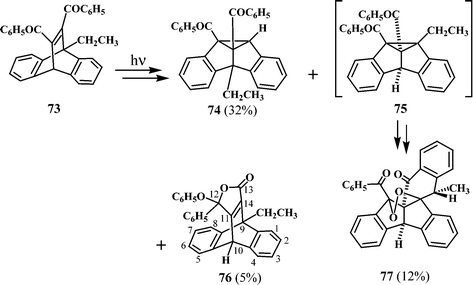 | ||
| Scheme 22 | ||
Irradiation of 73 should lead to both the 4b-ethyldibenzobarrelene 74 and the 8b-ethyl derivative 75. The formation of the peroxy compound 77 has been suggested to proceed through the pathway shown in Scheme 23, starting with the 8b-derivative 75.
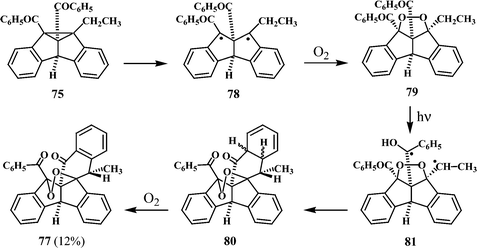 | ||
| Scheme 23 | ||
The formation of the lactone product 76 in the photolysis of 73 suggests that besides the usually observed dibenzocyclooctatetraenes and dibenzosemibullvalenes, the dibenzoylalkene type of rearrangement36 may also be operative in some cases. A reasonable pathway for the formation of 76 from 73 is indicated in Scheme 24. Initial excitation of 73, followed by intersystem crossing will give its triplet state, which can then interact with the adjacent phenyl group to give the diradical 82. Quenching of 82 with oxygen can lead to the hydroperoxy diradical 83, followed by a sequence of reorganisations leading to 76 (Scheme 24).
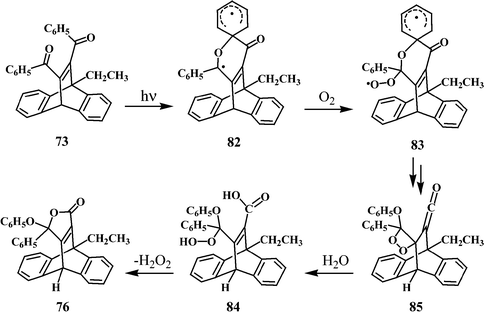 | ||
| Scheme 24 | ||
7 Polycyclic compounds derived through miscellaneous rearrangements
In this section we shall highlight the formation of some interesting heterocycles through the photolysis of certain bridgehead disubstituted dibenzobarrelenes. An interesting example is that of 11,12-dibenzoyl-9-hydroxymethyl-10-methyldibenzobarrelene 86, which on irradiation in acetone gives an 88% yield of the dibenzooxapropallane carbinol (89).37 The formation of 89 has been rationalised in terms of the regioselectively formed 4b-hydroxymethyl-8b-methyldibenzosemibullvalene (87) which undergoes further transformations as shown in Scheme 25. The interaction of the 4b-hydroxymethyl group with the 8e-benzoyl group, for example can lead to carbinol 88, which can undergo hydrogen atom abstraction to give the diradical intermediate 91. Further transformation of 91, through the cleavage of the cyclopropyl σ-bond will result in the enol 90, which can subsequently give 89 (Scheme 25).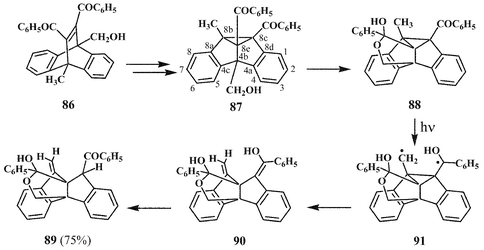 | ||
| Scheme 25 | ||
A novel polycyclic ketone 96 (7%) was isolated in the photolysis in benzene of 11,12-dibenzoyl-9-acetyl-10-methyl-substituted dibenzobarrelene (92), along with the dibenzosemibullvalene, 93 (Scheme 26).27 The formation of the major product 93 (74%) is through the expected di-π-methane pathway, whereas the formation of 96 is through a different mode of reaction of the starting dibenzobarrelene 92. The excited state of 92 can abstract a δ-hydrogen atom from the adjacent methyl group of the acetyl substituent to give the 1,5-diradical intermediate 94, which can subsequently couple to give the carbinol derivative 95. Elimination of water from 95 will lead to the observed product 96.
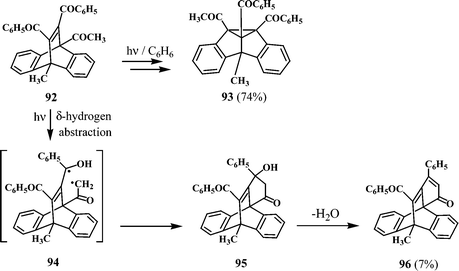 | ||
| Scheme 26 | ||
Interestingly, the dibenzosemibullvalene 93 formed from the dibenzobarrelene 92, undergoes a facile reaction with methanol to give a hexacyclic acetal derivative 99 (Scheme 27).27 It is assumed that methanol adds to the acetyl carbonyl of the dibenzosemibullvalene 93 to give a hemiacetal derivative 97 which on elimination of water would lead to the oxonium ion intermediate 98. Further addition of methanol to 98 will give the acetal derivative 99 (Scheme 27).
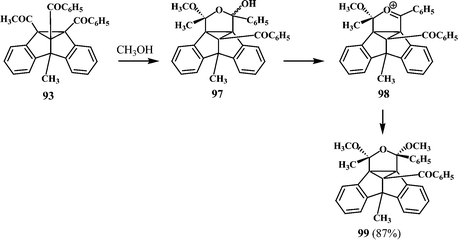 | ||
| Scheme 27 | ||
Depending on the number and nature of the substituents, some of the dibenzosemibullvalenes (primary photoproducts) undergo reaction with the solvent molecules leading to unusual products through interesting rearrangements. For instance, the 4b-acetoxy-substituted semibullvalene (14b) on treatment with methanolic potassium hydroxide gave the dibenzocyclooctatrienone 102 and a probable mechanism of this interesting reaction is shown in Scheme 28.11
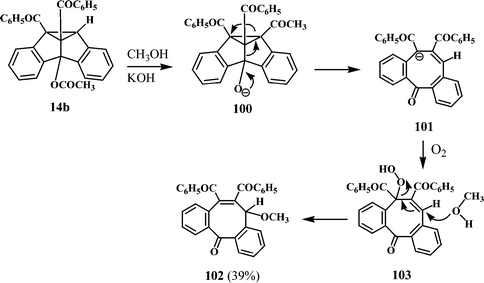 | ||
| Scheme 28 | ||
8 Conclusions
This report highlights the potential use of several dibenzoyl-substituted dibenzobarrelenes for the generation of different synthons, which may be useful for the synthesis of complex molecules. The synthesis of these novel synthons could be achieved through the di-π-methane rearrangement of appropriately substituted dibenzobarrelenes in one pot reactions. It is noteworthy that structurally different polycyclic compounds could be obtained with high regio- and stereoselectivity and in most cases in good yields under mild conditions. Systems like dibenzosemibullvalenes, dibenzocyclooctatetraenes, peroxy compounds and dibenzopentalenofurans may be of special interest to synthetic chemists. Although there have been several reports on the photochemistry of dibenzobarrelenes and di-π-methane rearrangement, this report is unique in highlighting the synthetic potential of the photochemistry of substituted dibenzobarrelenes.Acknowledgments
This is contribution No. RRLT-PPD(PRU)-183 from the Regional Research Laboratory, Trivandrum. The authors thank Dr. Nigam P. Rath for his help in the X-ray structure determination of several compounds mentioned in this report. We thank the Council of Scientific and Industrial Research (CSIR) and the Department of Science and Technology (DST), Government of India for financial support. One of the authors (MVG) thanks the Jawaharlal Nehru Centre for Advanced Scientific Research (JNCASR), Bangalore for support of this work.References
- H. E. Zimmerman and G. L. Grunewald, J. Am. Chem. Soc., 1966, 88, 183–184 CrossRef CAS.
- H. E. Zimmerman, in Organic Photochemistry, ed. A. Padwa, Marcel Dekker, New York, 1991, vol. 11, ch. 1, pp. 1–36 Search PubMed.
- H. E. Zimmerman and D. Armesto, Chem. Rev., 1996, 96, 3065–3112 CrossRef CAS.
- D. Armesto, M. J. Ortiz and A. R. Agarrabeitia, in Photochemistry of Organic Molecules in Isotropic and Anisotropic Media, ed. V. Ramamurthy and K. S. Shanze, Marcel Dekker, 2003, ch. 1, pp. 1–41 Search PubMed.
- M. Demuth, in Organic Photochemistry, ed. A. Padwa, Marcel Dekker, New York, 1991, vol. 11, ch. 2, pp. 37–109 Search PubMed.
- E. Ciganek, J. Am. Chem. Soc., 1966, 88, 2882–2883 CrossRef.
- P. W. Rabideau, J. B. Hamilton and L. Friedman, J. Am. Chem. Soc., 1968, 90, 4465–4466 CrossRef CAS.
- J. R. Scheffer, and J. Yang, in CRC Handbook of Organic Photochemistry and Photobiology, ed. W. M. Horspool and P. S. Sean, CRC Press, Boca Raton, FL, USA, 1995, ch. 16, pp. 204–221 Search PubMed.
- M. C. Sajimon, D. Ramaiah, S. Ajayakumar, N. P. Rath and M. V. George, Tetrahedron, 2000, 56, 5421–5428 CrossRef CAS.
- C. V. Kumar, B. A. R. C. Murty, S. Lahiri, E. Chackachery, J. C. Scaiano and M. V. George, J. Org. Chem., 1984, 49, 4923–4929 CrossRef CAS.
- B. A. R. C. Murty, S. Pratapan, C. V. Kumar, P. K. Das and M. V. George, J. Org. Chem., 1985, 50, 2533–2538 CrossRef CAS.
- M. C. Sajimon, D. Ramaiah, M. Muneer, E. S. Ajithkumar, N. P. Rath and M. V. George, J. Org. Chem., 1999, 64, 6347–6352 CrossRef CAS.
- J. Chen, J. R. Scheffer and J. Trotter, Tetrahedron, 1992, 48, 3251–3274 CrossRef CAS.
- S. V. Evans, M. Garcia-Garibay, N. Onkaram, J. R. Scheffer, J. Trotter and F. Wireko, J. Am. Chem. Soc., 1986, 108, 5648–5650 CrossRef CAS.
- M. A. Garcia-Garibay, J. R. Scheffer, J. Trotter and F. Wireko, J. Am. Chem. Soc., 1989, 111, 4985–4986 CrossRef CAS.
- J. N. Gamlin, R. Jones, M. Leibovitch, B. Patrick, J. R. Scheffer and J. Trotter, Acc. Chem. Res., 1996, 29, 203–209 CrossRef CAS.
- D. Gudmunsdottir and J. R. Scheffer, Tetrahedron Lett., 1990, 31, 6807–6810 CrossRef.
- H. Ihmels, M. Schneider and M. Waidelich, Org. Lett., 2002, 4, 3247–3250 CrossRef CAS.
- S. Pratapan, K. Ashok, D. R. Cyr, P. K. Das and M. V. George, J. Org. Chem., 1987, 52, 5512–5517 CrossRef CAS.
- C. V. Asokan, S. A. Kumar, S. Das, N. P. Rath and M. V. George, J. Org. Chem., 1991, 56, 5890–5893 CrossRef CAS.
- P. R. Pokkuluri, J. R. Scheffer and J. Trotter, J. Am. Chem. Soc., 1990, 112, 3676–3677 CrossRef CAS.
- J. Chen, P. R. Pokkuluri, J. R. Scheffer and J. Trotter, J. Photochem. Photobiol., A, 1991, 57, 21–26 CrossRef CAS.
- S. A. Kumar, C. S. Rajesh, S. Das, N. P. Rath and M. V. George, J. Photochem. Photobiol., A, 1995, 86, 177–183 CrossRef.
- H. Ihmels, C. J. Mohrschladt, J. W. Grimme and H. Quast, Synthesis, 2001,(8), 1175–1180 CrossRef CAS.
- S. Pratapan, K. Ashok, K. R. Gopidas, N. P. Rath, P. K. Das and M. V. George, J. Org. Chem., 1990, 55, 1304–1308 CrossRef CAS.
- M. Muneer, D. Ramaiah, E. S. Ajithkumar, M. C. Sajimon, N. P. Rath and M. V. George, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, C55, 996–1000 CrossRef CAS.
- M. C. Sajimon, D. Ramaiah, M. Muneer, N. P. Rath and M. V. George, J. Photochem. Photobiol., A, 2000, 136, 209–218 CrossRef CAS.
- E. Wenkert, Acc. Chem. Res., 1980, 13, 27–31 CrossRef CAS.
- D. Griller and K. U. Ingold, Acc. Chem. Res., 1980, 13, 317–323 CrossRef CAS.
- G. Quinkert, Angew. Chem., Int. Ed. Engl., 1972, 12, 1072–1087 CrossRef.
- M. C. Sajimon, D. Ramaiah, K. G. Thomas and M. V. George, J. Org. Chem., 2001, 66, 3182–3187 CrossRef CAS.
- R. G. Paddick, K. E. Richards and G. J. Wright, Aust. J. Chem., 1976, 29, 1005–1015 CAS and references cited therein.
- D. Ramaiah, S. A. Kumar, C. V. Asokan, T. Mathew, S. Das, N. P. Rath and M. V. George, J. Org. Chem., 1996, 61, 5468–5473 CrossRef CAS . See also: S. A. Kumar, T. Mathew, S. Das, N. P. Rath and M. V. George, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1996, C52, 2797–2800 Search PubMed.
- P. R. Pokkuluri, J. R. Scheffer and J. Trotter, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1993, C49, 2014–2018 CrossRef CAS.
- T. Mathew, S. A. Kumar, S. Das, N. P. Rath and M. V. George, J. Photochem. Photobiol., A, 1996, 95, 137–141 CrossRef CAS.
- H. E. Zimmerman, H. G. C. Dürr, R. S. Givens and R. G. Lewis, J. Am. Chem. Soc., 1967, 89, 1863–1874 CrossRef CAS.
- S. A. Kumar, C. V. Asokan, S. Das, J. A. Willur, N. P. Rath and M. V. George, J. Photochem. Photobiol., A, 1993, 71, 27–31 CrossRef.
Footnotes |
| † Dedicated to Professor P. T. Narasimhan on his 75th birthday. |
| ‡ Throughout, we have used the commonly accepted trivial names such as dibenzobarrelene, dibenzosemibullvalene and dibenzocyclooctatetraene, instead of their long and complicated scientific names, for easy reading. |
| § Steady state irradiation experiments, in most cases, were carried out in a Rayonet Photochemical Reactor (350 nm), with pyrex vessels or with a Hanovia 450 W medium-pressure mercury lamp with pyrex filters in a quartz-jacketed immersion well. |
| This journal is © The Royal Society of Chemistry 2005 |