Synthetic glycopeptides and glycoproteins as tools for biology
Matthew R.
Pratt
a and
Carolyn R.
Bertozzi
*abcd
aDepartment of Chemistry, University of California, California 94720
bDepartment of Molecular and Cell Biology, University of California, California 94720. E-mail: bertozzi@cchem.berkeley.edu; Fax: (510) 643-2628; Tel: (510) 643-1682
cHoward Hughes Medical Institute, University of California, California
dCenter for Advanced Materials, Materials Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, California
First published on 13th December 2004
Abstract
Investigations into the roles of protein glycosylation have revealed functions such as modulating protein structure and localization, cell–cell recognition, and signaling in multicellular systems. However, detailed studies of these events are hampered by the heterogeneous nature of biosynthetic glycoproteins that typically exist in numerous glycoforms. Research into protein glycosylation, therefore, has benefited from homogeneous, structurally-defined glycoproteins obtained by chemical synthesis. This tutorial review focuses on recent applications of homogeneous synthetic glycopeptides and glycoproteins for studies of structure and function. In addition, the future of synthetic glycopeptides and glycoproteins as therapeutics is discussed.
 Matthew Pratt | Matthew Pratt was born in Mesa, Arizona in 1976. He received his BS in biochemistry and mathematics from the University of Arizona in 1999. While an undergraduate, he worked for Prof. Robin Polt on glycosylation methodology and the synthesis of glycosylated analogs of enkephalin. He then entered the PhD program at the University of California Berkeley under the direction of Prof. Carolyn Bertozzi. His current research focuses on the chemical synthesis of glycoproteins and glycopeptide mimetics, developing assays for glycosyltransferase activity and inhibitor screens, and designing chemical tools to understand O-linked glycosylation of proteins. |
 Carolyn Bertozzi | Prof. Carolyn Bertozzi received her PhD in Chemistry from UC Berkeley in 1993, working with Professor Mark Bednarski on the synthesis and biological activity of C-glycosides. She pursued postdoctoral research at UCSF with Professor Steven Rosen, studying the activity of endothelial oligosaccharides in promoting leukocyte adhesion at sites of inflammation. Prof. Bertozzi returned to Berkeley as a member of the faculty in 1996, where she is now Professor of Chemistry and Molecular Biology and a member of the Howard Hughes Medical Institute. Her research focuses on understanding and controlling changes in cell surface glycosylation associated with cancer, inflammation and bacterial infection. Prof. Bertozzi is a member of several Scientific Advisory Boards of biotechnology and pharmaceutical companies, and a co-founder of Thios Pharmaceuticals. She also serves on the editorial advisory boards of numerous journals, including J. Med. Chem., J. Org. Chem., Acc. Chem. Res. and J. Am. Chem. Soc. Prof. Bertozzi's awards include the Irving Sigal Young Investigator Award of the Protein Society, the ACS Award in Pure Chemistry, the Merck Academic Development Program Award, the Glaxo Wellcome Scholars' Award, the Presidential Early Career Award in Science and Engineering, the MacArthur Foundation Fellowship, the Camille Dreyfus Teacher-Scholar Award, the Arthur C. Cope Scholar Award, the Horace S. Isbell Award in Carbohydrate Chemistry, the Alfred P. Sloan Research Fellowship, the Donald Sterling Noyce Prize for Excellence in Undergraduate Teaching and the UC Berkeley Distinguished Teaching Award. She is also an elected member of the American Academy of Arts and Sciences. |
Introduction
Oligosaccharides attached to proteins or lipids are major determinants of cell surfaces. The various combinations of the naturally occurring monosaccharides joined through numerous possible linkages and branch points, have the potential to generate a bewilderingly complex set of oligosaccharide structures. It can be argued that because of their enormous potential for encoding molecular information, oligosaccharides have evolved from basic structural elements of the cell to modulators of complex processes in higher organisms. These processes can include protein folding and trafficking, immune recognition, and developmental regulation.1Glycosylation of proteins clearly allows biological systems to augment the information contained in a genome. For example, the single gene product tissue plasminogen activator (tPA) can exist in more than 100 discrete glycoforms with a distribution of activities.2 The potential utility of complex posttranslational modifications such as glycosylation is the ability to expand protein structure and function exponentially. Glycosylation represents a level of complexity that is not under direct genetic control and may be necessary for many complex processes in higher organisms. As an example, O-linked fucose found on epidermal growth factor (EGF)-like repeats in the developmental switch Notch has been shown to be necessary for correct signaling.3 Furthermore, the N-acetylglucosaminyltransferase Fringe, which modifies these O-linked fucose residues, was shown to modulate developmental patterning of wing cells in Drosophilamelanogaster.4 This discovery underscores the importance of even simple monosaccharide glycosylation patterns in multicellular biology.
Despite the obvious importance of oligosaccharides in a variety of biological processes, progress towards understanding their specific functions has been limited by their complexity and heterogeneity.5 Oligosaccharides are products of a template-independent biosynthetic pathway and, therefore, direct genetic methods for the expression of homogeneous glycosylation patterns do not exist. At best, the sites of glycosylation can be added or removed to probe the effects on function. Some genetic and biochemical modifications to the termini of glycans can produce limited structural definition,6,7 but, as with natively expressed glycoproteins, the structures obtained remain heterogeneous and difficult to characterize.
The only way to access glycopeptides and glycoproteins of defined structure is through chemical and enzymatic synthesis. The synthesis of glycopeptides and glycoproteins from readily available components is therefore an important goal. Unlike peptide and nucleic acid chemistry, carbohydrate chemistry is complicated by structural branching and varied stereochemistry found in large oligosaccharides. Furthermore, the formation of glycosidic bonds is a delicate matter requiring strictly anhydrous conditions that are incompatible with unprotected peptides and proteins. Despite these challenges, glycopeptides and glycoproteins with pendant glycosylation ranging from monosaccharides to large glycans have been prepared through chemical synthesis, chemoselective ligation and chemoenzymatic transformations. These tools and approaches have been reviewed extensively elsewhere.8,9 This review focuses on recent applications of synthetic glycopeptides and glycoproteins to studies of their structure and function, and as prospects for developing therapeutic agents.
Mucin-type O-linked glycosylation
In eukaryotes, the most prevalent type of O-linked glycosylation is mucin-type glycosylation, where N-acetylgalactosamine (GalNAc) is linked in an α-anomeric configuration to the β-hydroxyl group of either a serine or a threonine residue of the polypeptide (Fig. 1).10 Mucins are glycoproteins with dense clusters of this type of glycosylation resulting in highly extended protein conformations. Individual mucin-type glycans can occur at singular sites on non-mucin glycoproteins. Other types of O-linked glycosylation include glycosaminoglycans, such as heparin and chondrotin sulfate, which are attached to the polypeptide chain through β-linked xylose residues. These structures are found on proteoglycans, which are principal components of the extracellular matrix. β-O-N-Acetylglucosamine (GlcNAc) is found as a dynamic modification of serine and threonine residues of cytosolic and nuclear proteins (Fig. 1).11 Another form of O-linked glycosylation is α-O-linked fucose found on EGF-like repeats on the transmembrane protein Notch and its cognate ligands.12 Likewise, β-O-linked glucose13 and α-O-linked mannose14 have been described although their functions are less well understood.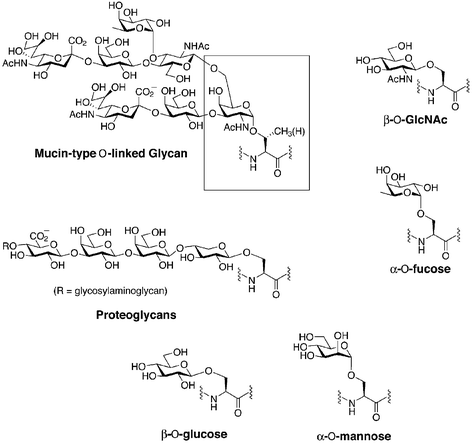 | ||
| Fig. 1 Six major classes of O-linked glycans. The Tn antigen of mucin-type O-linked glycans is outlined in the box. | ||
The α-O-GalNAc-Ser/Thr structure, commonly referred to as the Tn antigen, forms a biosynthetic foundation for eight core structures resulting from glycosylation at the C-3 and/or C-6 hydroxyl groups of GalNAc (Fig. 2). Of these, cores 1 and 2 are the most common core structures in mucin-type glycoproteins. Cores 3 and 4 are less abundant and are confined to mucins. The core structures can be elaborated with other monosaccharides such as sialic acid, fucose, and/or repeating units of Galβ1,4GlcNAc. Together with other modifications, such as sulfation, these elaborations give rise to highly complex structures often containing important recognition elements involved in cell–cell recognition.1
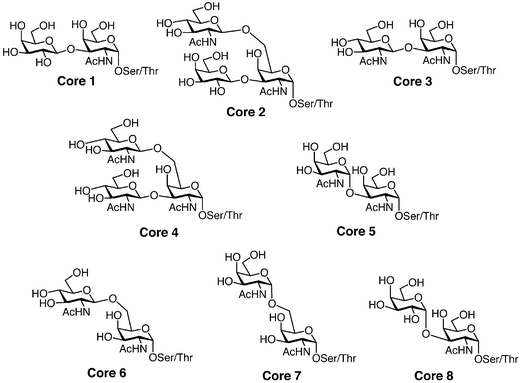 | ||
| Fig. 2 Eight known O-linked mucin-type core structures. | ||
Synthesis of mucin-type glycopeptides is commonly accomplished by incorporation of a suitably protected O-glycosyl amino acid into a polypeptide by solid-phase peptide synthesis (SPPS). Of the two standard methods, 9-fluorenylmethoxycarbonyl (Fmoc)-based chemistry is more often employed than tert-butyloxycarbonyl (Boc)-based chemistry for the SPPS of glycopeptides. The sequential removal of base-labile Fmoc protecting groups for peptide elongation is compatible with the presence of acid-sensitive glycosidic bonds and avoids repeated exposure to trifluoroacetic acid and final deprotection with hydrogen fluoride, common to Boc-based methods. The hydroxyl groups of the carbohydrates are typically protected as acetyl or benzoyl esters that can be removed by treatment with sodium methoxide or hydrazine after the peptide has been cleaved from the solid support. Obtaining the appropriate O-glycosyl amino acid is the major challenge to glycopeptide synthesis.
Although the assembly of peptides containing simple α-GalNAc residues is now relatively straightforward, the routine construction of glycopeptides bearing more elaborate O-linked glycans remains a tremendous challenge. The chief obstacle in the synthesis of complex O-glycosyl amino acids is obtaining high α-selectivity in the formation of the O-Ser/Thr mucin-type linkage. Even with simple monosaccharide donors, establishing α-selective conditions for the glycosylation reaction can be trying, and this variability is exaggerated when dealing with large oligosaccharide donors to prepare complex structures found on native mucins. The α-O-GalNAc-Ser/Thr building blocks required for SPPS of mucin-type glycopeptides are generally prepared by glycosylation on the appropriate serine or threonine derivative. The use of a 2-azidogalactose halo- or thioglycoside donor ensures high α-selectivity in the glycosylation reaction (Fig. 3). Conversion of the 2-azido group to an N-acetamido group provides the Fmoc-protected GalNAc-Ser/Thr amino acids. Since these building blocks are now commercially available, simple O-linked glycopeptides are readily accessible to those with experience in SPPS. The size limitation on synthetic glycopeptides is imposed by the technical constraints of SPPS.
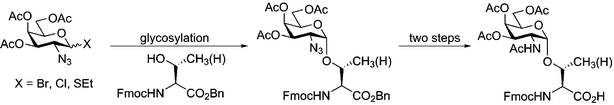 | ||
| Fig. 3 Synthesis of α-O-GalNAc-serine and threonine building blocks for SPPS of O-linked glycopeptides. | ||
Structural effects of O-linked glycosylation
Because of the density of O-linked glycosylation in mucin domains, the peptide backbone has been postulated to exist in an extended conformation. Electron microscopy, atomic force microscopy, and light scattering analysis of cell-surface mucin glycoproteins have supported this hypothesis.15–18 To address the structural effects of mucin-type glycosylation at the molecular level, several studies have been performed with small synthetic glycopeptides. Most notably, Live, Danishefsky and coworkers have reported nuclear magnetic resonance (NMR) solution structures of a mucin fragment from CD43.19 Complex glycosyl amino acids bearing mono-, di-, and trisaccharides corresponding to Tn, β-O-GalNAc, TF, and 2,6-STF antigens were incorporated into peptide 1 using SPPS (Fig. 4). Comparison of the unglycosylated peptide to the four different glycoforms shown in Fig. 4 suggested that the peptide is extended and ordered upon glycosylation with α-GalNAc, which is consistent with other conformational studies of O-linked glycopeptides and glycoproteins.20–22 Replacement of the natural α-GalNAc residue with β-GalNAc resulted in a disordered structure similar to that of the unglycosylated peptide. Furthermore, the distal sugar residues did not influence the overall peptide conformation. This suggests that it is the clusters of α-GalNAc on the mucin scaffold, and not the peripheral glycan structures, that induce the extended backbone characteristic of mucin glycoproteins. In the future, chemical synthesis should allow for the production of full O-linked glycoproteins for structural characterization, however, this remains a considerable challenge.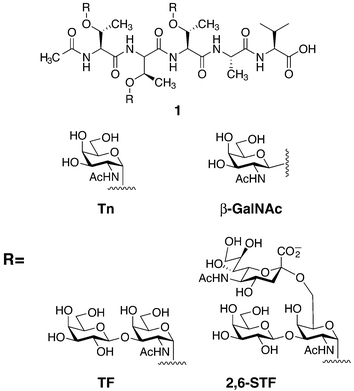 | ||
| Fig. 4 Glycopeptides prepared by solid-phase peptide synthesis (SPPS) bearing the tumor-associated antigens Tn, TF, and 2,6-sialyl TF (STF) on 1. | ||
Functional consequences of O-linked glycosylation
As mentioned above, glycoproteins, including those of the mucin-type, are involved in a variety of biological processes. However, the specific contributions of the glycans themselves often remain elusive. One area in which synthetic glycopeptides have been used to great effect is in studies of the selectin family of adhesion proteins. The selectins are C-type lectins that mediate the early stages of leukocyte homing to sites of inflammation. They are cell-surface receptors that bind, by their lectin domains, to cell surface glycoprotein counter-receptors such as GlyCAM-1 (glycosylation dependent cellular adhesion molecule), CD34, ESL-1 (E-selectin ligand 1), and PSGL-1 (P-selectin glycoprotein ligand 1).23 The molecular basis of these interactions has been of great interest, as the selectins are attractive targets for anti-inflammatory therapeutics. For example, PSGL-1-deficient mice are severely impaired in early immune cell recruitment and P-selectin-mediated leukocyte rolling.24 The binding of P-selectin to PSGL-1 requires the N-terminus of PSGL-1 bearing an O-linked sialyl Lewis x (sLex) glycan at Thr57 and one or more sulfated Tyr residues (Tyr46, Tyr48, Tyr51).25 To elucidate the individual contributions of these modifications to binding, a chemoenzymatic synthesis of a collection of N-terminal PSGL-1 fragments was established by Cummings and coworkers (Fig. 5a).26,27 Glycopeptide 2 was prepared by SPPS, and appropriate glycosyltransferases were used to elaborate the core monosaccharide to give an sLex hexasaccharide motif. Enzymatic sulfation of the three important tyrosine residues with tyrosylprotein sulfotransferase-1 (TPST-1)28 provided the fully modified peptide 3, which was shown to bind P-selectin nearly as well as full-length recombinant PSGL-1. More recently, the tyrosine sulfate residues were installed on the peptide using SPPS and the building block Fmoc-Tyr(SO3)-OH, to generate all possible combinations of mono-, di-, and trisulfated peptides.26 These and related PSGL-1 glycopeptides were compared for binding to P-selectin, revealing the individual contributions of each modification. These data, combined with crystal structures of E- and P-selectin lectin domains bound to a fragment of PSGL-1, provided a functional model of P-selectin binding (Fig. 6).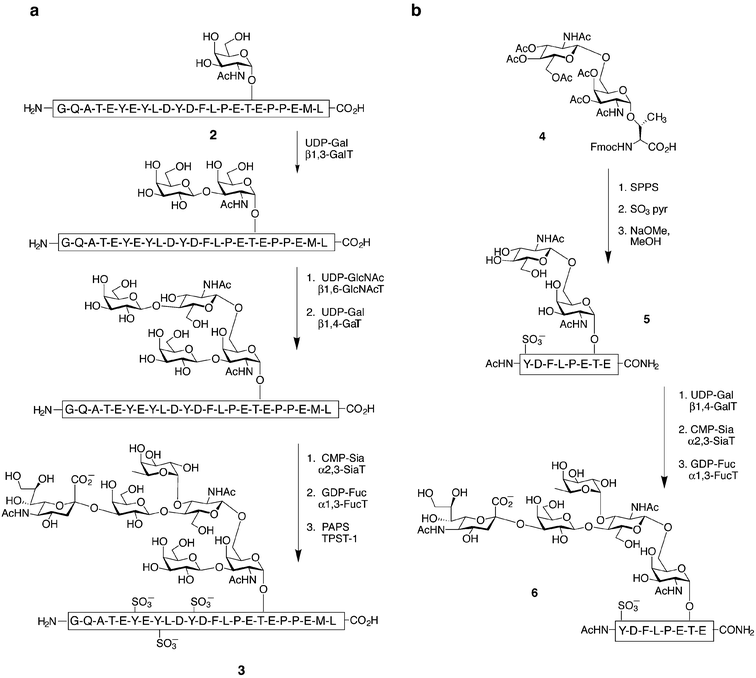 | ||
| Fig. 5 Chemoenzymatic syntheses of a sulfate- and sialyl Lewis x (sLex)-modified PSGL-1 glycopeptide perfomed by Cummings (a) and Wong (b) and their coworkers. Synthetic glycopeptides (2 or 5) were sequentially treated with the appropriate sugar donors and glycosyltransferases; galactosyl transferase (GalT), N-acetylglucosaminyltransferase (GlcNAcT), sialyltransferase (SiaT), fucosyltransferase (FucT), 3′-phosphoadenosine-5′-phosphosulfate (PAPS) and tyrosylprotein sulfotransferase-1 (TPST-1). | ||
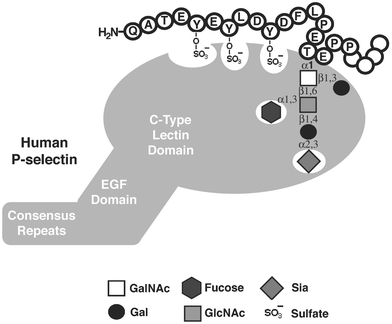 | ||
| Fig. 6 Essential determinants of PSGL-1 for binding P-selectin. P-selectin is an endothelial adhesion molecule comprising a C-type lectin domain, an epidermal growth factor (EGF) domain, and a series of consensus repeats similar to complement regulatory proteins. P-selectin binding to the N-terminus of PSGL-1 requires sulfotyrosine residues and an sLex glycan. The sialic acid and fucose residues of sLex interact strongly with the lectin domain. | ||
Wong and coworkers accomplished a similar chemoenzymatic synthesis of PSGL-1 fragment 6 (Fig. 5b).29 Enzymatic glycosylations were used to transform a monosulfated glycopeptide (5) carrying an α-O-linked disaccharide rather than a simple monosaccharide. Although this route required the synthesis of a more complicated disaccharide glycosyl amino acid (4), it is advantageous in that it does not require either the core 1 β1,3-galactosyltransferase (GalT) or the core 2 β1,6-N-acetylglucosaminyltransferase (GlcNAcT), both of which are not commercially available, to create the sLex hexasaccharide moiety. The synthetic glycopeptides generated by the above method are interesting prospects for anti-inflammatory therapy, as they bind and inhibit P-selectin with potencies superior to simple sLex glycans.
In addition to functioning as adhesion ligands, protein-bound glycans can greatly impact the immunogenic properties of many antigens.30 Many viral envelope proteins are glycosylated and use this characteristic to avoid immune detection.31,32 Tumor cells often display dramatic changes in glycosylation patterns, and these tumor-associated structures may be recognized by the immune system as tumor-specific antigens.33 In order for these antigens to elicit an adaptive immune response, they must be processed by antigen presenting cells, such as dendritic cells (DCs), and displayed to T cells in the context of a complex with major histocompatability (MHC) molecules. The T cells that recognize the glycopeptide antigen are stimulated to proliferate and can then assist in the immune reaction against the virus-infected cell or tumor. T cells that recognize a particular antigen can be cultured in vivo (termed a T cell hybridoma) and used to study the specificity of their receptors for related antigens. Despite the clear ability of glycans to modulate protein structure and function, processing of glycoproteins by antigen presenting cells for presentation to T cells has not been well studied. Recently, synthetic and enzymatically elaborated glycopeptides based on the tumor antigen MUC1, a cell surface glycoprotein, were used to probe the processing and presentation of glycopeptides by DCs.34 It was shown that DCs endocytose MUC1 glycopeptides of various lengths, transport them to compartments for processing into smaller glycopeptides, and present them on MHC II molecules without removal of the glycans. This suggests that a repertoire of carbohydrate-specific T cells can also be elicited against glycoprotein antigens.
To probe the fine specificity of T cells against glycopeptides, Meldal and Werdelin tested peptides 7 and 8 and glycopeptides 10–16 for cross-reactivity with a T cell clone specific for glycopeptide 9 (Fig. 7).35 The T cells proved to be extremely specific in their recognition of the α-GalNAc residue, displaying only mild cross-reactivity with glycopeptide 10, where the α-GalNAc was bound to Ser rather than Thr. Remarkably, glycopeptide 13, with only the subtle stereochemical change from α-GalNAc to α-GlcNAc, showed no cross-reactivity. Thus, it can be concluded that glycopeptides can elicit T cell activation in a carbohydrate-dependent manner that is extremely specific for the structure of the carbohydrate moiety. Because many pathogenic microbes utilized glycosylation to evade the immune system, the knowledge that T cells can recognize glycopeptides in a glycosylation specific manner is important for a complete understanding of the immune response.
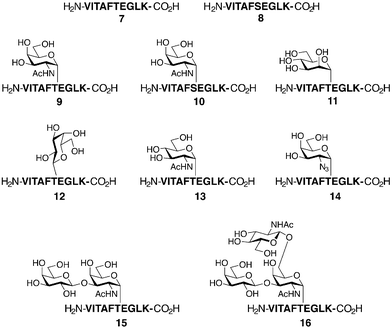 | ||
| Fig. 7 Synthetic glycopeptides used to probe antigen processing and presentation. Peptides 7 and 8 and glycopeptides 9–16 were prepared by SPPS. T-cell hybridomas were raised against glycopeptide 9 bearing an α-GalNAc residue. All other (glyco)peptides were used to probe the specificity of the above hybridomas. | ||
Because of the aberrant glycosylation patterns characteristic of some types of cancer, there has been increasing interest in generating antitumor vaccines based upon these abnormal glycan structures. For example, Danishefsky and coworkers synthesized several glycopeptides represented by 1 (Fig. 4) and 17 (Fig. 8).36 These glycopeptides contained clustered carbohydrates corresponding to the Tn, TF, and 2,6-STF antigens described earlier, as well as the Lewis y (Ley) antigen (Fig. 8). Glycopeptides represented by 1 and 17 generate robust antibody responses that cross-react with the same antigen expressed on tumor cells.37 Although it is difficult to determine with certainty the factors that contribute to immunogenicity, it appears that a mucin-like oligomeric display of the carbohydrate antigens is critical. Only antibodies raised against the Ley-elaborated peptide 17 reacted with both clustered Ley-mucin glycoproteins and monomeric Ley-ceramide. These clustered glycopeptides and others are undergoing evaluation as vaccines for several types of cancer.36
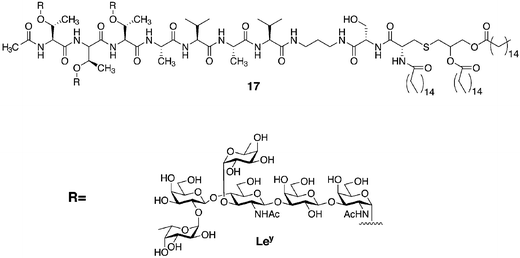 | ||
| Fig. 8 Glycopeptides prepared by SPPS bearing the tumor-associated antigen Lewis y (Ley). | ||
The methods described above for the construction of O-linked glycopeptides permit access to structurally diverse but relatively short glycopeptide fragments (∼20 amino acids). Naturally occurring mucin-type oligosaccharides are typically present on proteins that far exceed this size. To surpass the size limits inherent in linear SPPS, the coupling of peptide fragments by native chemical ligation (NCL) technology has found widespread use.38 The ligation of two unprotected peptide segments, one bearing a C-terminal thioester and the other an N-terminal cysteine residue, affords the product peptide with a native amide bond at the ligation site. NCL is efficient and highly chemoselective, and the reaction conditions are entirely compatible with glycans and native proteins. For these reasons, the extension of NCL to glycoprotein synthesis presents an ideal solution for accessing large glycoprotein structures.
Lymphotactin (Lptn) is a 93-residue chemokine that serves as a potent chemoattractant for both T cells and natural killer cells.39 With a small mucin-like domain located at its C-terminus, Lptn is unusual, for relatively few chemokines are extensively O-glycosylated. Lptn is readily dissected by the NCL strategy into two synthetic peptides: a 47-residue peptide α-thioester (18) and a 46-residue glycopeptide (19) with eight α-GalNAc residues (Fig. 9a). The thioester (18) was synthesized using traditional Boc-based SPPS methods,40 and Fmoc-based synthesis with α-O-GalNAc-Ser/Thr afforded the lymphotactin mucin domain. Ligation of the two fragments cleanly gave the glycosylated chemokine, which was biologically active in a standard calcium mobilization assay.39 Synthesis of this glycoprotein by NCL has provided milligram quantities of homogeneous Lptn for structural and functional studies. Since Lptn has immunostimulatory properties, this chemokine may find therapeutic use in the future.
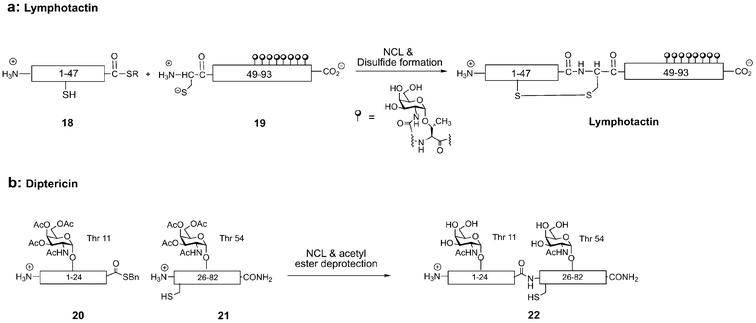 | ||
| Fig. 9 Native chemical ligation (NCL) of peptide thioesters to N-terminal cysteinyl peptides provides full-length glycoproteins lymphotactin (a) and diptericin (b). | ||
A chemically defined version of diptericin, an 82-residue antimicrobial glycoprotein from insects, has also been prepared by NCL (Fig. 9b). Containing a proline-rich sequence similar to the antimicrobial peptide drosocin and an attacin-like domain, this modular antimicrobial peptide carries potential O-linked glycosylation sites at Thr11 and Thr54.41 Diptericin entirely lacks cysteine, thus Gly25 was strategically changed to the cysteine residue required for the NCL reaction. Positioned between the drosocin- and attacin-like domains, this disconnection also made possible investigation of the isolated domains for biological activity. For generation of the acid- and base-sensitive N-terminal glycopeptide-α-thioester 20, conventional Boc- and Fmoc-based methods to prepare the thioester could not be used. To overcome this obstacle, Fmoc-based SPPS was performed on a sulfonamide “safety-catch” resin developed by Ellman and coworkers that allowed the release of peptide thioesters under mild conditions by nucleophilic addition of thiols.42 Glycopeptide thioesters for NCL can be routinely prepared by this method. Removal of side-chain protecting groups yielded glycopeptide-α-thioester 20, which was ligated to the glycopeptide fragment 21 generated by Fmoc-based SPPS. NCL efficiently produced the full-length glycoprotein 22, which inhibited bacterial growth with an IC50 of 2.70 ± 0.30 μM, similar to the potency of synthetic native diptericin previously prepared in our laboratory.43 Glycopeptides such as diptericin appear to have broad spectrum activity against numerous bacteria, and are thus attractive lead compounds for new antibiotics that might avoid resistance.
N-linked glycosylation
An older and more prevalent form of glycosylation, N-linked glycosylation, is found in a wide range of organisms ranging from Archae to mammals and other eukaryotes.44,45N-Glycosylation is a modification performed cotranslationally (during the translation of mRNA to protein) and is available to any secreted or membrane-bound protein containing the triplet amino acid sequence AsnXaaSer/Thr (where Xaa is any amino acid except Pro). An oligosaccharide is transferred to the amide side chain of Asn, from a dolichol phosphate glycosyl donor, by the action of membrane-bound oligosaccharyl transferase in the endoplasmic reticulum (ER).5,46 The fully translated glycoprotein is then subjected to glycan trimming and processing and further glycan elaboration in the ER and Golgi apparatus. The Man3GlcNAc2(β-N)Asn structure is the ubiquitous pentasaccharide core found in all N-linked glycans (Fig. 10). This core oligosaccharide can be extended in three broadly defined fashions to produce complex (A), high-mannose (B) and hybrid (C) type glycans. As with O-linked glycans, differing numbers of repeating units of Gal/GalNAc and GlcNAc, addition of sialic acid and/or fucose, and other modifications such as sulfation lead to a vast array of highly complex structures.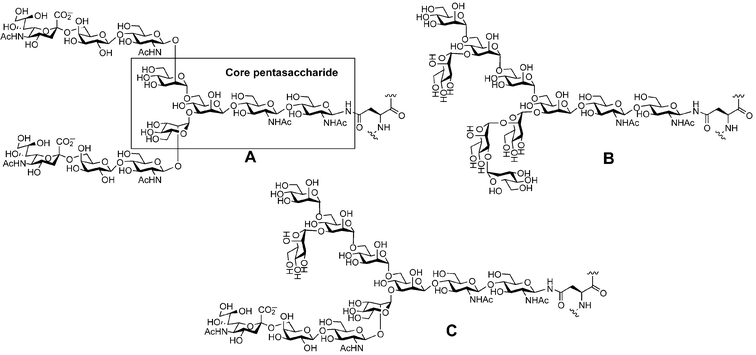 | ||
| Fig. 10 The three classes of N-linked glycans: (A) complex, (B) high mannose, (C) hybrid. | ||
The functions of N-linked glycosylation are wide-ranging and not understood in the case of every protein. However, they can be divided into two types: intra- and extracellular. Intracellularly, the broad function of N-linked glycans is protein folding and trafficking.46 This is exemplified by the protein folding quality control mechanism of the cell that involves a chaperone system found in the ER of nearly all eukaryotes, the calnexin–calreticulin cycle. Calnexin and calreticulin are related ER lectins that interact with N-linked structures bearing a single glucose residue that is exposed by the trimming action of glucosidases I and II.47 Calnexin or calreticulin binding slows the trafficking process and allows for proper protein folding and disulfide bond isomerization.48 While the glycoprotein is retained in the ER, glucosidase II removes the last glucose residue of N-linked glycans and causes release of the protein from the lectin-like chaperones. In a poorly understood process, a glucosyltransferase simultaneously acts as a folding “sensor”, apparently replacing a glucose residue only on improperly folded proteins, causing their binding to calnexin and calreticulin and return to the folding cycle of the ER.49
Extracellularly, N-linked glycans can function as structural elements and as ligands for receptors. Structurally, the large and flexible N-linked glycans increase protein stability by restricting the conformational flexibility of the underlying protein without sacrificing the net entropy of the system. A thermodynamic study performed by Robertson and co-workers with an ovomucoid protein domain demonstrated that two glycans found on Asn10 and Asn52 increased the melting temperature of the 68-residue polypeptide by 4.8 °C.50 An N-linked glycan can also affect the local structure and stability of a protein, as revealed by an NMR structure of a soluble form of human CD2 solved by Wagner and coworkers.51 CD2 is a cell surface glycoprotein present on T lymphocytes and natural killer cells, and the attachment of an N-linked glycan at Asn65 is necessary for its binding to CD58. As measured from nuclear Overhauser effects (NOEs), the protein-proximal GlcNAc-GlcNAc disaccharide was in close contact with a cluster of charged and polar residues located on one face of a β-sheet. The proper orientation of this sheet within CD2 was required for folding of the CD58 binding site. In this case, as well as others,52 the glycan acts in concert with the polypeptide to orchestrate the overall structure and function of the protein.
N-Linked glycans can also function extracellularly as ligands for carbohydrate receptors, as is highlighted by the glycoprotein growth factors and hormones. For example, erythropoietin (EPO) is synthesized by the kidneys and circulated in the blood to stimulate red cell proliferation and differentiation in bone marrow. The carbohydrates on EPO consist of one O-linked and three N-linked glycans to make up 40% of the protein's total weight. Fukuda and co-workers have shown that variation in the carbohydrate content of the N-linked glycans alters the serum half-life of EPO and thus alters its activity in vivo.53 Asialo-erythropoietin has no measurable activity in vivo due to rapid clearance from the bloodstream. EPO glycoforms containing N-acetyllactosamine (LacNAc) repeats were similarly cleared from serum circulation in a rapid fashion. In both cases, exposed galactose residues were recognized by the hepatic asialoglycoprotein receptor, and the hormone was internalized by endocytosis and degraded in the lysosome. Thus, EPO must be glycosylated in a very specific fashion to retain activity in vivo.
Structural effects of N-linked glycosylation
To understand the influence of an N-linked glycan on local peptide conformation, Imperiali and coworkers undertook the synthesis of several differentially glycosylated fragments of the extracellular domain of the nicotinic acetylcholine receptor (nAChR) (Fig. 11).54 The transglycosylation reaction enacted by Mucor hiemalis endo-β-N-acetylglucosaminidase (Endo-M) was utilized as an attractive methodology for the production of complex glycopeptides. The nAChR domain 23 and its simple glycoforms 24 and 25 were prepared by traditional SPPS. Glycopeptide 26 was synthesized by Endo-M-mediated transglycosylation as described above, with 24 serving as the glycosyl acceptor. The distal sugar residues of glycopeptide 26 were then enzymatically cleaved to produce glycopeptides 27 and 28. NMR studies revealed several NOEs between the Asn residue and the proximal GlcNAc residue in glycopeptide 26. These signals were much weaker in glycopeptides 24 and 25, suggesting that distal sugar residues may rigidify the proximal core disaccharide. The addition of glycans also increased disulfide bond formation in the peptide, and the rate of cis/trans proline isomerization of 23 was two-fold faster than those of glycosylated derivatives 24–28. This study implies that N-linked glycosylation can alter both protein stability and the dynamic process of protein folding.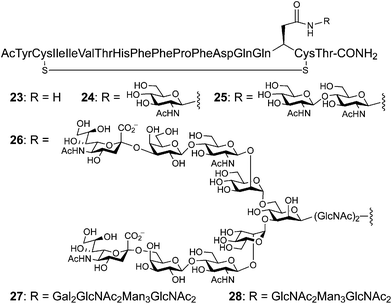 | ||
| Fig. 11 Peptide fragments from the extracellular domain of the nicotinic acetylcholinesterase receptor (nAChR) (23, 24 and 25) prepared by SPPS. Several glycoforms were constructed by the sequential action of Endo-M on 24 to produce 26 followed glycosidase digestion to afford 27 and 28. | ||
In addition to the chemoenzymatic methology described above, the convergent coupling of synthetic glycosylamines to the aspartyl side chains of unprotected peptides has been utilized to explore the effects of N-linked glycosylation on peptide conformation. Danishefsky and coworkers have applied this approach to short peptides bearing very large N-linked oligosaccharides.55 Pentasaccharide 29 was synthesized using the “glycal assembly method”,56 converted to glycosylamine 30, and then coupled to a pentapeptide to produce glycopeptide 31 in good yield (Fig. 12). These synthetic achievements have enabled landmark studies of the stereochemical communication between the carbohydrate and peptide domains. NMR studies undertaken by Live and coworkers compared two glycopeptides, differing only in the absolute stereochemistry of the amino acids (L-peptide vs.D-peptide).55 Both peptides adopted a type I β-turn, but there were measurable differences between the stereochemically “matched” peptide and the “mismatched”. Thus, communication between the carbohydrate and polypeptide domains of a glycoprotein is not solely based upon the bulk of the carbohydrate. Rather, specific interactions between the polypeptide and the carbohydrate are governed by their precise structures.
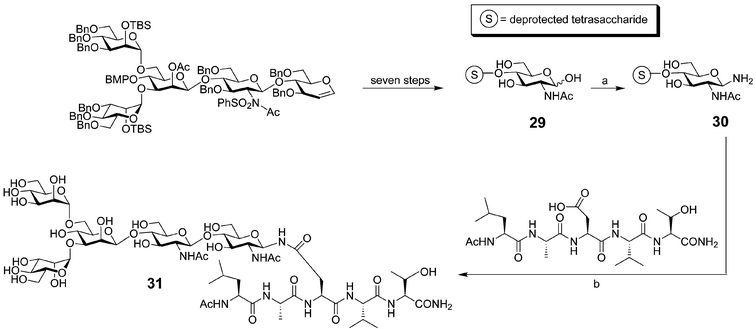 | ||
| Fig. 12 Synthesis of a pentasaccharide-modified glycopeptide (31): a) (NH4)HCO3, H2O (95%); b) amide formation: HOBt and HATU in DMF (40% yield). | ||
Functional consequences of N-linked glycosylation
Chemoenzymatic synthesis was again used for the preparation and study of an N-linked glycopeptide hormone, calcitonin, a 32-amino acid calcium-regulating hormone used as a therapeutic agent for hypercalcemia, Paget's disease, and osteoporosis.57 The transglycosylation reaction catalyzed by Endo-M was used to elaborate a synthetic glycoprotein with a complex carbohydrate. To study the consequences of glycosylation at a potential N-linked site, a synthetic glycopeptide was prepared containing a single GlcNAc residue on the Asn of the triplet sequence. Transglycosylation by Endo-M using a di-sialo transferrin glycosyl amino acid (STF-GP) as the glycosyl donor gave the desired glycosylated calcitonin in 8.5% isolated yield (Fig. 13). The glycosylated version of the hormone had increased biological activity in vitro, confirming the importance of the carbohydrate moiety for the function of the underlying glycoprotein. Because of their large size, N-linked glycoproteins present a larger challenge for organic synthesis. However, very large N-linked glycopeptides have been synthesized by specialized groups and should allow for more complex structural and functional studies to be undertaken in the future.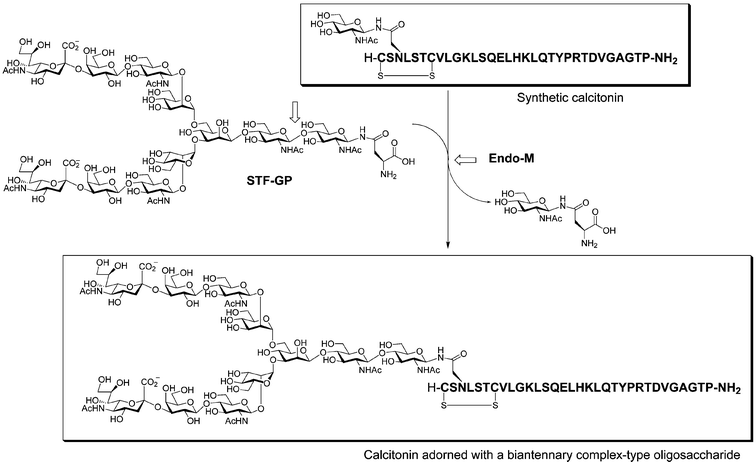 | ||
| Fig. 13 Synthesis of a calcitonin derivative with a complex-type N-linked glycan by transglycosylation. Endo-M transfers the biantennary glycan from the donor glycosyl amino acid (STF-GP) derived from transferrin, liberating GlcNAc-Asn. | ||
Unnatural glycopeptides as therapeutics
Endogenous cerebral peptides are involved in the control of many aspects of brain function, cognition and perception. Unfortunately, the therapeutic use of brain-derived peptides as pharmaceutical agents has been retarded because peptides do not readily penetrate the blood–brain barrier. Many opioid peptides of this type have been isolated and structurally characterized, most of which contain the same tetrapeptide Tyr-Gly-Gly-Phe N-terminal pharmacophore (enkephalin).58 Using the enkephalin scaffold as a starting point many analogues have been prepared, including peptide 32 (Fig. 14), with different in vitro affinities for opioid receptors.59 Attempts to use peptide 32 in an in vivo setting have been fruitless, however, due to the blood–brain barrier mentioned above. Polt and coworkers have overcome this limitation by synthesizing a glycosylated form of 32, peptide 33, where a glucose monosaccharide has been appended in a β-linkage to the C-terminal serine residue.60 The addition of this one monosaccharide allows the enkephalin peptide 33 to produce analgesic effects similar to morphine, even when administered peripherally (intravenously).61 This improved performance in vivo has been postulated to occur due to uptake of the peptide therapeutic by an unknown mechanism.62,63 Further studies by the same group are underway to understand the mechanism of transport, as well as optimize the peptide/carbohydrate partnership for maximum delivery and potency.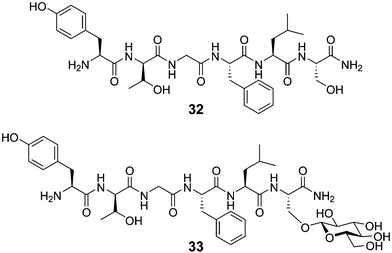 | ||
| Fig. 14 Structures of peptide 32 and the corresponding glycopeptide 33. | ||
Summary and outlook
Despite the difficulties inherent in the merger of carbohydrate and peptide/protein chemistries, the examples described in this review clearly indicate that remarkable progress generating synthetic glycopeptides and glycoproteins has been made. Chemical synthesis of homogeneous materials has allowed for a diverse array of carbohydrate structures to be prepared and their functions, from local structure perturbations to immune recognition, to be probed in a systematic fashion. These techniques in combination with others reviewed elsewhere are complementary and should provide for an even greater understanding of protein glycosylation in the future.Acknowledgements
The authors would like to thank HCH for his careful reading of this manuscript.References
- Essentials of Glycobiology, ed. A. Varki, R. D. Cummings, J. Esko, H. Freeze, G. W. Hart and J. Marth, Cold Spring Harbor Labs, Cold Spring Harbor, NY, 1999 Search PubMed.
- R. B. Parekh, R. A. Dwek, J. R. Thomas, G. Opdenakker, T. W. Rademacher, A. J. Wittwer, S. C. Howard, R. Nelson, N. R. Siegel, M. G. Jennings, N. Harakas. and J. Feder, Biochemistry, 1989, 28, 7644 CrossRef CAS.
- S. Shi and P. Stanley, Proc. Natl. Acad. Sci. USA, 2003, 100, 5234 CrossRef CAS.
- M. E. Fortini, Nature, 2000, 406, 357 CrossRef CAS.
- P. M. Rudd and R. A. Dwek, Crit. Rev. Biochem. Mol. Biol., 1997, 32, 1 CAS.
- T. S. Raju, J. B. Briggs, S. M. Chamow, M. E. Winkler and A. J. S. Jones, Biochemistry, 2001, 40, 8868 CrossRef CAS.
- S. Weikert, D. Papac, J. Briggs, D. Cowfer, S. Tom, M. Gawlitzek, J. Lofgren, S. Mehta, V. Chisholm, N. Modi, S. Eppler, K. Carroll, S. Chamow, D. Peers, P. Berman and L. Krummen, Nat. Biotechnol., 1999, 17, 1116 CrossRef CAS.
- B. G. Davis, Chem. Rev., 2002, 102, 579 CrossRef CAS.
- M. J. Grogan, M. R. Pratt, L. A. Marcaurelle and C. R. Bertozzi, Annu. Rev. Biochem., 2002, 71, 593 CrossRef CAS.
- G. J. Strous and J. Dekker, Crit. Rev. Biochem. Mol. Biol., 1992, 27, 57 CAS.
- F. I. Comer and G. W. Hart, J. Biol. Chem., 2000, 275, 29179 CrossRef CAS.
- R. S. Haltiwanger, Curr. Opin. Struct. Biol., 2002, 12, 593 CrossRef CAS.
- L. Shao, Y. Luo, D. J. Moloney and R. S. Haltiwanger, Glycobiology, 2002, 12, 763 CrossRef CAS.
- S. Strahl-Bolsinger, M. Gentzsch and W. Tanner, Biochim. Biophys. Acta, 1999, 1426, 297 CrossRef CAS.
- A. M. Fong, H. P. Erickson, J. P. Zachariah, S. Poon, N. J. Schamberg, T. Imai and D. D. Patel, J. Biol. Chem., 2000, 275, 3781 CrossRef CAS.
- F. Li, H. P. Erickson, J. A. James, K. L. Moore, R. D. Cummings and R. P. McEver, J. Biol. Chem., 1996, 271, 6342 CrossRef CAS.
- T. J. McMaster, M. Berry, A. P. Corfield and M. J. Miles, Biophys. J., 1999, 77, 533 CAS.
- R. Shogren, T. A. Gerken and N. Jentoft, Biochemistry, 1989, 28, 5525 CrossRef CAS.
- D. H. Live, L. J. Williams, S. D. Kuduk, J. B. Schwarz, P. W. Glunz, X. T. Chen, D. Sames, R. A. Kumar and S. J. Danishefsky, Proc. Natl. Acad. Sci. USA, 1999, 96, 3489 CrossRef CAS.
- X. Huang, J. J. Barchi, Jr., F. D. Lung, P. P. Roller, P. L. Nara, J. Muschik and R. R. Garrity, Biochemistry, 1997, 36, 10846 CrossRef CAS.
- R. Liang, A. H. Andreotti and D. Kahne, J. Am. Chem. Soc., 1995, 117, 10395 CrossRef CAS.
- T. A. Gerken, K. J. Butenhof and R. Shogren, Biochemistry, 1989, 28, 5536 CrossRef CAS.
- D. Vestweber and J. E. Blanks, Physiol. Rev., 1999, 79, 181 Search PubMed.
- J. Yang, T. Hirata, K. Croce, G. Merrill-Skoloff, B. Tchernychev, E. Williams, R. Flaumenhaft, B. C. Furie and B. Furie, J. Exp. Med., 1999, 190, 1769 CrossRef CAS.
- R. P. McEver and R. D. Cummings, J. Clin. Invest., 1997, 100, 485 CAS.
- A. Leppanen, P. Mehta, Y. B. Ouyang, T. Ju, J. Helin, K. L. Moore, I. van Die, W. M. Canfield, R. P. McEver and R. D. Cummings, J. Biol. Chem., 1999, 274, 24838 CrossRef CAS.
- A. Leppanen, S. P. White, J. Helin, R. P. McEver and R. D. Cummings, J. Biol. Chem., 2000, 275, 39569 CrossRef CAS.
- Y. Ouyang, W. S. Lane and K. L. Moore, Proc. Natl. Acad. Sci. USA, 1998, 95, 2896 CrossRef CAS.
- K. M. Koeller, M. E. Smith and C. H. Wong, Bioorg. Med. Chem., 2000, 8, 1017 CrossRef CAS.
- I. Brockhausen, J. Schutzbach and W. Kuhns, Acta Anat. (Basel), 1998, 161, 36 Search PubMed.
- S. Olofsson, APMIS Suppl., 1992, 27, 84 Search PubMed.
- E. O. Freed and M. A. Martin, J. Biol. Chem., 1995, 270, 23883 CrossRef CAS.
- Y. J. Kim and A. Varki, Glycoconjugate J., 1997, 14, 569 CrossRef CAS.
- A. M. Vlad, S. Muller, M. Cudic, H. Paulsen, L. Otvos, Jr., F. G. Hanisch and O. J. Finn, J. Exp. Med., 2002, 196, 1435 CrossRef CAS.
- T. Jensen, P. Hansen, L. Galli-Stampino, S. Mouritsen, K. Frische, E. Meinjohanns, M. Meldal and O. Werdelin, J. Immunol., 1997, 158, 3769 CAS.
- S. J. Danishefsky and J. R. Allen, Angew. Chem., Int. Ed., 2000, 39, 837 CrossRef CAS.
- G. F. Springer, Science, 1984, 224, 1198 CAS.
- P. E. Dawson and S. B. Kent, Annu. Rev. Biochem., 2000, 69, 923 CrossRef CAS.
- L. A. Marcaurelle, L. S. Mizoue, J. Wilken, L. Oldham, S. B. H. Kent, T. M. Handel and C. R. Bertozzi, Chem. Eur. J., 2001, 7, 111 CrossRef.
- H. Hojo and S. Aimoto, Bull. Chem. Soc. Jpn., 1991, 64, 111 CAS.
- P. Bulet, G. Hegy, J. Lambert, A. van Dorsselaer, J. A. Hoffmann and C. Hetru, Biochemistry, 1995, 34, 7394 CrossRef CAS.
- Y. Shin, K. A. Winans, B. J. Backes, S. B. H. Kent, J. A. Ellman and C. R. Bertozzi, J. Am. Chem. Soc., 1999, 121, 11684 CrossRef CAS.
- K. A. Winans, D. S. King, V. R. Rao and C. R. Bertozzi, Biochemistry, 1999, 38, 11700 CrossRef CAS.
- R. Kornfeld and S. Kornfeld, Annu. Rev. Biochem., 1985, 54, 631 CrossRef CAS.
- J. Lechner and F. Wieland, Annu. Rev. Biochem., 1989, 58, 173 CrossRef CAS.
- A. Helenius and M. Aebi, Science, 2001, 291, 2364 CrossRef CAS.
- C. Hammond, I. Braakman and A. Helenius, Proc. Natl. Acad. Sci. USA, 1994, 91, 913 CAS.
- J. B. Huppa and H. L. Ploegh, Cell, 1998, 92, 145 CrossRef CAS.
- K. S. Cannon and A. Helenius, J. Biol. Chem., 1999, 274, 7537 CrossRef CAS.
- G. T. DeKoster and A. D. Robertson, Biochemistry, 1997, 36, 2323 CrossRef CAS.
- D. F. Wyss, J. S. Choi, J. Li, M. H. Knoppers, K. J. Willis, A. R. Arulanandam, A. Smolyar, E. L. Reinherz and G. Wagner, Science, 1995, 269, 1273 CAS.
- K. C. Garcia, M. Degano, R. L. Stanfield, A. Brunmark, M. R. Jackson, P. A. Peterson, L. Teyton and I. A. Wilson, Science, 1996, 274, 209 CrossRef CAS.
- M. N. Fukuda, H. Sasaki, L. Lopez and M. Fukuda, Blood, 1989, 73, 84 CAS.
- S. E. O’Connor and B. Imperiali, Chem. Biol., 1996, 3, 803 CrossRef CAS.
- D. H. Live, Z. G. Wang, U. Iserloh and S. J. Danishefsky, Org. Lett., 2001, 3, 851 CrossRef CAS.
- Z.-G. Wang, X. Zhang, M. Visser, D. Live, A. Zatorski, U. Iserloh, K. O. Lloyd and S. J. Danishefsky, Angew. Chem., Int. Ed., 2001, 40, 1728 CrossRef CAS.
- K. Haneda, T. Inazu, M. Mizuno, R. Iguchi, K. Yamamoto, H. Kumagai, S. Aimoto, H. Suzuki and T. Noda, Bioorg. Med. Chem. Lett., 1998, 8, 1303 CrossRef CAS.
- F. L. Strand, Endorphins, Chemistry, Physiology, Pharmacology, and Clinical Relevance, Marcel Dekker, New York, NY, 1982 Search PubMed.
- V. J. Hruby and C. Gehrig, Med. Res. Rev., 1989, 9, 343 CAS.
- S. A. Mitchell, M. R. Pratt, V. J. Hruby and R. Polt, J. Org. Chem., 2001, 66, 2327 CrossRef.
- E. J. Bilsky, R. D. Egleton, S. A. Mitchell, M. M. Palian, P. Davis, J. D. Huber, H. Jones, H. I. Yamamura, J. Janders, T. P. Davis, F. Porreca, V. J. Hruby and R. Polt, J. Med. Chem., 2000, 43, 2586 CrossRef CAS.
- R. D. Egleton, S. A. Mitchell, J. D. Huber, J. Janders, D. Stropova, R. Polt, H. I. Yamamura, V. J. Hruby and T. P. Davis, Brain Res., 2000, 881, 37 CrossRef CAS.
- R. Polt, F. Porreca, L. Z. Szabo, E. J. Bilsky, P. Davis, T. J. Abbruscato, T. P. Davis, R. Horvath, H. I. Yamamura and V. J. Hruby, Proc. Natl. Acad. Sci. USA, 1994, 91, 7114 CAS.
| This journal is © The Royal Society of Chemistry 2005 |