Salts of extended tetrathiafulvalene analogues: relationships between molecular structure, electrochemical properties and solid state organisation
Pierre
Frère
*a and
Peter J.
Skabara
b
aGroupe Systèmes Conjugués Linéaires, CIMMA, UMR-CNRS 6200, 2 Boulevard Lavoisier, 49045 Angers Cedex France. E-mail: pierre.frere@univ-angers.fr; Fax: 33 (0)2 41 73 54 05
bDepartment of Chemistry, University of Manchester, Oxford Road, Manchester, UK M13 9PL
First published on 9th December 2004
Abstract
By considering the structures of many salts derived from extended TTF analogues, relationships between the molecular architecture of the donors with their electrochemical properties and their stacking mode in the salts are presented in this critical review. Three categories of donors corresponding to their extension modes have been considered. Firstly, for linearly extended TTFs the crucial role of the spacer in modifying the electrochemical properties and the packing mode in the salts is presented. Secondly, bidimentional extension of the donors obtained by linking several dithiafulvenyl units on a TTF core led to materials with increased dimensionality. Finally, the last class corresponds to the fusion, directly or across a benzene ring, of TTF frameworks. The former are the base of many salts with metallic behaviour. (148 references.)
 Pierre Frère Pierre Frère | Pierre Frère was born in Laval, France, in 1961. He studied physics and chemistry at the University of Nantes. He passed the national competitive exam of “Agregation” in Chemistry in 1986 and received his PhD degree in organic chemistry from the University of Angers in 1993, under the supervision of Professor A. Gorgues. He then became Maître de Conférences at the University of Angers, where he was promoted to Professor in 1999. His current research interests cover organic materials, especially oligothiophenes and polythiophenes for semiconducting and conducting applications and molecular conductors based on tetrathiafulvalene analogues. |
 Peter J. Skabara Peter J. Skabara | Peter Skabara was born in Edinburgh, Scotland, in 1968. He obtained his first degree (BSc Hons in Chemistry) from Queen Mary and Westfield College, University of London in 1991. After completing his doctoral studies under the supervision of Professor Martin Bryce at the University of Durham (1994), he then went on to work in the group of Professor Klaus Müllen at the Max-Planck Institute for Polymer Research in Mainz, Germany. In 1995, he was appointed as Lecturer in Organic Chemistry at Sheffield Hallam University and moved to his present position as Lecturer in Inorganic Materials Chemistry at the University of Manchester in 2000. His research activities involve the synthesis and characterisation of electroactive molecules and macromolecules, with particular emphasis on polythiophenes and tetrathiafulvalenes. He is the very proud father of 4 daughters (to date). |
Introduction
The rational design of new organic molecular materials requires intensive experimental investigations in order to elucidate the relationship existing between molecular structures and solid state properties. In this field, cation radical salts of tetrathiafulvalene (TTF) derivatives are the subject of intense research activities owing to their unusual and exciting electric and magnetic properties.1–4 Such behaviour results from the combination of specific electrochemical and structural properties associated with the chemical structure of the TTF core. On one hand, TTF derivatives constitute a well known class of reversible redox systems leading to cation radical and dication species, highly stabilised by the aromatic character of the 1,3-dithiolium rings (Fig. 1). Secondly, radical cation salts are characterised, in the majority of cases, by segregated stacks of donors and anions (organic or mineral). The short intermolecular distances within a column, corresponding to an overlap of the π orbitals combined with a mixed valence state, allow the delocalisation of electrons and thus conduction along the stacking direction. The main limitation in the transport properties for these materials resides in their strong monodimensional character which leads to a metal–insulator transition at low temperature (Peierls transition), due to the localisation of charge density resulting from subtle lattice distortions.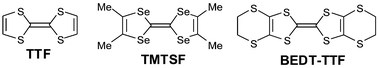 | ||
| Chart 1 | ||
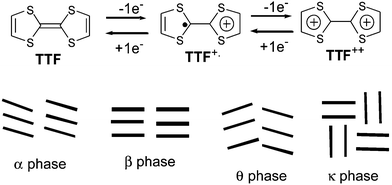 | ||
| Fig. 1 Reversible oxidation processes of TTF (top) and main crystallographic arrangements observed in salts based on BEDT-TTF (bottom). | ||
Extensive studies concerning the modification of the TTF framework have been elaborated in order to control the intermolecular organisation in the solid state and thus to improve the properties of the materials.5–7 Nevertheless, these crystalline materials are the result of the interplay of different factors which are often difficult to separate and identify. A slight structural modification of a given molecule may have significant effects on its electrochemical properties and intermolecular interactions, which may in turn influence the packing in the solid state. In order to promote interstack contacts, the most basic strategy is to manipulate the TTF core either by changing the nature of the chalcogen atoms, or by grafting appropriate substituents onto the dithiole rings. Following this approach, TTF derivatives such as TMTSF and the famous BEDT-TTF have led to several superconducting materials.8 Although the structural modification of these new donors did not implicate strong variation in electrochemical behaviours, the packing modes in the resulting salts were strongly modified. Thus with BEDT-TTF, in addition to the classical crystalline phases that involve stacks of donors (namely α, β and θ phases), the increase in number of S—S contacts involving the external sulfur atoms induced a new phase called the κ-phase, which is characterised by a two-dimensional arrangement of dimers (Fig. 1).9–11
Another strongly developed trend has involved the design of extended analogues of TTF by the incorporation of a π-conjugated system and/or by grafting chalcogen-rich heterocycles onto the TTF core. The expected properties for these new donors concern both the following electronic and crystallographic reasoning:
—stabilisation of the oxidised states by delocalization of the charges and easier access to polycationic states due to a diminution of coulombic repulsion,
—enhanced dimensionality of the materials by increasing the number of π–π and/or chalcogen—chalcogen interactions.
By contrast with the non-extended TTF derivatives, the large diversity of the molecular structures created by the extension of the TTF cores is also responsible for changing the basic electrochemical properties and for manifesting original packing modes in the salts.
On the other hand, as outlined by recent reviews, the interest of the TTF family exceeds the field of organic conductors. Modified TTF derivatives are increasingly implicated in the development of new applications such as cation sensors, macromolecular and supramolecular systems, molecular switches, molecular rectification, non linear optical materials and organic semiconductors.4,12–15 For these new areas of research, the diversity and flexibility of designed extended TTFs offer a powerful opportunity to control the electrochemical and physical properties of the π-electron systems in order to adapt them for different applications.
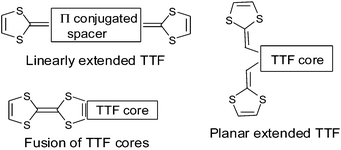
In preceding reviews, Müllen in 199416 and Bryce in 19956 described the structural approaches to increase the dimensionality of the materials by using extended TTFs. On the other hand, Roncali in 199717 reviewed the electronic properties of linear extended TTFs incorporating thiophene rings. Taking into consideration the large number of crystallographic structures published to date, the purpose of this article is to review salient features by showing how the topology of the extended TTF derivatives can act on their electrochemical properties and stacking modes in the corresponding salts. This review will mainly study π-extended TTF derivatives which have led to salts (cation radical, dication or transfer charge complexes). In this respect and based on the different extension modes of the TTF cores, we have considered the following three classes of donors:
—linearly extended derivatives with π-conjugated spacer between the two 1,3-dithiole heterocycles,
—planar extended molecules with dithiafulvenyl groups grafted onto a TTF core,
—bisTTFs linked via fusion of two TTF cores.
 | ||
| Chart 2 | ||
For each group, after a brief synopsis of their synthetic preparation, the electrochemical properties then the stacking modes in the salts will be related to the molecular structures of the donors.
1 Linearly extended TTFs with π-conjugated spacers
The design of new TTF analogues by the incorporation of a π-conjugated unit between the two 1,3-dithiole rings has rapidly emerged as one of the main synthetic strategies to extend the TTF core. A large variety of π-conjugated systems have been explored, thus revealing the strong influence of the nature of the spacer for inducing changes both in the electrochemical properties of the donors and in the structure of the corresponding materials.1.1 Vinylogues of tetrathiafulvalene
The simplest modification, previously explored by Yoshida et al.18 consisted of replacing the central double bond of the TTF by unsubstituted polyene units. Many generic derivatives of TTF vinylogues 1, with different substituents R grafted on the 1,3-dithiole rings and with polyene units containing up to 10 sp2 carbon atoms, have been synthesized.19–22 In general, the synthetic pathways proceed by Wittig reactions from ylides W bearing the dithiafulvene systems onto the corresponding dialdehyde. Alternatively, the mono-protected derivatives 2 are reacted to give aldehydes 3, followed by a Wittig–Horner olefination with phosphonate anions P or by a McMurry coupling (Scheme 1).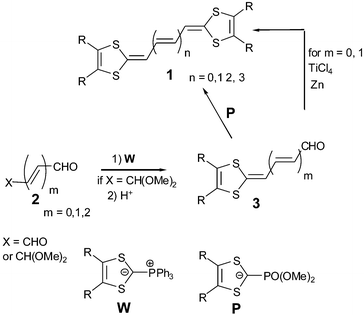 | ||
| Scheme 1 General synthesis of vinylogue TTFs 1. | ||
As for TTF derivatives, the cyclic voltamogramms (CV) of donors present two reversible oxidation processes corresponding to the successive formation of cation radical and dication species (Fig. 2). The values of the first oxidation potentials E1 depend essentially on the electron donating character of the substituents R grafted on the 1,3-dithiole rings, while elongation of the spacer provokes a decrease in the difference ΔE = E2 − E1 between the two oxidation potentials.18,21,22 The easier access to the dication state is indicative of the decrease in intramolecular coulombic repulsion between the two positive charges.23 Such evolution is associated with a strong diminution of the thermodynamic stability of radical cation 1+˙. Thus for compounds 1, coalescence of the two oxidation processes, corresponding to the direct access to the dication state, takes place when n = 2 in CH2Cl2 or n = 1 in CH3CN and shows that elongation of the polyene unit does not favour the formation of cation radical salts.
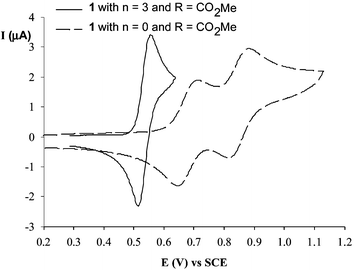 | ||
| Fig. 2 CVs of two compounds 1 (R = CO2Me) with n = 0 or n = 3 in CH2Cl2 from ref. 21. | ||
Even for the shorter derivatives 1 with n
= 0 and despite the very large variety of substituents R which have been employed, no cation radical or dication salts suitable for X-ray analysis have been obtained. Only the structure of the complex between 1a
(n
= 0, R = SMe) and tetracyanoquinodimethane (TCNQ) has been described and allows a comparison of the geometries of 1a in the neutral and the +1 states (Fig. 3).24 Neutral molecule 1a is characterised by a fully planar extended TTF core in a transoid conformation while the four methyl groups are located in the perpendicular plane. As observed for tetraalkylsulfanyl-TTF, the conformation adopted by the methanesulfanyl groups is consistent with the fact that the sulfur atoms do not participate through a mesomeric effect to the π-donor ability of the molecule, since their lone pair electrons are not directed in the perpendicular plane of the 1,3-dithiole rings.25 In the 1a.TCNQ complex a slight torsion of the donors appears between the two dithiole units. Within the spacer unit, the oxidation is characterised by the shortening of the central bond lengths from 1.434 Å for 1a to 1.402 Å for 1a+˙ while the two others bonds increase from 1.355 Å to 1.380 Å, indicating a delocalisation of the π-electrons centred on the conjugated system. This result is also confirmed by solution ESR studies of various cation radicals of compounds 1 with n
= 0, which show that the spin population is located on the S2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–CCS2 part of the molecules.24 The structure of the complex consists of independent columns with alternation of the 1a+˙ and TCNQ−˙ ions and confers to the complex an insulating character. It also exhibits diamagnetic behaviour, indicating a strong correlation between anion and cation radicals.
C–CCS2 part of the molecules.24 The structure of the complex consists of independent columns with alternation of the 1a+˙ and TCNQ−˙ ions and confers to the complex an insulating character. It also exhibits diamagnetic behaviour, indicating a strong correlation between anion and cation radicals.
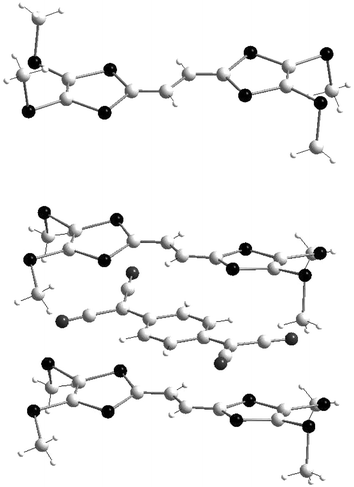 | ||
| Fig. 3 Molecular structures of 1a and 1a+˙ in 1a.TCNQ redrawn from ref 24, copyright (1996) with permission from the American Chemical Society. | ||
Compounds 4 involving a cyclohexene unit as spacer, which can be seen as a cis-fixed configuration of the polyene units, have also led to cation radical salts.26 The synthesis proceeds in two steps from diketone 5 by a two fold olefination with phosphonate P followed by a retrocycloaddition reaction at ca. 220 °C (Scheme 2).
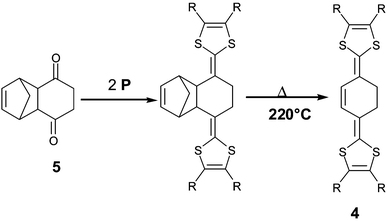 | ||
| Scheme 2 Synthesis of compounds 4. | ||
Donors 4 are oxidized to cation radical and dication species in two reversible oxidation steps. The difference ΔE between the two oxidation potentials (ca. 0.1 V) is similar to that observed for vinylogues 1 with n = 1.
With derivative 4a substituted by methyl groups on the 1,3-dithiole rings, Yamashita et al. obtained two isomorphous cation radical salts (4a)2 PF6 and (4a)2 AsF6 which constitute rare examples of vinylogue TTFs crystallising in a mixed valence state. The molecular structure of the donors shows good planarity. The structures, very similar to tetramethyl-TTF salts, are characterized by the stacking of molecules along the c axis with interplanar distances around 3.66 Å (Fig. 4). The columns of donors are separated by the anions, thus procuring to the salts a mono-dimensional character. The conductivities of the salts around 10 S cm−1 at room temperature rapidly decrease upon cooling.
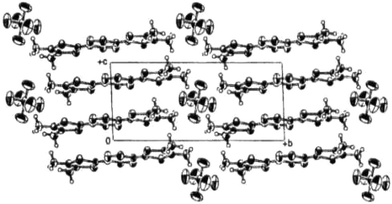 | ||
| Fig. 4 Crystal structure of (4a)2.PF6 reprinted from ref. 26, copyright (2002), with permission from Taylor & Francis Ltd. (http://tandf.co.uk/journals). | ||
1.2 Conformational changes in aryl substituted TTF vinylogues
The last decade has seen the emergence of a new series of TTF vinylogues 6 substituted by aryl groups upon the central part. These compounds are synthesized from oxidative coupling of the corresponding 1,4-dithiafulvene 7 (Scheme 3).27–29 | ||
| Scheme 3 Synthesis of vinylogue TTFs 6. | ||
As shown in several X-ray structures of neutral 6, the steric hindrance due to substitution of the hydrogen atoms of the vinylogue unit by aryl groups, provokes a distortion of the molecules which present the twisted conformations shown in Fig. 5 (left).29,30 After oxidation to radical cation or dication species, strong conformational changes occur and the extended TTF core becomes planar, whilst the aryl groups are sited almost perpendicular to the plane of the heterocycles.30,31
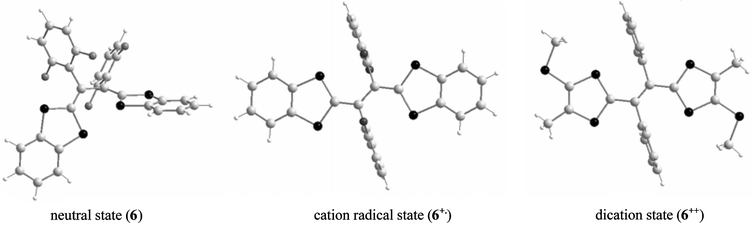 | ||
| Fig. 5 Molecular structures of derivatives 6 in neutral and oxidized states. Compounds 6 and 6++++++++˙ redrawn from ref. 30, 6++++++++++++++++ redrawn from ref. 31, copyright (1998) with permission from IUCr. | ||
The electrochemical properties and, in particular, the stability of the cation radical vary with geometric and electronic factors arising from the aryl groups. Lorcy–Hapiot et al. have shown that the dramatic modification of the electrochemical properties is the consequence of the strong conformational change which takes place during the oxidation processes.29,32 The first electron transfer, associated with the strongest conformational change, is more difficult than the second one for which only a little change of the structure is required. For unsubsituted or para-substituted benzene units as aryl groups, an inversion of the potentials takes place and the compounds are directly oxidised to the dications through a reversible two electron oxidation process. On the contrary, with ortho- or the electron donating p-NMe2 substituents, the +1 state is stabilized and the CVs present two reversible single electron oxidation waves. Hence, the composition of the salt obtained by electrochemical or chemical oxidation is strongly dependent on the nature of the aryl groups.
In earlier work, Lorcy et al. have obtained X-ray structures of dication salts with derivatives bearing unsubstituted or para-substituted aryl groups31,33 while Yamashita et al. have isolated cation radical salts with ortho-substituents.30,34 The conformations of the molecules 6 in the oxidised states are similar for radical cation and dication species (Fig. 5). However, the bond lengths for the spacer are characteristic of the +1 or the +2 species. For dications, the central bond around 1.35 Å is close to a double bond while the two others represent expected single bond lengths around 1.44 Å, indicating that the canonical form 6++, with the positive charges localized on the dithiole rings, has an important contribution (Scheme 4). For the radical cations, the three bond lengths are closer at ca. 1.4 Å, as expected for a delocalized spacer unit.
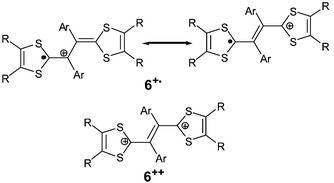 | ||
| Scheme 4 Main mesomeric forms of radical cation and dication species of 6. | ||
Very recently, control of the steric strain has been achieved by connecting the two dithiole rings of compounds 8 with bridging units.35 With a short link (m = 3, 4), the oxidation directly proceeds by a two electron transfer process, independent of the nature of the aryl groups. Although the X-ray structures of 8 in the neutral state are similar to their acyclic analogues 6, the dications 8++ with short links (m = 3, 4) adopt non-planar conformations for which the topology is imposed by the tension of the link and not by the steric hindrance of the aryl groups. These systems demonstrate a fascinating molecular clip trigger action, actuated by electron transfer.
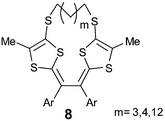 | ||
| Chart 3 | ||
The cation radical salts obtained from derivatives 6 always give a 1 ∶ 1 stoichiometry and consequently present semiconducting behaviour with conductivities in the region of 10−5 S cm−1 to 10−2 S cm−1, depending both on the substituents R borne by the dithiafulvene groups and on the anion.
 | ||
| Chart 4 | ||
The bulky aryl groups located in the perpendicular plane relative to the extended systems prevent a direct superposition of two molecules in the crystals. Yamashita and co-workers have shown that the stacking modes are essentially driven by the substituents R, with some variations due to the nature of the counteranions.36
With R–R =
–(CHCH)2– as substituents, the benzene cores fused with the 1,3-dithiole rings allow the formation of π−π interactions by developing a stacking mode whereby a molecule bridges two others as indicated in Fig. 6. This overlapping mode minimises the steric repulsion and leads to the establishment of two-dimensional networks. Such a structure is clearly demonstrated by the stacking mode of salt 6a.PF6 which features intermolecular distances of 3.6 Å between the benzodithiole moieties. Along the b axis, the donors form ribbons separated from each others by anions (Fig. 6).
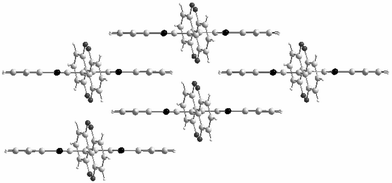 | ||
| Fig. 6 Crystal structure of 6a.PF6 redrawn from ref. 30, copyright (1998) with permission from the Royal Society of Chemistry. | ||
For the same derivative 6a, a change in the counteranion affects the overlapping mode of radical cations as shown in Fig. 7, but the structures are always driven by the π-stacking between the benzodithiole moieties.The replacement of PF6− anions by FeCl4− procures a slight shift of the radical species and there is no longer a direct superposition of the benzene cores. The decrease in π−π interactions induces a slight increase of the intermolecular distances to 3.8 Å. With the bulkier ReO4− anion, the radicals overlap through rotations of almost 90° between paired molecules, thus forming an original two dimensional zigzag structure.35
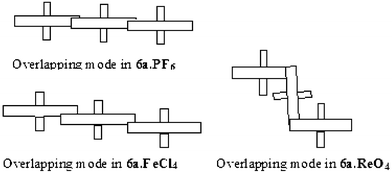 | ||
| Fig. 7 Variation of the overlapping modes for 6a as a function of the counteranions. | ||
Although salt 6b.Au(CN)2.PhCl is always characterized by the overlapping mode of the radicals, in 6c.PF6 the structure is driven by hydrogen bonding between the OH groups and the F atoms of the anions and no stacking between the benzodithiole moieties is observed.37
The replacement of benzo by ethylenedithio substituents on the 1,3-dithiole rings leads to the disruption of the π−π overlapping mode between the radicals. 6d.PF6.(H2O)836 and 6e.ReO4.(H2O)837 show similar grid-like structures (Fig. 8) characterised by a large volume occupied by eight water molecules. In the two isomorphous crystals 6f.Au(CN)2 and 6g.Au(CN)2, the radicals are separated by the anions by forming mixed organic–inorganic columns.34
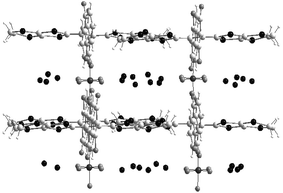 | ||
| Fig. 8 Crystal structure of 6e.ReO4.(H2O)8 redrawn from ref. 37, copyright (2001) with permission from Elsevier. | ||
Several dication salts have been described.31,33,38 Here also, the role of the substituents R for organizing the packing mode is clearly evident. Hence, in the presence of akylsulfanyl groups, the distances separating two dithiafulvalene rings are very large and often anions are inserted between two dications. By contrast, for several benzo derivatives with CuCl42− and Cu2Cl62− anions, π−π stacking of the dication is observed.38
1.3 Conformational changes for extended TTFs with anthracene cores as spacers
Much attention has been devoted to the study of extended TTFs built with various π-quinoid spacer units. The simplest p-quinodimethane analogues 9 developed by Yamashita and co-workers have the strongest donor ability amongst TTF analogues with oxidation potentials which can be less than 0 V/SCE.39 This behavior is due to the high stability of oxidized species which consist of two or three aromatic units, corresponding to benzene and dithiolium rings (Scheme 5). For the unsubstituted derivative 9a the oxidation was described to proceed in two single electron steps, whilst for the benzo derivatives 9b only a single two electron process was observed.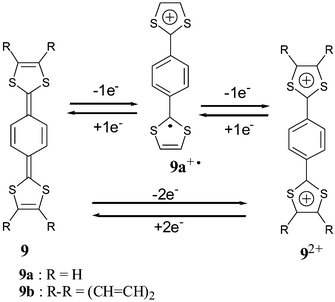 | ||
| Scheme 5 Oxidation of compounds 9. | ||
Owing to the very low stability of p-quinodimethane analogues 9, more stable derivatives such as 9,10-bis(1,3-dithiol-2-ylidene)-9,10-dihydroanthracene 10 have been synthesised (Scheme 6). Symmetrical derivatives can be obtained by a two fold olefination of anthraquinone 11 with phosphonate anions P.40,41 An alternative route, which offers the possibility of non-symmetrical systems, proceeds in two steps from anthranone 12. The ketones 14, which correspond to a mono-olefination of 11, are synthesised by reaction of dithiolium salts13 with anthranone 12.41
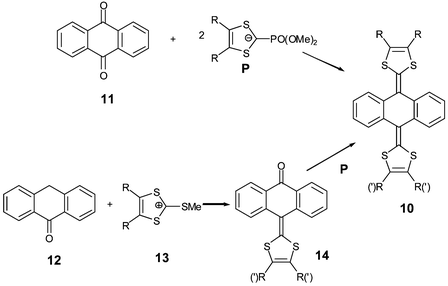 | ||
| Scheme 6 General synthesis of compounds 10. | ||
Theoretical calculations, together with X-ray crystallographic evidence, confirm that the compounds 10 in the neutral state always adopt the non-planar butterfly or saddle-like conformation presented in Fig. 9 (left).42,43 The central ring of anthracene adopts a boat conformation with the two dithiafulvenyl arms pointing upwards and the benzene rings downwards. Such a conformation allows a reduction in steric hindrance between the sulfur atoms of the 1,3-dithiole rings and the hydrogen atoms in the peri positions of the anthracene system. Bryce and co-workers have developed and studied several series of these derivatives, denoted as ‘Molecular Saddles’, by varying the substituents grafted on both the dithiole and anthracene units.43–49
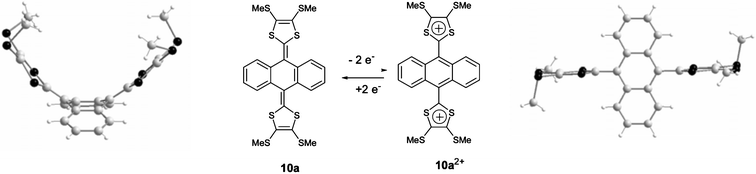 | ||
| Fig. 9 Molecular structure of 10a for the neutral state (left) redrawn from 42 and the dication state (right) redrawn from ref. 48, copyright (2001) with permission from Wiley-VCH. | ||
All the donors 10 are directly oxidised to the corresponding dications and the oxidation is accompanied by a marked structural change, as shown in Fig. 9 (right) for derivative 10a.48 The analysis of the structure of the dications is consistent with the formation of fully aromatic anthracene units and the bond lengths in the two dithiolium rings are characteristic of those observed for dications of TTF derivatives such as the vinylogues 6++. The dithiolium groups are connected to the spacer by single bonds (d ≈ 1.47 Å) and the dihedral angles between the anthracene and dithiolium planes are around 75–85°, showing that the two positive charges localised within the dithiolium rings are not conjugated across the spacer.
The electrochemical properties of 10 are driven by the conformational change accompanying the oxidation process. The CVs of 10 show a quasi-reversible two-electron oxidation peak in the range 0.3–0.8 V/SCE, depending on the substituents R grafted on the 1,3-dithiole rings. The high stability of the dication state, due to the tremendous gain in aromaticity, induces facile removal of the second electron, compared to the first, and thus leads to the classical inverted potentials in a single two-electron process. The separation around 300 mV between the anodic and cathodic peaks, which increases by cooling the solution,41 represents the slow kinetics of the conformational change during the reduction process (Fig. 10 for 10b).
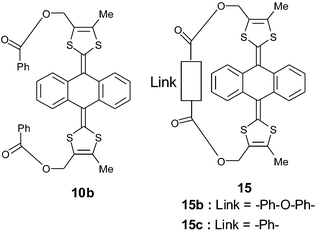
Moreover, Bryce and co-workers have shown that for derivatives 15, which are bridged intramolecularly across the two 1,3-dithiole rings, the length and the flexibility of the link induce a strong effect on the reversibility of the oxidation process, as shown in Fig. 10.46,49
 | ||
| Chart 5 | ||
The insertion of an oxygen bridging unit between the phenyl groups of 10b to give 15b, or linking the ester groups of 15b by a common phenylene unit to give 15c, results in the concomitant increase in the oxidation potentials and reduction in the separation between the anodic and cathodic peaks. Comparison of the X-ray structures of the non-bridged 10 and bridged 15 derivatives, both for neutral and dication species, shows that the bridge produces marked differences on the structure of the dication. In particular, the anthracene core in 15 no longer has the fully planar structure which is observed for the non-bridged derivatives 10, but adopts a slight boat conformation. Nevertheless, this non-planar conformation is not as pronounced as the one observed in the neutral state. In conclusion, the strong tension imposed on the structure by a short bridge destabilises the dication species and restricts the conformational change between the neutral and oxidised states, thus increasing the speed of the reduction process.
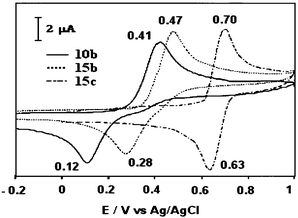 | ||
| Fig. 10 CVs of compounds 10b, 15b and 15c from ref. 46, copyright (2001) with permission from the American Chemical Society. | ||
Donors 10 interact with electron acceptors such as TCNQ to give charge transfer salts of which several were suitable for X-ray analysis. Fig. 11 compares the stacking modes observed for three complexes with different stoichiometries. Derivative 10c affords a complex with an unusual 1 : 4 stoichiometry 10c.(TCNQ)4 in which the donors are in the dication form 10c2+ while the acceptors possess a mixed valence (TCNQ)42−.50 This structure is characterised by continuous stacks of the organic anions with intermolecular distances around 3.36 Å. Such a distance allows overlap of the π-orbitals of TCNQ allowing delocalisation of electrons along the perpendicular axes of the columns. By contrast, the non-planar conformation of the donors discourages the efficient overlap of π-orbitals and the dications are regularly intercalated between the columns of acceptors by presenting only weak S—S interactions. The paramagnetic character of the material and its room temperature conductivity σ = 60 S cm−1 are associated exclusively to the mixed valence stacks of TCNQ molecules.
 | ||
| Chart 6 | ||
With 10d the 1 ∶ 2 stoichiometry 10d.(TCNQ)2 for the complex is indicative of the formation of dication 10d2+ and radical anion TCNQ.−.44 The conformation adopted by the donor 10d and the bond lengths observed in the structure of TCNQ confirm the oxidation states for the donor and the acceptor. Here also, the anions stack by forming columns of TCNQ with weak interactions, which are separated by dications. However, in contrast with the preceding structure, the radical anions form dimers with intermolecular distances of 3.15 Å within a dimer and 3.56 Å between two dimers. The magnetic and electric properties are not given, but for derivative 10c a similar 1 ∶ 2 complex with 2,5-dibromo-TCNQ is indicated to be insulating and diamagnetic.50 The dimers of TCNQ.− are known to have diamagnetic behaviour due to the pairing of spins.
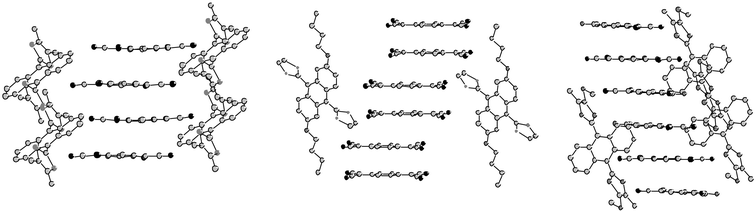 | ||
| Fig. 11 Stacking modes for three complexes of donors 10 with TCNQ: (left) 10c2+.(TCNQ)42− redrawn from ref. 50, copyright (1990) with permission from Wiley-VCH; (middle) 10d2+.(TCNQ−)2 redrawn from ref. 44, copyright (2000) with permission from Wiley-VCH; (right) 10e2+.TCNQ(16−)2.H2O redrawn from ref. 51, copyright (1993) with permission from IUCr. | ||
For derivative 10e, reaction with TCNQ gave the complex 10e2+.TCNQ(16−)2.H2O.51 The formation of 16− corresponds to the oxidation of TCNQ2− by oxygen,52 indicating that the reduction of TCNQ by 10e must attain the dianionic species. Trimers built by the intercalation of neutral TCNQ between two molecules of 16− form stacks separated by the dications.
Replacing TCNQ by the weaker π-acceptor fluorene derivative 17, Bryce and co-workers obtained complex 10f2+.(17−)2.2MeCN which represents the first example where a full charge transfer between fluorene 17 and a TTF derivative has been observed. This result is probably due to the greater donor ability of 10f than that of tetramethyl-TTF.53 The analysis of the structure of the anion 17− shows good planarity between the fluorene core and the dicyanomethylene group, whilst the bond lengths are consistent with complete delocalisation of the π-electron over the entire molecule. As shown in the TCNQ complex, the fluorene anions stack with intermolecular distances between the planes formed by the rings at ca. 3.3 Å. The dications, featuring only weak interactions, are inserted between the anion columns. With the related derivative 10g, the reaction with fluorene 17 in dioxane gave, after a long time (about 10 months), the salt 10g2+.(18−)2.3C4H8O2.54 The generation of anion 18− from 17 is not clearly established. By contrast with other complexes, the structure does not present any columns of acceptors but the crystal packing corresponds to a succession of alternating layers of dications and anions.
Several dication salts with inorganic anions have also been obtained by electrocrystallisation or by oxidation with iodine vapour.46,48,49 Owing to the topology of the dications, the donors do not self-assemble into stacks and there are no significant S—S contacts.
1.4 Limited conformational changes for extended TTFs with naphthathiadiazole cores as spacers
Another strategy developed by Yamashita and co-workers consisted of replacing the benzene rings with heterocycles such as pyrazine 1955,56 or 1,2,5-thiadiazole 20,5721,582259 units (Scheme 7). The substitution of the sp2 carbon atoms in the peri position by sp2 nitrogen atoms decreases steric hindrance, thereby promoting dramatic changes in structural and electrochemical properties compared to 10. These compounds are obtained by the same synthetic pathways used for 10 and 4 from the respective diketones 23a–d and the phosphonate anions P bearing the dithiole rings. As shown by X-ray crystallography, the molecular structures of compounds 19,552057 and 2158 are planar (Fig. 12 for 21) and the analysis of the structures reveals that the distances separating the sulfur and nitrogen atoms (d = 2.73–2.85 Å) are significantly inferior to the sum of the van der Waals radii (S + N = 3.35 Å). Such short contacts are indicative of attractive interactions which stabilise the planar conformations.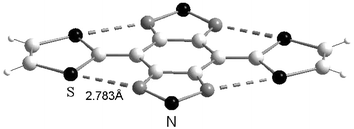 | ||
| Fig. 12 X-ray structure of 21a (R = H) redrawn from ref. 58, copyright (1992) with permission from the American Chemical Society. The S—N interactions are presented by dotted lines. | ||
 | ||
| Scheme 7 General synthesis of compounds 19, 20, 21 and 22. | ||
CVs of 19, 20 and 21 present two reversible single-electron oxidation waves corresponding to the successive formation of the radical cation and dication. By contrast with 10, a possible access to the +1 oxidation state is interpreted by the expected non-conformational change upon oxidation. Consequently, cation radical salts are accessible and several salts presenting electrical conductivities around 0.2–0.5 S cm−1 for 19 and 16–68 S cm−1 for 20 have been reported, although no structure has ever been described. For derivative 21, physical studies were essentially devoted to the conductivity and mobility of charge carriers for the neutral state.58 Indeed, single crystals of 21a (R = H) exhibit a significant conductivity of 8.3 × 10−4 S cm−1. This unusual electrical behaviour for a neutral organic material results from the strong π interactions between the molecules which are uniformly stacked with interplanar distances of 3.46 Å.
For compounds 22, which have a hybrid structure of fused benzene and 1,2,5-thiadiazole rings, structural and electrochemical properties are derived from those of derivatives 10 and 21. In the neutral state, the structure of 22 presents the same butterfly or saddle conformation adopted by 10.59 Nevertheless, the short distances d = 2.78 Å separating the sulfur and nitrogen atoms reveal the formation of S—N interactions between the dithiole and thiadiazole rings. Depending on the nature of the substituents grafted on the dithiole rings, the CVs of 22 show one or two close reversible oxidation waves. The occurrence of two independent oxidation processes for some derivatives is in agreement with the formation of a cation radical in the first step. For derivatives having a single oxidation peak, the width at half maximum of 45–65 mV is larger than the 28.5 mV expected for an ideal two electron transfer, indicating that the oxidation process also involves two very close one-electron steps. The difference (ΔE) between the oxidation potentials corresponding to the formation of the radical cation and dication was calculated from Myers and Shain's method60 and gave values of 12–90 mV. Such results indicate that the cation radicals are not very stable in solution and have a tendency to undergo disproportionation to the dication and neutral species. Nevertheless, in contrast with compounds 10, their stability is sufficient to lead to cation radical salts in the solid state. Derivative 22a led to cation radical and dication salts suitable for X-ray analysis, thereby allowing a comparison of the structures for the different oxidised species (Fig. 13).61 In the +1 state, the molecule still adopts a butterfly conformation stabilised by S—N intramolecular interactions thus indicating that no important conformational change exists between the neutral and +1 states. For the dication, two structures with varying anions have been described. In the presence of large anions such as SbF6−61 or the complex Au(mnt)2−,62 the structure of the dication (conformation b) is close to that of 10++ with the two dithiafulvene groups twisted by 45–46° from the planar naphthathiadiazole spacer. The second conformation is an intermediate between the preceding dication and radical cation structures. Apart from one dithiole ring, which is twisted by 45° from the plane of the spacer group, the molecule is very close to the conformation of the +1 state.
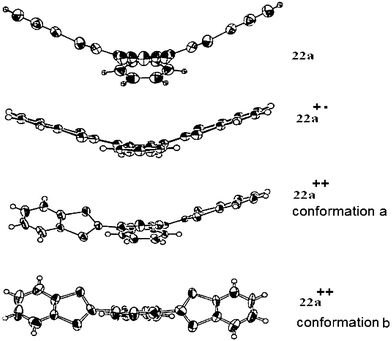 | ||
| Fig. 13 Evolution of the molecular structures of derivative 22a with the oxidation states from ref. 61, copyright (1996), with permission from Taylor & Francis Ltd. (http://tandf.co.uk/journals). | ||
We can conclude that, in spite of the steric hindrance imposed by the benzene ring, the S—N intramolecular interactions of the cation radical impart stability and delay the conformational change, thus avoiding the potential inversion observed for compounds 10. Consequently, by comparison with 10, the thermodynamic stability of the +1 state of 22 increases and in the solid state several cation radicals have been obtained with 1 ∶ 1 stoichiometry 22+˙.Y− (with 22a and Y− = ClO4− and PF6−)59 or in mixed valence states, such as (22)2+˙.Y−(solvent) (with 22b and 22c; Y = PF6−, AsF6−, BF4−; solvent = tetrahydrofuran THF, 2,5-dihydrofuran DHF, 1,3-dioxolane DO).63–65
The crystal structures of 22a+˙.Y− are characterised by the stacking motif of the donors, which, in spite of their non-planar conformation, form columns with intermolecular distances around 3.60–3.65 Å. With PF6− counteranions, intercolumnar interactions are established by means of superimposed benzene rings as presented in Fig. 14. This material exhibits semiconducting behaviour with surprisingly high electrical conductivity (8.3 S cm−1) for a 1 ∶ 1 stoichiometric salt which is attributable to facile access to the dication state due to the small difference between the two oxidation processes.59
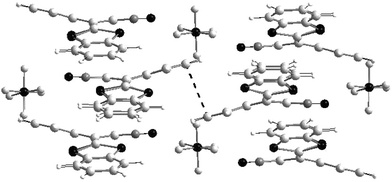 | ||
| Fig. 14 Stacking mode of cation radical in 22a+˙.PF6− salt redrawn from ref. 59, copyright (1994) with permission from Wiley-VCH. The π−π intercolumnar interactions are presented by a dotted line. | ||
The overlap mode of donors in the mixed valence salts (22)2+˙.Y−(solvent) is shown in Fig. 15. The cations always stack along the b axis with intermolecular distances around 3.7 Å and form columns of donors which are separated along the a axis by the insertion of solvent molecules.66 On the other hand, along the c axis, short S—S contacts between the sulfur atoms of ethylenedithio groups are established. These materials exhibit metallic behaviour with electrical conductivities (at room temperature) of 25–60 S cm−1 along the b axis and show a metal–insulator (MI) transition at low temperature. Optical and structural studies revealed that below the MI transition temperature, crystals (22c)2+˙.PF6−(DHF) and (22c)2+˙.BF4−(THF) undergo a decrease in the symmetry of the space groups from P21/a to P/a or P2/a. Such behaviour indicates a dimerisation of the donors corresponding to the classical Peierls–Hubbard type MI transition which characterises the quasi 1D systems.67
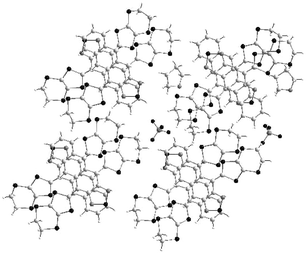 | ||
| Fig. 15 X-ray structure of the (22c)2+˙.PF6−(solvent) redrawn from ref. 67, copyright (2000) with permission from the Royal Society of Chemistry. | ||
The comparison between the structures of compounds 10 and 22 provides irrefutable evidence for the importance of intramolecular interactions obtained by the insertion of heteroatoms into the spacer. This strategy modifies the structural and electrochemical properties of the extended TTFs and consequently changes the properties of the corresponding materials.
1.5 Extended TTFs with thiopheno or furano-quinonoid spacers
In a similar strategy, Takahashi et al. prepared extended-TTFs 24 by incorporating dihydrothiophene or selenophene units as spacers.68 The low stability of 24 prevented the isolation of analysable cation radical salts and more stable derivatives 25,692670 were prepared by fusing the central heterocycles with benzene or pyrazine rings. The synthesis of 24 proceeds, in the last step, by dehydrogenation of 27 with chloranil. Compounds 25, 26 and 27 are synthesised in two steps by trimethylphosphite-mediated coupling of anhydride 28a,b,c with an excess of 2-thioxo-1,3-dithiole 29 (Scheme 8).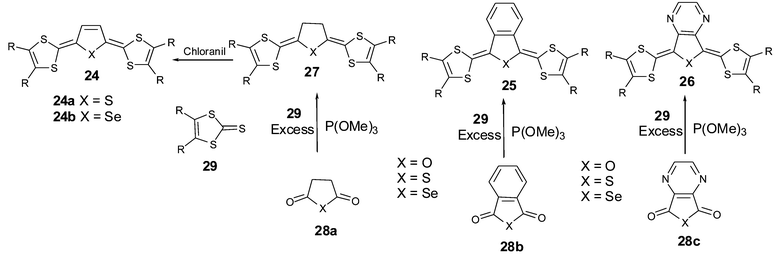 | ||
| Scheme 8 General synthesis of extended TTF with thiopheno or furano-quinonoid spacers. | ||
Each of the donors 24, 25, and 26 are reversibly oxidised into radical cations then dications by two well separated single-electron processes.68 Their oxidation potentials are always inferior to those of the parent TTFs bearing the same substituents R. The stability of the radical cation is due to the contribution of the mesomeric form A with the central aromatic core (Scheme 9). The first oxidation potentials of unsubstituted derivatives 24a (E1 = 0.11 V/SCE) and 24b (E1 = 0.18 V/SCE) are higher than for the p-quinodimethane analogue 9a (E1 = −0.11 V/SCE), due to the decrease in stabilisation of the +1 state by the less aromatic thiophene or selenophene spacers compared to benzene ring. The oxidation potentials of compounds 24, 25 and 26 with the same heteroatoms X and the same substituents R follow the sequence E24 < E25 < E26. The higher oxidation potentials for 26 are attributed to the electron withdrawing effect of the pyrazine ring. In both series, the furan derivatives have slightly better donor ability than the thiophene analogues. This behaviour is an indication of a major contribution from mesomer B in stabilising the radical cation, since if mesomer A predominated the superior aromatic character of the thiophene ring should allow easier access to the +1 state. On the other hand, the differences between the two oxidation potentials ΔE = E2 − E1 of 0.2 V for 24 and 0.3 V for 25 and 26 are smaller than those for TTF analogues, as expected by the decrease in coulombic repulsion between the positive charges of the dication species. It can be noted that ΔE values are greater than those of the model extended TTFs developed by Yamashita, indicating a higher thermodynamic stability of the +1 state.
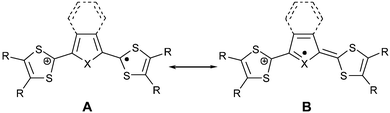 | ||
| Scheme 9 Main mesomeric forms of radical cations 24+˙ and 25+˙. | ||
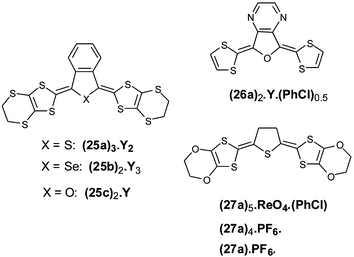
For compounds 25 bearing ethylenedithio groups on the dithiole rings, electrocrystallisation led to different salts with the composition varying with the nature of the heteroatom in the spacer.71–73 Thus, derivative 25b with selenium gave salts with the composition (25b)2.Y3 (with Y− = ReO4−, ClO4− and BF4−) corresponding to an average charge of +3/2 per donor. Replacement of the selenium atom by sulfur (25a) then oxygen (25c) decreases the global charge to +2/3 in (25a)3.Y2 (Y− = ReO4−) and +1/2 in (25c)2.Y (Y− = PF6−, AsF6−, SbF6−).71–73 The structures of the salts are described from two, three or four crystallographically independent molecules which stack by forming dimeric, trimeric or tetrameric sequences. Thus, in the salt (25c)2.PF6, the four independent molecules stack in a ABCDDCBA manner by presenting five overlapping modes as indicated in Fig. 16. The intra-tetramer contacts are shorter than the two inter-tetramer ones in which only the central parts of the molecules participate. The weak conductivity of 7.3 S cm−1 for this mixed valence salt is a consequence of the irregular stacking of the donors. In the salts (25a)3.(ReO4)2 and (25c)2.SbF6(PhCl)0.5 the dimerisation or trimerisation of the donors also induces low conductivities (0.1 and 0.4 S cm−1 respectively).
 | ||
| Chart 7 | ||
For analogues 26 incorporating a pyrazine ring, only the unsubstituted derivative 26a with an oxygen atom in the spacer led to a mixed valence salt. The materials (26a)2.Y.(PhCl)0.5 (Y− = AsF6− or PF6−) possess rt conductivities of 40 S cm−1 and 4 S cm−1.74 The two crystallographically independent molecules are fully planar with short intramolecular S—N contacts. The two molecules stack alternately along the a axis with interplanar distances of 3.47–3.48 Å; several S—S intercolumnar contacts are also observed, as shown in Fig. 17.
 | ||
| Fig. 16 Stacking mode of the donors in (25c)2.PF6 from ref. 71, copyright (1998) with permission from the Chemical Society of Japan. | ||
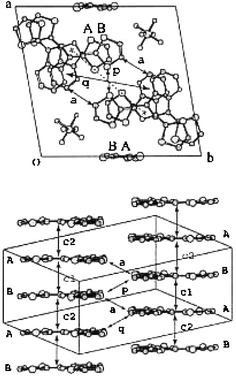 | ||
| Fig. 17 Structure of (26a)2.PF6.(PhCl)0.5 from ref. 74, copyright (2001) with permission from the Royal Society of Chemistry. | ||
The intermediate 27a with a tetrahydrothiophene unit as spacer proved to be a good candidate for the electrocrystallisation of cation radical salts. Thus, with ethylenedioxy groups as substituents, salts with composition (27a)5.(ReO4)2(PhCl), (27a)4.PF6 and 27a.PF6 have been isolated. The first two of these examples are mixed valence salts and exhibit metallic properties down 30 K and 200 K respectively.75 Independent of their stoichiometry, the two salts present very similar structures, characterised by regular stacking of the donor with interplanar distances of 3.5 Å. An array of intrastack intermolecular C–H—O contacts seem to play an important role in the metallic nature of the salt (27a)4.PF6.
1.6 Extended TTF analogues with aromatic systems as spacer units
A further category of extended TTFs, widely studied over the last decade, consists of inserting aromatic conjugated spacer units. The aim of this approach was to drive new packing modes in the CT salts through π-stacking between aromatic rings of the donor molecules. An important parameter for the design of these extended TTFs was in the selection of the aromatic spacer which plays a crucial role for the electrochemical properties and consequently on the corresponding materials obtained.Such compounds can be synthesised by a manifold of olefination reactions on formylated conjugated spacers, either by Wittig–Horner reaction with phosphonate anions P40 or by triethylphosphite mediated coupling with 2-thioxo-1,3-dithiole derivatives 2976 (Scheme 10).
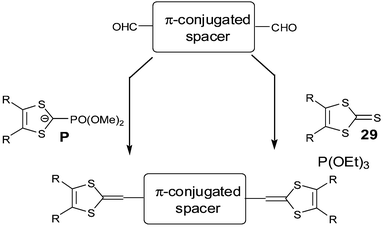 | ||
| Scheme 10 General synthesis of linear extended TTFs. | ||
 | ||
| Chart 8 | ||
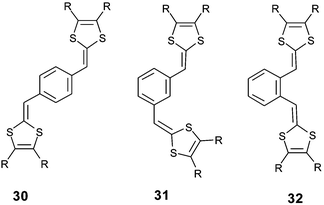
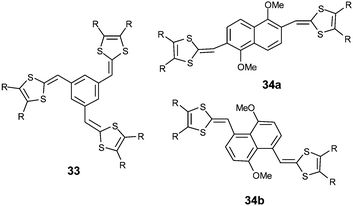
With meta-isomers 31, the application of successive scans with a positive limit beyond the first oxidation peak led to the emergence of a new redox system at lower potentials, corresponding to the formation of a polymer. The absence of conjugation associated with the meta orientation allows the simultaneous quasi-oxidation of the two electroactive dithiafulvenyl sites and leads to the formation of a bis(radical cation). Intermolecular coupling of the radical cations can take place by the same mechanism developed to form TTF vinylogues 6, thereby allowing electropolymerisation on the anode (Scheme 11). The CVs of the resulting polymers show perfect reversible oxidation peaks due to the oxidation of the non-conjugated TTF vinylogues.78 Such polymerizations have been described for 1,3,5-substituted derivatives 3380 and for compounds 34 built by insertion of a naphthalene core as spacer.81
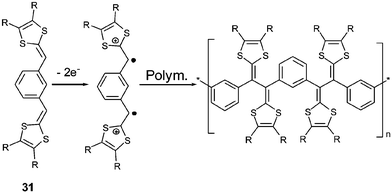 | ||
| Scheme 11 Electropolymerisation of compounds 31. | ||
 | ||
| Chart 9 | ||
For para-isomers 30, the oxidation proceeds in two steps to give the cation radical and dication species sequentially.82 Due to the conjugation of the system across the benzene core, the first oxidation potentials are lower than for the meta-isomers carrying the same substituents on the dithiole rings. The high difference ΔE = 300–330 mV between the two oxidation potentials is indicative of restricted accessibility to the dication. According to the high resonance energy of the benzene core, the formation of the dication associated with the p-quinoid form (Scheme 12) of the spacer involves the unfavorable loss of aromaticity. For the same reason, the radical cation is not stabilised and can eventually dimerise. Lorcy et al. have shown that preparative electrolysis (by applying a controlled potential beyond the second oxidation potential) gave macrocyclic 35 products up to 10 bis(dithiafulvenyl)benzene units.83
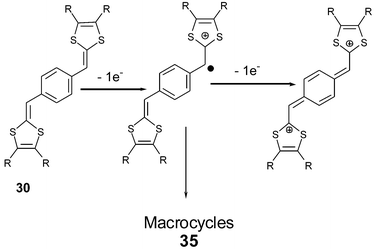 | ||
| Scheme 12 Oxidation of compounds 30. | ||
The ortho-isomers 32 only present an irreversible oxidation peak. Upon successive scans, or by electrolysis, a rapid intramolecular cyclisation reaction leads to compounds 36 (Scheme 13).84 The complete conversion of 32 into 36 requires less than one faraday per mole and a mechanism involving a protonic catalyst induced by the oxidation has been proposed.
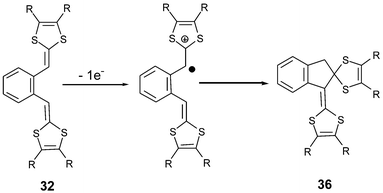 | ||
| Scheme 13 Intramolecular cyclisation reaction of cation radical 32+˙. | ||
The insertion of less aromatic spacers, such as furan (37), thiophene (38) or pyrrole (39) derivatives, has been achieved independently by three groups.85–87 The heteroatoms of the spacer units influence both the structure and the electrochemical properties of the molecules. Concerning the structure of the molecules, several X-ray studies have revealed that in contrast with pyrrole derivatives 39, which present a non-planar conformation, the planar δ-syn conformation adopted by the furan and thiophene derivatives is stabilised by two S—O or S—S intramolecular interactions between sulfurs of the dithiole rings and oxygen or sulfur atoms of the spacer (Fig. 18).88
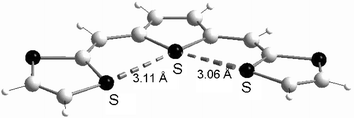 | ||
| Fig. 18 Molecular structure of 38a (R = H) redrawn from ref. 88, copyright (1994) with permission from Wiley-VCH. The intramolecular S—S contacts are presented by dotted lines. | ||
 | ||
| Chart 10 | ||
Contrary to compounds 30 which are irreversibly oxidised, the CVs of 37–39 present two reversible single-electron oxidation waves indicating the formation of stable cation radical and dication species. As expected, the oxidation potentials are strongly dependent on the electron releasing character of the substituents R on the dithiole rings. The comparison of oxidation potentials for derivatives bearing the same substituents R shows that the values of 37, 38 and 39 are lower than those obtained for compounds 30. As expected, the less aromatic spacers allow better electronic delocalisation (Scheme 14), leading to higher stability of the oxidised species. Nevertheless, the aromatic character is not the only parameter that determines the variation of the donor ability of the molecules with the spacer. Indeed, the values for the first oxidation potentials follow the sequence 39 < 37 < 38 whilst the aromaticity of the five-memberes heterocycles is furan < pyrrole < thiophene. Cava et al. have postulated that the greater tendency of the nitrogen atom to stabilise the positive charge in the mesomer structure B was able to explain the improved stabilisation of the cation radical 39+˙.86 Cation radical salts for the furan and thiophene derivatives have been isolated (see the following section) and the analysis of the structures was in agreement with the semi-quinoid B form (Scheme 14). On the other hand, the difference ΔE between the first and the second oxidation potentials is greater for pyrrole (230–280 mV) than for furan and thiophene derivatives (80–150 mV). Such behaviour can be interpreted by an increase of intramolecular coulombic repulsion between the positive charges when the central nitrogen atom develops a higher affinity to localise a charge in the middle of the molecule.
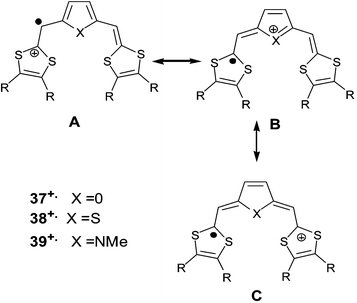 | ||
| Scheme 14 Main mesomeric forms of radical cations 37+˙, 38+˙ and 39+˙. | ||
The influence of the spacer on the donor ability of the molecules has been enhanced by the insertion of electron-releasing alkoxy groups on the 3,4 positions of the thiophene ring.89 Compounds 40 that combine the donor effects of ethylenedioxythiophene (EDOT) and dithiafulvenyl units lead to strong donor molecules with oxidation potentials close to 0.1V/ECS. The participation of the oxygen atoms in localising the positive charges in the middle of the oxidised molecule is clearly demonstrated by the higher ΔE values obtained for compounds 40, compared to 38, bearing the same substituents R.
Elongation of the spacer group has been achieved by the insertion of short oligothiophene units between the dithiafulvalene rings (41).88,90 Nevertheless, by comparison with vinylogues of TTF 1 with identical substituents R, the extension of the conjugated systems did not decrease the first oxidation potential or did not provoke a bathochromic shift of the absorption maximum in solution spectra. Such behaviour was interpreted by the rotational disorder inherent in the oligothiophene chains which, by not providing improved π-electron delocalisation along the conjugated chain, does not increase the HOMO levels of the molecules.88
An increase in the linear extension of these hybrid molecules must promote rigidification of the spacer and further design needs to be implemented to enforce planarity of the molecules. Several series have been developed using sp2 C covalent links between the heterocyles, such as compounds 42,91 or by the insertion of fused thiophene units, as seen in thienothiophene derivatives 43 or 44.92
 | ||
| Chart 11 | ||
Compared to the bithiophene system, prevention of the torsion of the spacer led to a decrease of the first oxidation potentials and to a red shift of the absorption bands. Both the intrinsic rigidity of the spacer and intramolecular S—S interactions increase the rigid character of the extended TTF. Recently, self-rigidification of the spacer was achieved in compounds 45, by exploiting the intramolecular interactions developed from bis-EDOT units between the sulfur and oxygen atoms.93 As shown by the X-ray structure, the molecule adopts a fully planar conformation within the conjugated framework stabilised by several S—O interactions between the EDOT units and by S—S interactions between the thiophene and dithiole rings. All these interactions are confirmed by the small distances separating the oxygen and sulfur atoms which are less than the sum of the van der Waals radii for the corresponding atoms (Fig. 19). The combination of the electron-releasing effect of the ethylenedioxy groups with the full self-rigidification of the molecules gives rise to an extended TTF derivative with strong π-donor properties.
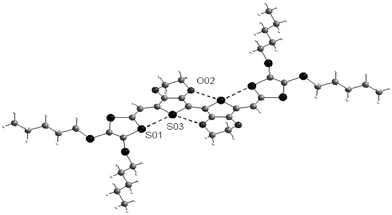 | ||
| Fig. 19 Structure of 45 (R = SC5H11) from ref. 93, copyright (2003) with permission from Elsevier. The S—O and S—S interactions are shown by dotted lines. | ||
Nevertheless, the prospect for further extension of these systems is limited by the synthetic difficulties encountered for developing longer spacer units.
Combining the vinylogue and aromatic approaches Roncali et al. have synthesized further large hybrid derivatives 46,9447,954896 and 49.97 The insertion of ethylenic bonds between the heterocycles was expected to increase the rigidity of the molecules and to reduce the resonance energy of the spacer, thus favouring good electronic delocalisation. For all these systems, S—O or S—S 1,5-intramolecular interactions between heteroatoms of the spacer and sulfur atoms of the dithiole rings stabilise the planar conformation and contribute by enhancing the rigidity of the molecules.
 | ||
| Chart 12 | ||
The optical study of these hybrid compounds demonstrates the efficiency of this approach since, for the same series of substituents R, extension of the spacer is accompanied by a regular bathochromic shift of the maximum wavelength λmax corresponding to the reduction of the HOMO–LUMO gap. The efficient electronic delocalisation within these systems can also explain their electrochemical behavior and in particular the dication states are easily accessed. Compounds 46 and 47 with heteroarylenevinylene (HV) units (n = 1) present a first reversible oxidation wave with a width at half maximum of 40 mV, which indicates two close single-electronic transfers (ΔE = 10 mV). The addition of a supplementary HV unit favors the direct oxidation to the dication through a simultaneous two-electron transfer. Moreover, two other oxidation waves, corresponding to the formation of the radical trication and the tetracation, appear at higher potentials. Compounds 48 also present a reversible two-electron oxidation wave corresponding to the formation of the dication.
For both series, the comparison of optical and electrochemical data allows the following conclusions to be made:
—for a given spacer, the increase of the electron donor character of the substituents R grafted on the dithiole rings provokes a decrease of the first oxidation potential E1 and a red shift of the maximum wavelength λmax,
—for a given substituent R, the extension of the conjugated spacer provokes a bathochromic shift of λmax but without a significant reduction of E1.
Such behaviour for these hybrid systems indicates that the dithiafulvenyl groups have a major contribution to the HOMO level, which is strongly affected by the nature of the substituents R, while the LUMO level is essentially dependent upon the length of the spacer. The electrochemical reduction of compounds 46 with n = 2 shows that all the derivatives are reduced at the same potential of −1.20 V/SCE, independent of the substituents R.94
The extension of the spacer has also been achieved by using thienylenevinylene (TV) units substituted on the β positions of the thiophene by n-hexyl groups in order to increase the solubility of the molecules 50.98 With this approach, the longest extended TTF analogues, with the spacer containing up to 70 sp2 carbons (n = 5), have been synthesized. The electrochemical behaviour of these derivatives for oxidation and reduction processes is in agreement with the preceding conclusion. Compounds 50 are directly oxidised to the dications by a two-electron oxidation at a potential Eox1 = 0.28V (vs. Ag/AgCl) and is independent of the length of the spacer. Concerning the reduction, the first reversible wave corresponds to a two-electron transfer for the formation of the dianion and the reduction potentials increase with the length of the spacer from Ered1 = −1.60 V for 50a to Ered1 = −1.51 V for 50c (vs. Ag/AgCl). On the other hand, the lengthening of the conjugated spacer favours access to many redox states, both in oxidation and reduction processes. Derivatives such as 50c with twelve TV units can be charged up to the octacation and hexaanion species within a narrow potential range.
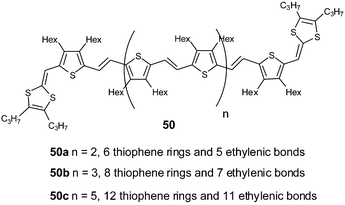 | ||
| Chart 13 | ||
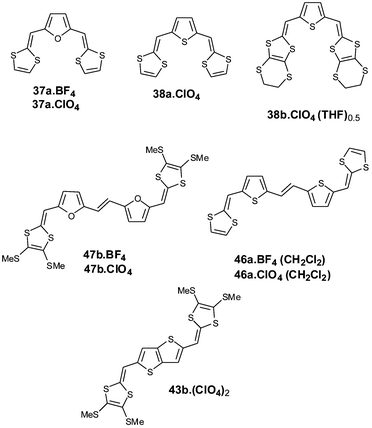 | ||
| Chart 14 | ||
A general characteristic for all these compounds in their oxidised states (cation radical or dication) is that they always adopt a δ syn planar conformation between the dithiafulvalenyl groups and the adjacent heteroaromatic rings. As for the neutral state, such conformations are stabilised by S—S or S—O intramolecular interactions. Thus, the heteroatoms of the spacer play a key role in the control of planarity (in both the neutral and oxidized states) by developing these intramolecular interactions and preventing strong conformational changes during the oxidation processes.
The comparison between the structures of the salts 37a+˙.Y− and 38a+˙.Y− (Y− = BF4− or ClO4−) clearly shows the role of the heteroatom in the spacer for driving the molecular packing in the solid.99 Both structures are characterised by a head to tail overlap of radical cations (Fig. 20). Nevertheless the stacking modes of the donors strongly depend on the central heteroaromatic core. With the furan ring, the radical cations 37a+˙ are uniformly stacked along the c axis with an overlap of the furan and 1,3-dithiole rings. Between the columns of donors the distances d = 3.76 Å separating the sulfur atoms are indicative of weak 2-D character for the materials. In spite of this 1 ∶ 1 stoichiometry, this salt has a conductivity of 4.0 × 10−2 S cm−1.
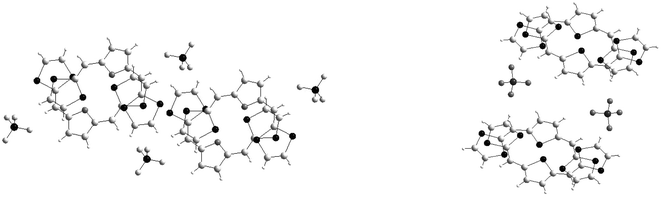 | ||
| Fig. 20 Overlapping mode of the radical cations in the structure 37a.BF4 (left) and 38a.ClO4 (right) redrawn from ref. 99, copyright (2001) with permission from the Royal Society of Chemistry. | ||
In the salt 38a+˙.ClO4− the thiophene ring induces a dimerisation of the radical cations. In each dimer, the strongest interactions occur between the sulfur atoms of the thiophene rings which present the shortest intermolecular distances whilst between the dithiole rings weaker contacts with distances are found. The dimers stack along the b axis through weak inter-dimer contacts and no inter-columnar interactions are observed. This salt possesses a conductivity three orders of magnitude lower than that for the preceding salt with a furan spacer, which is in good correlation with the absence of regular π-stacking and lower dimensionality of the material, as revealed by the X-ray structure.
 | ||
| Chart 15 | ||
Reports from Torrance on TTF series100 and Miller on oligothiophene series101 have shown that the radical cations of these two different families of donors have tendencies to form π-dimers. The consequences of dimerisation on the electronic properties of the two classes of materials are the subject of considerable interest. The hybrid extended TTFs constitute interesting models for the evaluation of S—S intermolecular contacts or π–π interactions for the formation of cation radical π-dimers. From the basic structure of the salt 38a.ClO4, which presents some tendency towards dimerisation, the strength of intermolecular interactions have been investigated by effecting structural modifications either on the dithiole rings or on the conjugated system.
Firstly, for derivative 38b, ethylenedithio groups have been grafted on the 1,3-dithiole rings.102 By analogy with BEDT–TTF, the increase in the number of sulfur atoms was expected to promote S—S intermolecular interactions. For example, the structure of the salt 38b.ClO4.(THF)0.5 is characterised by a strong dimerisation of the donors. In contrast with the preceding structure of the unsubstituted analogue, the dimer (38b2)2+ is assembled through a face-to-face stacking of the monomers (Fig. 21). The majority of the sulfur atoms participate in intermolecular S—S contacts with distances slightly smaller than the sum of the van der Waals radii. On the other hand, such stacking of the donors leads to short C—C intermolecular contacts, which are characteristic of the formation of a π-dimer. The strong dimerisation of the radical cation in the solid state is confirmed by the absence of an ESR signal due to spin pairing in the dimer. These results show that the dimer results from the combination of the S—S interactions associated with the dithiafulvalenyl groups, with overlap of the π-orbitals of the carbon atoms within the conjugated spacer. Along the a axis the dimers stack through weak interactions with interplanar distances of 3.70 Å. The introduction of peripheral sulfur atoms contributes to a higher degree of dimensionality in the material and the conductivity of this salt (10−3 S cm−1) is two orders of magnitude larger than that of the cation radical salt 38a.ClO4.
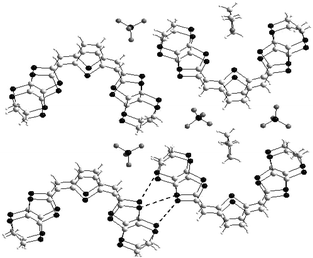 | ||
| Fig. 21 Structure of the dimer (38b2)2+ and X-ray structure of the salt 38b.ClO4(THF)0.5 redrawn from ref. 102, copyright (2002) with permission from Wiley-VCH. | ||
In a second approach with derivative 46a, the spacer has been lengthened by using a thienylenevinylene (TV) unit in order to promote stronger π−π interactions. The efficiency of the TV units to produce π-stacks has recently been demonstrated with oligothienylenevinylenes.102 Both crystalline materials are isostructural and are characterised by the formation of π-dimers (46a2)2+, through face to face overlap of the molecules and separated along the a axis by the anions and solvent molecules (Fig. 22). In the bc plane, the dimers are not connected, thus justifying the insulating character of this salt. Overlapping of the donors mainly generates intermolecular contacts between the carbon atoms of the spacer, whilst the only significant S—S contacts involve one sulfur atom from each of the dithiole rings. By comparison with the previous dimer, the contribution of the S—S interactions is less important, showing that the dimerisation process is closer to that of conjugated oligomers than that of TTF derivatives. The existence of π-dimers in short oligomers has been observed in the X-ray structures of oxidised diphenyl-bipyrrole 51103 or diphenyl-terthiophene 52104 derivatives, demonstrating the strong similarity in the structure of molecular materials derived from conjugated oligomers and extended TTF analogues.
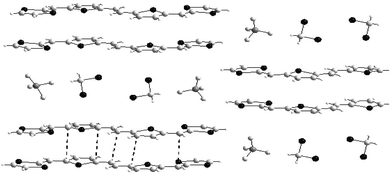 | ||
| Fig. 22 Packing mode of the dimers (46a2)2+ in 46a.BF4(CH2Cl2) redrawn from ref. 102, copyright (2002) with permission from Wiley-VCH. The π–π interactions are presented by dotted lines between dimmers. | ||
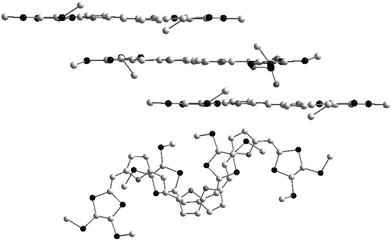
Finally, cation radical salts have been obtained from compounds 47b which possess furylenevinylene units as spacers.105 In contrast with the previous materials, the crystals give strong ESR signals, indicating that the radical cations do not dimerise in the solid state. The structure is built up from stacks of the donors aligned along the a axis (Fig. 23). Two of the methylthio groups are not located in the plane of the molecule, which prevents the direct alignment of the cations and thus the formation of π-dimers. The steric interactions between the SMe groups are minimised by a shift of the cations and the regular average distances between the molecules is d = 3.6 Å. Several short C—C intermolecular distances exist between the carbon atoms of the conjugated spacer indicating the formation of π–π interactions between the donors. The salt has a conductivity of 10−3 S cm−1.
A dication salt of compound 43b has been obtained recently by electrocrystallisation under constant current.92 The analysis of the bond lengths in the dication 43a is in full agreement with the quinoid structure postulated for the conjugated system (Scheme 15). The dication stacks along the b axis with lateral displacement of the molecules. The intermolecular distances of ca. 3.8 Å are too long to allow π–π interactions and only weak S—S contacts are observed. The intermolecular coulombic repulsion between the positive charges deters any close association between dication molecules.
 | ||
| Scheme 15 Structure of dication 43++. | ||
1.7 Magnetic properties of some linear extended TTF analogues
In contrast to the comprehensive studies devoted to the electrochemical properties of extended TTFs, reports on the magnetic properties of these materials has been under-represented since it is difficult to establish a relationship between the molecular structure and the magnetic behaviours of the extended donors. Nevertheless, data for vinylogues 1a24 and 1b,106 compounds 24c107 and 37b107 containing dihydrofuran or furan units as spacers are avalaible (Scheme 16) and their comparison shows a dependency upon the extension of the linker unit. As expected, cation radical species in solution give well resolved ESR/ENDOR spectra. The signal of each cation radical presents hyperfine structure due to coupling with protons. At the periphery of the spectra, 33S satellites can be discerned. Assignment of the coupling constants to protons and 33S nuclei is based on spin population calculated by the McLachan procedure.108 The smallest aH values for the external protons are indicative of the low spin population in the substituents and show that the spin distribution for extended TTFs mainly reside in the spacer. On the other hand, the coupling constants aS essentially concern the 33S nuclei of the dithiole rings. The elongation of the spacer from 1a,b to 37b is accompanied by a decrease of aS which is an indication of the decrease in the spin population at the sulfur centre due to a localisation on the central π-system. The expansion of the spacer also provokes a decrease of the g factor from g = 2.0081 for 1b to g = 2.0052 for compound 37b.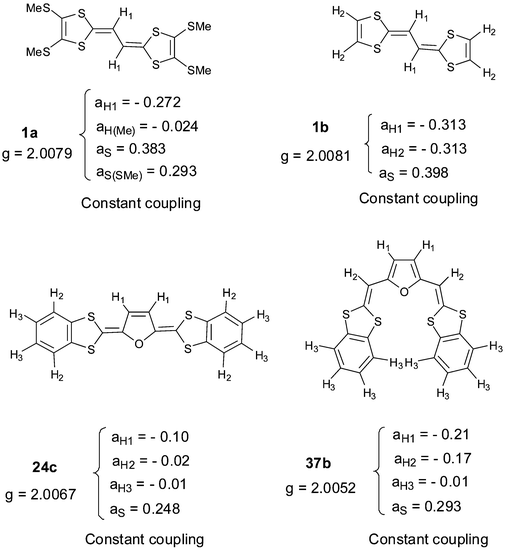 | ||
| Scheme 16 Magnetic properties of 1a, 1b, 24c and 37b from refs 24, 106 and 107. | ||
2 Highly sulfur-rich extended TTF derivatives with 2D-dimensional arrangement
The ability of sulfur rich radical cation molecules to self-assemble by S—S intramolecular contacts and π–π interactions has been covered in the preceding sections. An alternative strategy for increasing the dimensionality of materials has been to develop highly extended and sulfur-rich TTF derivatives by linking several 1,3-dithiole-2-ylidene moieties in a two or three-dimensional arrangement. In this respect, two families (A and B) can be considered, in which the 1,4-dithiafulvenyl fragments are linked together by a central core (conjugated or not) or that they are grafted onto the dithiole rings of a TTF core (extended or not).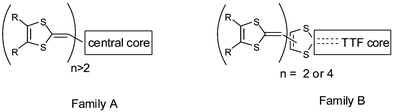 | ||
| Chart 16 | ||
2.1 Multiple 1,3-dithiole-2-ylidene fragments on a central core
A first approach in this trend involved the generation of dendralene-TTF derivatives 53 and 54 by adding dithiafulvenyl fragments upon the ethylene units of a TTF vinylogue core. This methodology was initially developed by Yoshida et al.109 then by Bryce et al.110,111 who completed the structural and electrochemical studies. The synthesis proceeds by combining formylation and Wittig olefination reactions, as depicted in Scheme 17. More recently, Cava et al. elaborated new dendralene analogues by replacing several sulfur atoms by selenium or tellurium.112,113 An X-ray structure of the [3]dendralene 53 derivative showed that two 1,3-dithiole rings are conjugated through a butadiene unit by forming a quasi-planar vinylogue system, whilst the third dithiafulvalene group is located almost perpendicularly to this plane (Fig. 24).110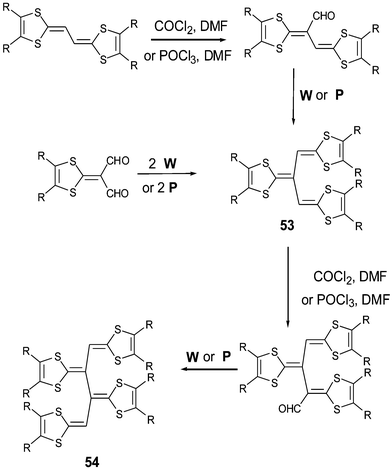 | ||
| Scheme 17 General synthesis of compounds 53 and 54. | ||
![X-ray structure of [3]dendralene 53
(R = Me), redrawn from ref. 110.](/image/article/2005/CS/b316392j/b316392j-f24.gif) | ||
| Fig. 24 X-ray structure of [3]dendralene 53 (R = Me), redrawn from ref. 110. | ||
The CVs of 53 present two single-electron reversible oxidation waves due to the oxidation of the vinylogue unit of the molecules, generating the radical cation then dication, followed by a quasi reversible peak at considerably higher potential assigned to the oxidation of the final dithiafulvalenyl unit. The values for the first two oxidation potentials are about 140 mV lower than those of TTF vinylogues 1 (n = 0) bearing the same R substituents. The electron donating character of the third dithiafulvalene group contributes by increasing the HOMO level of the molecule. The [4]dendralene 54 shows similar behaviour, the first two reversible oxidation processes are at lower potentials than 53. However, the third quasi-reversible peak, which corresponds to a two-electron transfer, is strongly shifted to less anodic values indicating easier oxidation of the last two dithiafulvenyl groups.
The extension of the molecule has also been achieved by grafting a 1,3-dithiole-2-ylidene moiety on a bithiophene unit thus forming the rigid extended TTFs 55.91 Theoretical calculations have shown that a planar conformation stabilised by S—S intramolecular interactions could be expected for the structure of 55.114 The CV of 55 presents two reversible single-electron waves relative to the formation of the radical cation and dication, followed by two irreversible peaks corresponding to the formation of the trication and tetracation. By comparison with the [3]-dendralene 53 bearing the same substituents (SMe), the lower value of 100mV for the first oxidation potential and the narrower range of the potential window to obtain the tetracation species in 55 (700 mV for 55 and 840 mV to oxidise 53 to the trication) is consistent with the large and two-dimensional extension of the molecule.
 | ||
| Chart 17 | ||
On the other hand, several 1,3-dithiole-2-ylidene fragments were also grafted on various cores leading to structures such as radialenes 56,11557116 or 1,3-indane derivatives 58.117 The X-ray structure of the [4]-radialenes 56 shows that the four dithiole rings are located in a plane,115 but the two other derivatives present a distorted molecular structure. The electrochemical behaviour of 56 and 58 are characterised by a first oxidation wave corresponding to the direct formation of the dication, followed at much higher potentials by a third (and fourth for [4]-radialenes 56) irreversible peaks relating to the oxidation of the subsequent dithiafulvenyl groups. Concerning [6]-radialenes 57, the oxidation proceeds in three steps, but repetitive scans lead to a modification of the CV with only one oxidation wave which appears at considerable higher potential. This behaviour indicates a slow conformational change within the structure of the molecule.
For derivative 56, the formation of single crystals of a radical cation salt have been reported to have a conductivity of 1.0 S cm−1, but the structure of this salt have never been described.118 For the other compounds, no characterisable material has been obtained.
2.2 Highly extended systems with dithiafulvenyl fragments grafted onto a TTF core
The second trend developed by Gorgues and co-workers consisted of grafting two or four dithiafulvenyl fragments onto the 1,3-dithiole rings of a central TTF or vinylogue TTF core, to give a large variety of highly extended TTFs 59–63.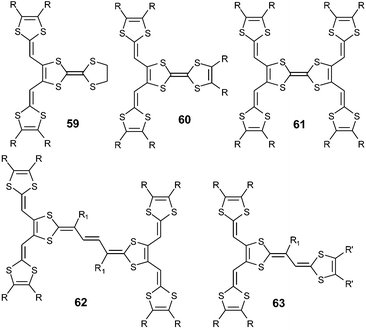 | ||
| Chart 18 | ||
The syntheses of 59–61119 and 62120 proceed in one step by two- or fourfold Wittig or Wittig–Horner olefinations, with the ylide W or phosphonate anions P and a TTF core previously functionalised with two or four aldehyde groups (Scheme 18). For compounds 63, the TTF vinylogue core is built in the last step by action of an excess of anions P on the aldehyde group of 64, which was in turn obtained by reacting trisformyl compound 65 with an excess of W.121 This procedure allowed the synthesis of a range of derivatives 63 substituted by different R and R′ groups. The olefination of 65 with an excess of P led directly to derivatives 63 bearing the same substituents.
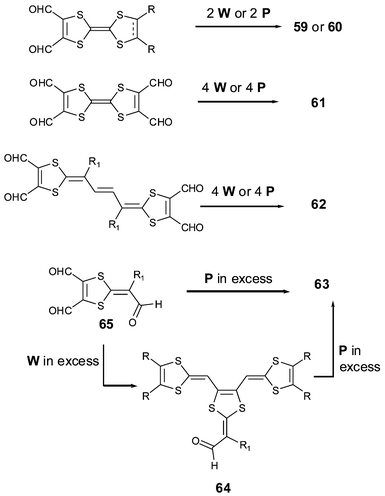 | ||
| Scheme 18 General synthesis of highly sulfur-rich extended TTF derivatives. | ||
In the neutral state, all these molecules are expected to adopt a planar conformation with the two vicinal dithiafulvalenyl arms in plane with the central dithiole ring through S—S 1,5-intramolecular interactions. The X-ray structure of compound 66 (Fig. 25), which represents the same vicinal substitution on a dithioleylidene moiety, demonstrates unambiguously that intramolecular interactions predominate (dS—S = 3.065 Å) between the sulfur atoms of the coplanar heterocycles.119 The S—S contacts are responsible for preventing conformational changes upon oxidation of the molecules (see below). These extended TTFs constitute remarkable examples of a two-dimensional arrangement with a large extension of the π-system.
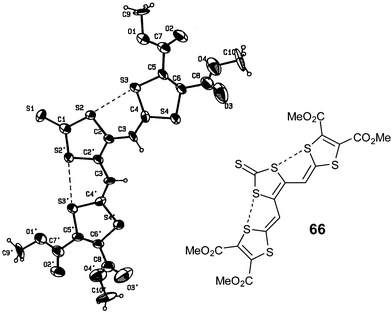 | ||
| Fig. 25 X-ray structure of compound 66 from ref. 119. The intramolecular interactions are shown by dotted lines. | ||
First oxidation potentials close to 0 V (vs. ECS) have been obtained for derivatives 61, 62 and 63 substituted by cyclohexyl (R–R =
[CH2]4) or benzo (R–R =
[CHCH]2) groups. A second remarkable tendency for these extended TTFs is their propensity to lead to stable multicationic states in a relatively small range of potentials. Thus, tetrakis derivatives 61 and 62 are oxidised to the dications in two close single-electron processes, followed by the formation of the tetracation in a two-electron transfer at a potential less than 1.0 V vs. SCE. The tris derivatives 63 are oxidised to stable trications at potentials around 1.0 V.
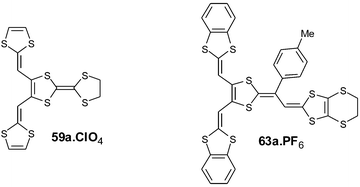
Electrooxidation of derivatives 59a and 63a under galvanostatic conditions gave single crystals of cation radical salts with the stoichiometries 59a.ClO4122 and 63a.PF6.121
 | ||
| Chart 19 | ||
The structure of radical cation 59a+˙ adopts the same planar conformation described for compound 66 and is stabilised by non bonded S—S contacts between the two vicinal dithiafulvalenyl arms and the central dithiole ring (Fig. 26).122 The structure of the salt is characterised by regular stacking of the donors in a head-to-tail mode which leads to the formation of several S—S intermolecular contacts. The columns interact through the lateral dithiafulvalenyl arms by forming slabs of donors separated from each other by the anions, resulting in an overall two-dimensional structure. The high rt conductivity of this 1 ∶ 1 material (0.4 S cm−1) is associated with a decrease in coulombic repulsion, allowing facile access to the dication state as already indicated for the salts resulting from compounds 22.59
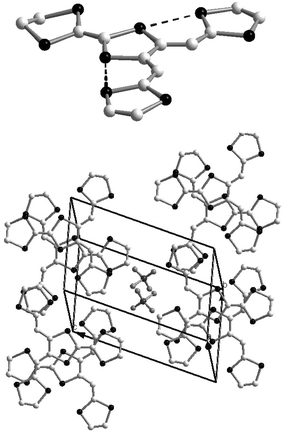 | ||
| Fig. 26 Structure of radical cation 59a+˙ and stacking mode in the salt 59aClO4 redrawn from data of ref. 122, copyright (1993) with permission from the American Chemical Society. | ||
The molecular structure of the radical cation 63a+˙ can be described as a juxtaposition of the structures of the aryl vinylogues TTF 6+˙ and 59a+˙. The para-tolyl unit is located perpendicular to the plane formed by the vinylogue part of 63, while the dithiafulvenyl arms present short S—S contacts which stabilise the planar extension of the molecule (Fig. 27).121 The structure of the salt 63a.PF6 has a strong similarity to that 6a.PF6 previously discussed.30 In order to minimise the steric interactions between the p-tolyl units, each cation bridges two others, as indicated in Fig. 27 (bottom), thus allowing π-stacking of the molecules with interplanar distances of ca. 3.6 Å. Short contacts with distances dS—S = 3.5 Å between the sulfur atoms of the ethylenedithio groups and the vicinal dithiafulvenyl fragments are observed. Consequently, each donor is in close contact with four neighbours located above and below and also with two others in the same plane, conferring to the material a strong two-dimensional character. This salt also presents a high rt conductivity of 0.1 S cm−1 in spite of the 1 ∶ 1 stoichiometry.
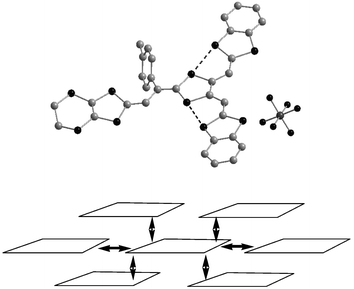 | ||
| Fig. 27 Structure of radical cation 63a+˙ and overlapping mode in the salt 63aPF6 redrawn from ref. 121, copyright (2001) with permission from Wiley-VCH. | ||
These two examples of materials demonstrate unambiguously the efficiency by which the spatial extension of the donors improves the dimensionality of the materials. Nevertheless, the formation of radical cation salts in a mixed-valence state, for which metallic behaviour should be expected, remains a challenge.
The possibility of obtaining the desired stoichiometry for these materials has been demonstrated by the formation of a mixed valence salt (60)2.ClO4. The X-ray structure, though poorly resolved, corresponds to an original 2-D network, but the poor quality and the fragility of the single crystals have so far prevented any electrical conductivity measurements.123
3 Extension of the TTF framework by the fusion of two TTF or extended TTF cores
The extension of the TTF framework has also been realised by the fusion of two TTF or extended TTF cores which can be achieved either by the direct fusion of two 1,3-dithiole rings or via a benzene ring. | ||
| Chart 20 | ||
3.1 Direct fusion of two TTF cores
The direct fusion of two TTF cores to build the basic framework BDT–TTP 67a (R′ = H), has been developed by Misaki and Mori over the last decade.124 This framework is the basic unit for several organic metals and, in the case of the vinylogous derivative 68a, an organic superconductor.125The synthesis of 67 with various R substituents proceeds from the dissymmetrical TTF 69 bearing two protected thiolate functionalities (Scheme 19). After deprotection, successive reactions lead to fused TTF-thioxo,1,3-dithiole 70. Phosphite-mediated cross-coupling with an oxo-1,3-dithiole derivative gives the fused TTF derivative 67. For vinylogue cores 68, the synthesis proceeds by the same reaction in the last step, whilst compound 71 is obtained by a Wittig–Horner olefination between the phosphonate 72 and aldehyde 3.126 Decarbomethoxylation reactions of the tetracarbomethoxy derivatives of 67 and 68 afford the unsubstituted compounds 67a and 68a.
 | ||
| Scheme 19 General synthesis of fused TTF derivatives. | ||
The CVs of 67 and 68 present four single-electron oxidation waves corresponding to the successive oxidation of the compounds to the tetracation species.126 The comparison of the oxidation potentials for derivatives 67a (E1 = 0.44 V, E2 = 0.62 V, E3 = 1.05 V, E4 = 1.13 V vs. SCE in benzonitrile) and 68a (E1 = 0.37 V, E2 = 0.50 V, E3 = 0.81 V, E4 = 1.05 V) with TTF (E1 = 0.35 V, E2 = 0.77 V) and vinylogue TTF 1 (n = 0) (E1 = 0.29 V, E2 = 0.49 V) shows the evolution of the redox properties for the different cores. The first oxidation potentials for 67 and 68 are higher than those of TTF and 1 bearing the same R substituents, indicating that, contrary to the linearly extended TTF (see section 1), the π-donor ability is not enhanced by the fusion of the TTF cores. By contrast, the extension of the backbone results in a decrease of the intramolecular coulombic repulsion between the positive charges of the multicationic species. Indeed, the difference between the two first oxidation potentials follows the series 68 < 67 < 2 < TTF and the third and fourth oxidation potentials, corresponding to the formation of trication and tetracation species, are smaller for vinylogue 68 than for 67.
An extensive series of radical cation salts 67.Ax with a variety of substituents R and various anions A− have been obtained and most of these salts possess high rt conductivities and metallic properties down to liquid He temperatures.124,127 Using the same anion often led to salts covering a range of stoichiometries or phases with dramatically different properties. In this review, we have tried to highlight some representative examples that possess interesting features of the fused-TTF in comparison with TTF cores.
 | ||
| Chart 21 | ||
The fully planar unsubstituted donor 67a has given many metallic salts with a variety of counter anions. In most of the salts, including tetrahedral (ReO4−, ClO4−, BF4−), octahedral (PF6−, SbF6−) and linear (I3−, AuCN2−) anions, the β type structure is characterised by face to face stacking of the donors with an overlap mode where two adjacent molecules are displaced by a half unit along the molecular axis (Fig. 28).128 The short regular interplanar distances less than 3.6 Å lead to very strong intrastack interactions. On the other hand, intercolumnar contacts, associated with many S—S close contacts, are strong enough to allow, in most cases, the suppression of one-dimensional instabilities. It is remarkable to note that the β-type arrangement is always maintained by using anions with larger size and charge such as the clusters (Re6S6Cl8)2− or (Mo6Cl14)2− in the salts (67a)6.(Re6S6Cl8).(CH2ClCHCl2)2 and (67a)6.(Mo6Cl14).(CH2ClCHCl2)2.129 Regardless of the anion, the robust β-slabs of the salts 67a are a manifestation of the rigid spatial extension of the 67a core. The materials offer the opportunity of modifying the band filling (and thus the metallic character) of the salts by changing the size and the charge of the anions.
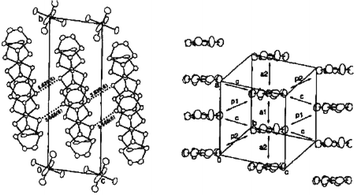 | ||
| Fig. 28 Packing mode of compounds 67a in β type salts reprinted from ref. 124, copyright (2002), with permission from Taylor & Francis Ltd. (http://tandf.co.uk/journals). | ||
Such a uniform β type arrangement is also observed for the vinylogue derivatives 68a in the structure of salts (68a)3.SbF6 and 68a(AuCN2)0.4,125 and also in the selenium containing salts (67b)2.AsF6130 and (68b)3.TaF6.13168a(AuCN2)0.4 is the only extended TTF salts which displays superconducting behaviour at temperatures below 4 K.
Other structural arrangements have been attempted by grafting R substituents on the dithiole ring. In particular, the famous κ-type structure, which has led to several superconducting materials of BEDT–TTF salts, has been the target. In the series of fused-TTFs, such arrangements are limited and they are mainly observed when cyclopentene or ethylenedioxy groups are used, as seen for the unsymmetrical derivative 67c in the salt 67c.(SbF6)0.4.132 The donors are organised in perpendicular dimers which form sheets separated by layers of anions. The salt is metallic down to 4.2 K.
Although in general only mixed valence salts of TTF derivatives provide metallic behaviour, the tetramethylsulfanyl derivative 67d is unique in that salts of 1 ∶ 1 composition exhibit metallic conductivity. This phenomenon was first observed for salt 67d.I3,133 and subsequent examples followed with anions C(CN)3−,134 FeBr1.8Cl2.2−135 and Fe0.9Ga0.1Cl4−,136 which present high conductivities up to 1000 S cm−1 and metallic properties down to 53 K. In contrast, the 1 ∶ 1 salts obtained from FeX4− or GaX4−(X = Cl− or Br−) anions are insulating.136 For the conducting salts the donors are regularly stacked, but in the insulating materials, the molecules dimerise through two overlapping modes as depicted in Fig. 29. These results demonstrate the variable conducting behaviour (insulating or metallic) which can be achieved using different anions in salts containing a common donor.135,136 Developing this trend, the control of the band filling in the materials has been achieved by using alloy metal salts 67d.MxM′1−xCl4.137 The variation in the degree of oxidation of donor 67d from +1 to +2, and thus the concomitant diminution of the band filling from 0.5 to 0, was accomplished by changing the ratio between monovalent (Fe, Ga) and divalent (Co, Mn) metals. All the salts present the same structure, characterised by an uniform stacking of the donors with very similar interplanar distances of around 3.40 Å. As the band filling deviates from 0.5, the conductivity gradually decreases and the salts become semiconducting.
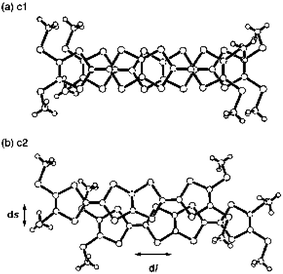 | ||
| Fig. 29 Overlapping mode of 67d in the insulating salt 67dGaCl4–(PhCl)0.5 reprinted from ref. 136, copyright (2001) with permission from the Royal Society of Chemistry. | ||
Very recently tris-fused TTF derivatives 73 have been synthesised by coupling of the oxo derivatives of compounds 70.138 X-ray structures show a high degree of planarity within the TTF frameworks. By comparison with the bis-fused TTF, the extension of the π-skeleton with a TTF core allows the attainment of a hexacation state over three oxidation steps, the last being irreversible. It is very likely that the first two-electron oxidation wave corresponds to the formation of the bis(radical cation). Oxidation of single crystals of 73 by iodine vapour led to materials with a 1 ∶ 1 composition (estimated from the S ∶ I ratio of the energy dispersion spectrum) and possessing conductivity values in the range 20–60 S cm−1.
 | ||
| Chart 22 | ||
3.2 Linear fusion of TTFs through a benzene core
The fusion of TTFs, achieved by the introduction of a benzene core between two 1,3-dithiole rings, has been developed by Müllen et al.16,139 Dimeric 74 and trimeric 75 derivatives were synthesised from the tetrathia-s-indacenedithione 76 and the 1,3-dithiole-2-thiomethyl anion 77 (Scheme 20). One or two equivalents of 77 react with 76 to lead to dimer 74 or thione 78 after addition of iodomethane and subsequent extrusion of dimethyldisulfide. Trimer 75 was obtained by a triethylphosphite mediated coupling of 78. The high degree of planarity in the extended TTF system has been clearly demonstrated by the X-ray structure of dimer 74 (R = SHex and R′ = Hex).16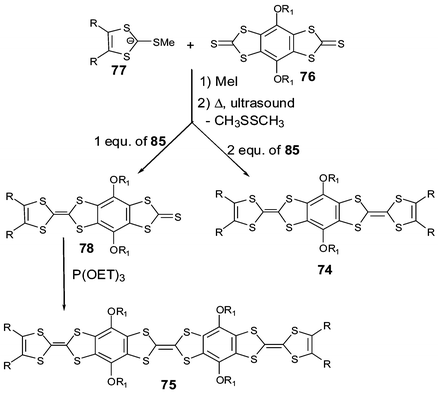 | ||
| Scheme 20 General synthesis of fused TTFs 74 and 75. | ||
Electrochemical studies reveal that the dimers 74 are oxidised up to the tetracation through four single-electron processes. The incorporation of an additional TTF unit in the trimer 75 allows a hexacation state to be achieved. In spite of the presence of the electron-releasing alkyloxy groups grafted onto the benzene cores, the values for the first oxidation potentials of 74 and 75 are similar to those of the fused-TTF 67 (bearing the same substituents R).16 This result is an indication that the radical cation is localised on a TTF unit. Theoretical calculations and spectroscopic investigations performed on the closely related derivatives 79 and 80 have shown that the unequal distribution of charge between the two TTF cores for radical cations 79+˙ and 80+˙ corresponds to the prerequisite conditions for mixed valence species and is characterised by intense and broad intervalence transitions in the near IR-region.140 On the other hand, access to polycationic states is favored by the extension of the molecules. The second oxidation potentials of trimers 74 are lower than for the parent TTFs 68 and the formation of trications and tetracations takes place at less anodic peaks for the trimers 75.
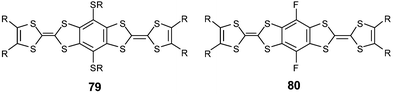 | ||
| Chart 23 | ||
Calculations performed on the doubly charged species of 80 indicated that the charges were symmetrically distributed between the two TTF cores and that the triplet state corresponding to the dication radical was more stabilised than the singlet state.140 The formation of the triplet state was confirmed by ESR spectroscopy which showed similar spectra for the single and doubly charged species. In fact, the formation of the unfavorable dication state, corresponding to the pairing of the two radicals, results in the loss of aromatic character of the central benzene ring by generating a quinoid structure (Scheme 21).
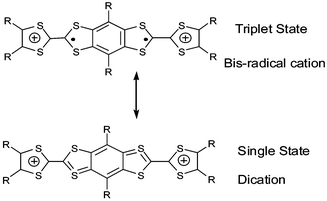 | ||
| Scheme 21 Main mesomeric forms of radical cations 80++. | ||
Radical cations suitable for X-ray analysis have been obtained from compounds 74a,b,c substituted by ethylenedithio groups on the dithiole rings and with varying R1 substituents grafted on the benzene core.16,141,142
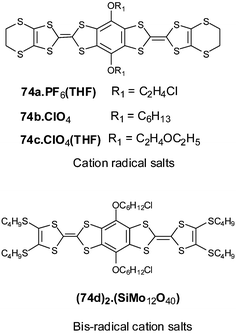 | ||
| Chart 24 | ||
The closely related structures of salts 74a.PF6(THF) and 74b.ClO4 feature face-to-face stacking of the donors to form columns (Fig. 30). The donors dimerise with an intra-dimer distance of 3.54 Å. In the dimer, the molecules overlap with a slight shift along the molecular axis. Between two dimers the stacking of the donors is associated with a larger shift of the molecules and the interdimer distances are 3.57 Å and 3.69 Å for 74a.PF6(THF) and 74b.ClO4 respectively. The columns of donors are connected by short S—S contacts between the sulfur atoms of the ethylenedithio groups and those of the terminal dithiole rings as indicated in Fig. 30. In the structure of the salt 74c.ClO4(THF), the packing mode of the donors is different; the molecules assemble within the stacks through a slight rotation between adjacent donors around an axis passing through the center of the benzene ring (Fig. 30). All these salts exhibit semiconducting properties and a conductivity of 10−2 S cm−1 is reported for 74a.PF6(THF).141
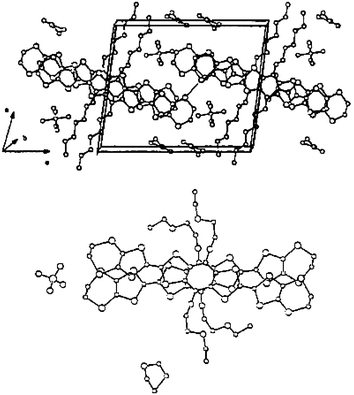 | ||
| Fig. 30 Different packing mode of donors 74a and 74c in salts 74a.PF6(THF) and 74c.ClO4(THF) reprinted from ref. 16, copyright (1994) with permission from Wiley-VCH. | ||
Electrocrystallisation of donor 74d in the presence of the large anion SiMo12O404− gave a CT salt with the composition (74d)2.(SiMo12O40), corresponding to a dicationic oxidation state of the donor.143 The intense ESR signal of the salt and the analysis of the bond lengths of the TTF units are in agreement with the formation of a bis(radical cation) as discussed above. It is interesting to note that the neutral, radical cation and bis(radical cation) species adopt very similar planar structures for the extended TTF system. The organic layers are constituted by two isolated groups of donors. Short S—S contacts are observed only between the molecules within the same group. A low conductivity of 10−4 S cm−1 is indicated for this salt.
3.3 Fused TTF systems with a perpendicular arrangement
Compounds 81144 and 82145 developed by Hudhomme et al. correspond to a spatial extension of fused TTFs obtained by linking, directly or via a benzene ring, a quinoid TTF unit on a TTF core. As indicated in Scheme 22, the laterally extended TTF unit is built in two steps from oxones 83 and 84. A three-fold Wittig–Horner olefination with phosphonate anion P is followed by a retro Diels–Alder reaction. | ||
| Scheme 22 Synthesis of fused TTF systems 81 and 82. | ||
The electrochemical properties of 81 combine the behaviour of a quinoidal TTF unit and the TTF core. The CVs display a two–electron reversible oxidation wave, corresponding to the formation of the dication for the extended unit, followed at a higher potential by two reversible single-electron waves for the TTF system.144 For compounds 82, the proximity of the two redox units induces a strong change in the electrochemical behaviour and the CVs show four sequential single-electron oxidation processes. The thermodynamic stability of the +1 state of 81 and 82 cannot be compared since no salts of these materials have yet been isolated.
Finally compound 87, corresponding to the fusion of two 9,10-bis(1,3-dithiole-2-ylidene)-9,10-dihydroanthracene derivatives in a perpendicular arrangement via a benzene core, has recently been synthesized by Bryce et al. by a two fold Wittig–Horner reaction between phosphonate anions P and the anthraquinone derivative 88 (Scheme 23).146 The formation of 88 proceeded from dichloromethyl derivative 90 by the generation of diene 89, trapped in situ with 1,4-naphthoquinone, followed by an aromatisation reaction in the presence of DDQ.147
For compound 87 in the neutral state, each extended TTF moiety adopts a similar saddle conformation described for molecules 10 in section 1.3. NMR experiments proved the existence of two isomers depending on the relative “up” or “down” orientations of the dithiafulvalenyl arms between the two extended TTF parts (up–up or down–down and up–down or down–up). The CV of 87 displays two reversible two-electron oxidation waves corresponding to the formation of a tetracation state by the successive oxidation to a dication in each extended TTF moiety. The conformational change accompanying the oxidation processes for 874+ leads to the formation of six planar aromatic units (anthracene and dithiolium rings) which are free to rotate with respect to each other. A comparison of the UV-vis spectra of the various oxidized states 87, 872+ and 874+ indicated an intramolecular charge transfer band at 650 nm for the dication 872+. The dication 872+ arises from the oxidation of only one extended TTF moiety and corresponds to a donor–π-acceptor system. The charge transfer band originates from interactions between the donor (neutral unit) and acceptor (dication unit) moieties.
 | ||
| Scheme 23 Synthesis of fused TTF systems 87 with a perpendicular arrangement. | ||
Conclusion and outlook
Over the last 20 years, the design of new extended TTFs has been investigated extensively by developing materials with increased dimensionality. The main goal in this theme has been the improvement in conducting or superconducting properties. During the first decade, intense research efforts led to the elaboration of a very high number of new derivatives with varied architectures, but examples of characterisable materials remained very limited. Due to significant developments in X-ray diffraction techniques, it has been possible to study smaller single crystals and even twinned samples. Therefore, the number of X-ray structures of salts based on extended TTFs published in the last six years has greatly increased. A valuable insight into the relationship between geometric and electronic structures of the donors and also on the molecular organisation in the solid state is thus provided.The stability of the oxidised species is governed by electronic (essentially electron donating effects of the substituents, aromatic or anti-aromatic character of the spacer units) and geometrical factors (steric effects or stabilising intramolecular interactions). A small modification of the molecular structure of the TTF precursor can invoke a dramatic change in the electrochemical properties by modifying the relative stability of the oxidised species. This particularity confers diverse electrochemical behavior to extended TTFs, which can induce new applications. In particular, the role of the heteroatom in the extended TTF framework in preventing conformational changes during the oxidation process has been clearly evidenced and leads to the classification of two categories of donors in relation to their structural difference between neutral and oxidised states.
In general, dramatic and reversible conformational changes observed upon oxidation are symptomatic of the stabilisation of the dication state to the detriment of the radical cations. Although this effect is not favourable for the development of conducting salts, such behaviour constitutes an example of molecular movement trigerred by electron transfer which represents an increased interest in the field of molecular electronics. The conformational change results from subtle compromises between steric repulsion and electron delocalisation, which vary between the neutral and oxidised states. The access to multicationic states for highly extended systems, with the possibility of controlling electrochemically several successive conformational changes, should therefore open up exciting new prospects and applications for these molecules.
On the contrary, when the conformation of the neutral and oxidised species are close, either because the TTF framework is not modified (fused systems) or because the conformation is stabilised by intramolecular interactions, oxidation of the donors affording cation radical salts is favoured. In the majority of cases, the linear or spatial extension of the TTF framework gave the expected materials with increased dimensionality. Nevertheless, apart from the fused systems which show a strong propensity to give mixed valence species, salts of extended TTFs often produce a 1 ∶ 1 stoichiometry and generally exhibit insulating or semiconducting properties. It must be noted that for each structure, the number of cation radical salts is often limited to one or two examples with classical tetrahedral or octahedral anions (ClO4−, BF4−, PF6−, …). From this point of view, the difficulty in obtaining single crystals of sufficient quality for analysis remains the main limitation in the development of conducting materials based on extended TTFs.
On the other hand, in hybrid oligomer–TTF derivatives the molecular organisation in the solid state for neutral or oxidised states adopts both types of packing mode generally observed for TTF derivatives and oligomers of conjugated systems. These last compounds, which have been studied as model systems for conjugated polymers, are now the basis of organic semiconductors used for applications such as field effect transistors or photovoltaic cells. The preceding results show that the combination of a TTF framework with a π-conjugated backbone should allow the development of a new class of semiconducting material. Already pointed out by Roncali,17 the potential of TTF derivatives for such applications has been definitively confirmed by very recent results showing that transistors can be made from a TTF core fused with two thiophene rings.148
The design of hybrid extended TTF-oligomers, in which a conjugated backbone separates several dithiole rings or incorporates several fused units, should furnish materials with a large variety of electronic properties and structural flexibility for adapting the materials to specific applications.
In summary, the synthesis of organic superconductors comprising cation radical salts of extended TTFs will remain a topical challenge. However, manipulation of the extended TTF framework to furnish new materials for applications such as organic semiconductors is an emerging and important area of investigation with exciting prospects.
References
-
Organic Superconducyors (including Fullerenes), ed.J. M. Williams, J. R. Ferraro, R. J. Thorn, K. D. Carlson, U. Geiser, H. H. Wang, A. M. Kini, M. H. Whanbo, Prentice Hall, New Jersey, 1992, Chapters 2–4 Search PubMed
.
-
(a) J. M. Williams, M. A. Beno, H. H. Wang, P. C. W. Leung, T. J. Emge, U. Geiser and K. D. Carlson, Acc. Chem. Res., 1985, 18, 261 CrossRef CAS
- M. R. Bryce, Chem. Soc. Rev., 1991, 20, 355 RSC
- J. L. Segura and N. Martin, Angew. Chem. Int. Ed. Engl., 2001, 40, 1372 CrossRef CAS
- For review devoted to the synthesis, see:
(a) G. Schukat and E. Fanghänel, Sulfur Reports, 2003, 24, 1–190 CAS
- M. R. Bryce, J. Mater. Chem., 1995, 5, 1481 RSC
- J. M. Fabre, J. Solid State Chem., 2002, 168, 367 CrossRef CAS
-
(a) K. Bechgaard, K. Carneiro, F. B. Rasmussen, M. Olsen, G. Rindford, C. S. Jacobsen, H. J. Pedersen and J. C. Scott, J. Am. Chem. Soc., 1981, 103, 2440 CrossRef CAS
- H. Urayama, G. Yamochi, G. Saito, K. Nozawa, T. Sugano, M. Kinoshita, S. Sato, K. Oshima, A. Kawamoto and J. Tanaka, Chem. Lett., 1988, 55
- S. A. French and C. R. A. Catlow, J. Phys. Chem. Solids, 2004, 65, 39 CrossRef CAS
- C. Rovira and J. Novoa, Chem. Eur. J., 1999, 5, 3689 CrossRef CAS
- M. R. Bryce, J. Mater. Chem., 2000, 10, 589 RSC
- M. B. Nielsen, C. Lomholt and J. Becher, Chem. Soc. Rev., 2000, 29, 153 RSC
- M. R. Bryce, Adv. Mater., 1999, 11, 11 CrossRef CAS
- T. Jorgensen, T. K. Hansen and J. Becher, Chem. Soc. Rev., 1994, 23, 41 RSC
- M. Adam and K. Müllen, Adv. Mater., 1994, 6, 439 CrossRef
- J. Roncali, J. Mater. Chem., 1997, 7, 2307 RSC
- T. Sugimoto, H. Awaji, I. Sugimoto, T. Kawase, S. Yoneda, Z. I. Yoshida, T. Kobayashi and H. Anzai, Chem. Mater., 1989, 1, 535 CrossRef CAS
- T. K. Hansen, M. V. Lakshmikantham, M. P. Cava, R. M. Metzger and J. Becher, J. Org. Chem., 1991, 56, 2720 CrossRef CAS
- M. R. Bryce, M. A. Coffin and W. Clegg, J. Org. Chem., 1992, 57, 1696 CrossRef CAS
- T. T. Nguyen, Y. Gouriou, M. Sallé, P. Frère, M. Jubault, A. Gorgues, L. Toupet and A. Riou, Bull. Soc. Chim. Fr., 1996, 133, 301 CAS
- A. J. Moore, M. R. Bryce, A. S. Batsanov, A. Green, J. A. K. Howard, M. A. McKervey, P. McGuigan, I. Ledoux, E. Orti, R. Viruela, P. M. Viruela and B. Tarbit, J. Mater. Chem., 1998, 8, 1173 RSC
- K. Deuchert and S. Hunig, Angew. Chem. Int. Ed. Engl., 1978, 17, 875 CrossRef
- M. R. Bryce, A. J. Moore, B. K. Tanner, R. Whitehead, W. Clegg, F. Gerson, A. Lamprecht and S. Pfenniger, Chem. Mater., 1996, 8, 1182 CrossRef CAS
- P. Hudhomme, S. Le Moustarder, C. Durand, N. Gallego-Planas, N. Mercier, P. Blanchard, E. Levillain, M. Allain, A. Gorgues and A. Riou, Chem. Eur. J., 2001, 7, 5070 CrossRef CAS
- Y. Yamashita, M. Tomura, M. Uruichi and K. Yakushi, Mol. Cryst. Liq. Cryst., 2002, 376, 19 CrossRef CAS
- D. Lorcy, R. Carlier, A. Robert, A. Tallec, P. Le Maguerres and L. Ouahab, J. Org. Chem., 1995, 60, 2443 CrossRef CAS
- A. Benahmed, P. Frère, J. Roncali, E. H. Elandaloussi, J. Orduna, J. Garin, M. Jubault and A. Gorgues, Tetrahedron Lett., 1995, 36, 2983 CrossRef CAS
- R. Carlier, P. Hapiot, D. Lorcy, A. Robert and A. Tallec, Electrochim. Acta, 2001, 46, 3269 CrossRef CAS
- Y. Yamashita, M. Tomura, M. B. Zaman and K. Imaeda, Chem. Commun., 1998, 1657 RSC
- C. Rimbaud, P. Le Maguerres, L. Ouahab, D. Lorcy and A. Robert, Acta Cryst., 1998, C54, 679 CAS
- N. Bellec, K. Boubekeur, R. Carlier, P. Hapiot, D. Lorcy and A. Tallec, J. Phys. Chem. A, 2000, 104, 9750 CrossRef CAS
- D. Lorcy, P. Le Maguerres, L. Ouahab, P. Delahes, R. Carlier, A. Tallec and A. Robert, Synth. Met., 1997, 86, 1831 CrossRef CAS
- Y. Yamashita, M. Tomura, S. Tanaka and K. Imaeda, Synth. Met., 1999, 102, 1730 CrossRef CAS
- M. Guerro, R. Carlier, K. Boubekeur, D. Lorcy and P. Hapiot, J. Am. Chem. Soc., 2003, 125, 3159 CrossRef CAS
- M. Tomura and Y. Yamashita, Cryst. Eng. Comm., 2000, 14 RSC
- Y. Yamashita, M. Tomura and K. Imaeda, Tetrahedron Lett., 2001, 42, 4191 CrossRef CAS
- Y. Yamashita and M. Tomura, J. Solid State Chem., 2002, 168, 427 CrossRef CAS
- Y. Yamashita, Y. Kobayashi and T. Miyashi, Angew. Chem. Int. Ed. Engl., 1989, 28, 1052 CrossRef
- K. Akiba, K. Ishikawa and N. Inamoto, Bull. Chem. Soc. Jpn., 1978, 51, 2674 CAS
- A. J. Moore and M. R. Bryce, J. Chem. Soc. Perkin Trans 1, 1991, 157 RSC
- A. S. Batsanov, M. R. Bryce, M. A. Coffin, A. Green, R. E. Hester, J. A. K. Howard, I. K. Lednev, N. Martin, A. J. Moore, J. N. Moore, E. Orti, L. Sanchez, M. Saviron, P. M. Viruela, R. Viruela and T. Q. Ye, Chem. Eur. J., 1998, 4, 2580 CrossRef CAS
- M. R. Bryce, T. Finn, A. J. Moore, A. S. Batsanov and J. A. K. Howard, Eur. J. Org. Chem., 2000, 51 CrossRef CAS
- M. R. Bryce, T. Finn, A. S. Batsanov, R. Kataky, J. A. K. Howard and S. B. Lyubchik, Eur. J. Org. Chem., 2000, 1199 CrossRef CAS
- N. Godbert, M. R. Bryce, S. Dahaoui, A. S. Batsanov, J. A. K. Howard and P. Hazendonk, Eur. J. Org. Chem., 2001, 749 CrossRef CAS
- N. Godbert, A. S. Batsanov, M. R. Bryce and J. A. K. Howard, J. Org. Chem., 2001, 66, 713 CrossRef CAS
- M. R. Bryce, A. S. Batsanov, T. Finn, T. K. Hansen, A. J. Moore, J. A. K. Howard, M. Kamenjicki, I. K. Lednev and S. A. Asher, Eur. J. Org. Chem., 2001, 933 CrossRef CAS
- A. E. Jones, C. A. Christensen, D. F. Perepichka, A. S. Batsanov, A. Beeby, P. J. Low, M. R. Bryce and A. W. Parker, Chem. Eur. J., 2001, 7, 973 CrossRef CAS
- C. A. Christensen, A. S. Batsanov, M. R. Bryce and J. A. K. Howard, J. Org. Chem., 2001, 66, 3313 CrossRef CAS
- M. R. Bryce, A. J. Moore, M. Hasan, G. J. Ashwell, A. T. Fraser, W. Clegg, M. B. Hursthouse and A. I. Karaulov, Angew. Chem. Int. Ed. Engl., 1990, 29, 1450 CrossRef
- S. Triki, L. Ouahab, D. Lorcy and A. Robert, Acta Cryst., 1993, C49, 1189 CAS
- M. R. Suchansky and R. P. Van Duyne, J. Am. Chem. Soc., 1976, 98, 250 CrossRef CAS
- D. F. Perepichka, M. R. Bryce, I. F. Perepichka, S. B. Lyubchik, C. A. Christensen, N. Godbert, A. S. Batsanov, E. Levillain, E. J. L. McInnes and J. P. Zhao, J. Am. Chem. Soc., 2002, 124, 14227 CrossRef CAS
- A. S. Batsanov, M. R. Bryce, S. B. Lyubchik and I. F. Perepichka, Acta. Crystallogr., Sect. E, 2002, 58 Part 10, O1106 CrossRef
- Y. Yamashita, S. Tanaka, K. Imaeda, H. Inokuchi and M. Sano, J. Chem. Soc., Chem. Commun., 1991, 1132 RSC
- Y. Yamashita, T. Suzuki and T. Miyashi, Chem. Lett., 1989, 1607 CAS
- Y. Yamashita, S. Tanaka, K. Imaeda, H. Inokuchi and M. Sano, Chem. Lett., 1992, 419 CAS
- Y. Yamashita, S. Tanaka, K. Imaeda, H. Inokuchi and M. Sano, J. Org. Chem., 1992, 57, 5517 CrossRef CAS
- Y. Yamashita, K. Ono, S. Tanaka, K. Imaeda and H. Inokuchi, Adv. Mater., 1994, 6, 295 CrossRef CAS
- R. L. Myers and I. Shain, Anal. Chem., 1969, 41, 980 CrossRef CAS
- K. Yakushi, J. Dong, M. Uruichi and Y. Yamashita, Mol. Cryst. Liq. Cryst., 1996, 284, 223 CAS
- M. Uruichi, K. Yakushi, Y. Yamashita and J. Qin, J. Mater. Chem., 1998, 8, 141 RSC
- Y. Yamashita, S. Tanaka and K. Imaeda, Synth. Met., 1995, 71, 1965 CrossRef CAS
- K. Imaeda, Y. Yamashita, S. Tanaka and H. Inokuchi, Synth. Met., 1995, 73, 107 CrossRef CAS
- Y. Yamashita, M. Tomura and K. Imaeda, Chem. Commun., 1996, 2021 RSC
- K. Yakushi, M. Uruichi and Y. Yamashita, Synth. Met., 2000, 109, 33 CrossRef CAS
- M. Uruichi, K. Yakushi and Y. Yamashita, J. Mater. Chem., 2000, 10, 2716 RSC
- K. Takahashi and K. Tomitani, J. Chem. Soc., Chem. Commun., 1995, 821 RSC
- K. Takahashi and I. Toshiro, Heterocycles, 1997, 45, 1051 CAS
- K. Takahashi and I. Toshiro, Mol. Cryst. Liq. Cryst., 2002, 380, 183 CAS
- K. Takahashi, I. Toshiro, T. Mori, H. Mori and S. Tanaka, Chem. Lett., 1998, 1147 CrossRef CAS
- K. Takahashi and I. Toshiro, J. Cryst. Growth, 2001, 229, 591 CrossRef
- I. Toshiro, T. Mori and K. Takahashi, Chem. Lett., 1997, 1013
- I. Toshiro, T. Mori and K. Takahashi, J. Mater. Chem., 2001, 11, 264 RSC
- K. Takahashi and T. Shirahata, Chem. Lett., 2001, 514 CrossRef CAS
- P. Leriche, S. Roquet, N. Pillerel, G. Mabon and P. Frère, Tetrahedron Lett., 2003, 44, 1623 CrossRef CAS
- M. Sallé, A. Belyasmine, A. Gorgues, M. Jubault and N. Soyer, Tetrahedron Lett., 1991, 32, 2897 CrossRef CAS
- P. Frère, A. Benahmed-Gasmi, J. Roncali, M. Jubault and A. Gorgues, J. Chim. Phys., 1995, 92, 863 CAS
- M. V. Lakshmikantham, M. P. Cava and P. J. Caroll, J. Org. Chem., 1984, 49, 7266728
- M. Fourmigué, I. Johannsen, K. Boubekeur, C. Nelson and P. Batail, J. Am. Chem. Soc., 1993, 115, 3752 CrossRef CAS
- S. Gonzalez, N. Martin, L. Sanchez, J. L. Segura, C. Seoane, I. Fonseca, F. H. Cano, J. Sedo, J. Vidal-Gancedo and C. Rovira, J. Org. Chem., 1999, 64, 3498 CrossRef CAS
- P. Hascoat, D. Lorcy, A. Robert, R. Carlier, A. Tallec, K. Boubekeur and P. Batail, J. Org. Chem., 1997, 62, 6086 CrossRef CAS
- D. Lorcy, D. Guérin, K. Boubekeur, R. Carlier, P. Hascoat, A. Tallec and A. Robert, J. Chem. Soc., Perkin Trans 1, 2000, 2719 RSC
- P. Frère, A. Gorgues, M. Jubault, A. Riou, Y. Gouriou and J. Roncali, Tetrahedron Lett., 1994, 35, 1991 CrossRef CAS
- U. Schöberl, J. Salbeck and J. Daub, Adv. Mater., 1992, 4, 41 CrossRef
- T. K. Hansen, M. V. Lakshmikantham, M. P. Cava, R. E. Niziurski-Mann, F. Jensen and J. Becher, J. Am. Chem. Soc., 1992, 114, 5035 CrossRef CAS
- A. Benahmed-Gasmi, P. Frère, B. Garrigues, A. Gorgues, M. Jubault, R. Carlier and F. Texier, Tetrahedron Lett., 1992, 33, 6457 CrossRef CAS
- J. Roncali, L. Rasmussen, C. Thobie - Gautier, P. Frère, H. Brisset, M. Sallé, J. Becher, O. Simonsen, T. K. Hansen, A. Benahmed-Gasmi, J. Orduna, J. Garin, M. Jubault and A. Gorgues, Adv. Mater., 1994, 6, 841 CrossRef CAS
- S. Akoudad, P. Frère, N. Mercier and J. Roncali, J. Org. Chem., 1999, 64, 4267 CrossRef CAS
- J. Roncali, M. Giffard, P. Frère, M. Jubault and A. Gorgues, J. Chem. Soc., Chem. Commun., 1993, 689 RSC
- H. Brisset, C. Thobie - Gautier, M. Jubault, A. Gorgues and J. Roncali, J. Chem. Soc., Chem. Commun., 1994, 1765 RSC
- P. Leriche, J. M. Raimundo, M. Turbiez, V. Monroche, M. Allain, F. X. Sauvage, J. Roncali, P. Frère and P. J. Skabara, J. Mater. Chem., 2003, 13, 1324 RSC
- P. Leriche, M. Turbiez, V. Monroche, P. Frère, P. Blanchard, P. J. Skabara and J. Roncali, Tetrahedron Lett., 2003, 44, 649 CrossRef CAS
- E. H. Elandaloussi, P. Frère, J. Roncali, P. Richomme, M. Jubault and A. Gorgues, Adv. Mater., 1995, 7, 390 CrossRef CAS
- E. H. Elandaloussi, P. Frère, A. Benahmed-Gasmi, A. Riou, A. Gorgues and J. Roncali, J. Mater. Chem., 1996, 6, 1859 RSC
- A. Benahmed-Gasmi, P. Frère, E. H. Elandaloussi, J. Roncali, J. Orduna, J. Garin, M. Jubault, A. Riou and A. Gorgues, Chem. Mater., 1996, 8, 2291 CrossRef CAS
- H. Brisset, S. Le Moustarder, P. Blanchard, B. Illien, A. Riou, J. Orduna, J. Garin and J. Roncali, J. Mater. Chem., 1997, 7, 2027 RSC
- I. Jestin, P. Frère, E. Levillain and J. Roncali, Adv. Mater., 1999, 11, 134 CrossRef CAS
- J. F. Favard, P. Frère, A. Riou, A. BenahmedGasmi, A. Gorgues, M. Jubault and J. Roncali, J. Mater. Chem., 1998, 8, 363 RSC
- J. B. Torrance, B. A. Scott, B. Welber, F. B. Kaufman and P. E. Seiden, Phys. Rev. B, 1979, 19, 730 CrossRef CAS
- L. L. Miller and K. R. Mann, Acc. Chem. Res., 1996, 29, 417 CrossRef CAS
- P. Frère, M. Allain, E. H. Elandaloussi, E. Levillain, F. X. Sauvage, A. Riou and J. Roncali, Chem. Eur. J., 2002, 8, 784 CrossRef CAS
- A. Merz, J. Kronberger, L. Dunsh, A. Neudeck, A. Petr and L. Parkanyi, Angew. Chem. Int. Ed. Engl., 1999, 38, 1442 CrossRef CAS
- D. D. Graf, R. G. Duan, J. P. Campbell, L. L. Miller and K. R. Mann, J. Am. Chem. Soc., 1997, 119, 5888 CrossRef CAS
- E. H. Elandaloussi, P. Frère, A. Riou and J. Roncali, New J. Chem, 1998, 22, 1051 RSC
- A. Terahara, H. Ohya-Nishiguchi, N. Hirota, H. Awaji, T. Kawase, S. Yoneda, T. Sugimoto and Z. I. Yoshida, Bull. Chem. Soc. Jpn., 1984, 57, 1760 CAS
- M. Scholz, G. Gescheidt, U. Schöberl and J. Daub, Magn. Reson. Chem., 1995, 33, S94 CAS
- A. D. McLachlan, Mol. Phys., 1960, 3, 233 CAS
- Y. Misaki, Y. Matsumura, T. Sugimoto and Z. I. Yoshida, Tetrahedron Lett., 1989, 30, 5289 CrossRef CAS
- M. A. Coffin, M. R. Bryce, A. S. Batsanov and J. A. K. Howard, J. Chem. Soc., Chem. Commun., 1993, 552 RSC
- M. R. Bryce, M. A. Coffin, P. J. Skabara, A. J. Moore, A. S. Batsanov and J. A. K. Howard, Chem. Eur. J., 2000, 6, 1955 CrossRef CAS
- R. R. Amaresh, D. Liu, T. Konovalova, M. V. Lakshmikantham, M. P. Cava and L. D. Kispert, J. Org. Chem., 2001, 66, 7757 CrossRef CAS
- D. Rajagopal, M. V. Lakshmikantham and M. P. Cava, Org. Lett., 2002, 4, 2581 CrossRef CAS
-
P. Frère and J. Roncali, Unpublished results.
- T. Sugimoto, H. Awaji, Y. Misaki and Z. I. Yoshida, J. Am. Chem. Soc., 1985, 107, 5792 CrossRef CAS
- T. Sugimoto, Y. Misaki, T. Kajita and Z. I. Yoshida, J. Am. Chem. Soc., 1987, 109, 4106 CrossRef CAS
- M. R. Bryce, M. A. Coffin, A. J. Moore, A. S. Batsanov and J. A. K. Howard, Tetrahedron, 1999, 55, 9915 CrossRef CAS
- Z. I. Yoshida and T. Sugimoto, Angew. Chem., Int. Ed.Engl., 1988, 27, 1573 CrossRef
- M. Sallé, A. Gorgues, M. Jubault, K. Boubekeur, P. Batail and R. Carlier, Bull. Soc. Chim. Fr., 1996, 133, 417 CAS
- A. Belyasmine, P. Frère, A. Gorgues, M. Jubault, G. Duguay and P. Hudhomme, Tetrahedron Lett., 1993, 34, 4005 CrossRef CAS
- P. Frère, K. Boubekeur, M. Jubault, P. Batail and A. Gorgues, Eur. J. Org. Chem., 2001, 3741 CrossRef CAS
- M. Sallé, M. Jubault, A. Gorgues, K. Boubekeur, M. Fourmigué, P. Batail and E. Canadell, Chem. Mater., 1993, 5, 1196 CrossRef CAS
-
M. Sallé and A. Gorgues, personal communication.
- Y. Misaki and T. Mori, Mol. Cryst. Liq. Cryst., 2002, 380, 69 CAS
- Y. Misaki, N. Higuchi, H. Fujiwara, T. Yamabe, T. Mori, H. Mori and S. Tanaka, Angew. Chem., Int. Ed. Engl., 1995, 34, 1222 CrossRef CAS
- Y. Misaki, T. Ohta, N. Higuchi, H. Fujiwara, T. Yamabe, T. Mori, H. Mori and S. Tanaka, J. Mater. Chem., 1995, 5, 1571 RSC
- T. Mori, T. Kawamoto, Y. Misaki, K. Kawakami, H. Fujiwara, T. Yamabe, H. Mori and S. Tanaka, Mol. Cryst. Liq. Cryst., 1996, 284, 271 CrossRef CAS
- Y. Misaki, H. Fujiwara, T. Yamabe, T. Mori, H. Mori and S. Tanaka, Chem. Lett., 1994, 1653 CAS
- A. Deluzet, P. Batail, Y. Misaki, P. Auban-Senzier and E. Canadell, Adv. Mater., 2000, 12, 436 CrossRef CAS
- Y. Misaki, T. Kochi, T. Yamabe and T. Mori, Adv. Mater., 1998, 10, 588 CrossRef CAS
- H. Fujiwara, Y. Misaki, T. Yamabe, T. Mori, H. Mori and S. Tanaka, J. Mater. Chem., 2000, 10, 1565 RSC
- Y. Misaki, S. Tanaka, M. Taniguchi, T. Yamabe and T. Mori, Adv. Mater., 1999, 11, 25 CrossRef CAS
- T. Mori, H. Inokuchi, Y. Misaki, T. Yamabe, H. Mori and S. Tanaka, Bull. Chem. Soc. Jpn., 1994, 67, 661 CAS
- T. Mori, T. Kawamoto, K. Iida, J. Yamaura, T. Enoki, Y. Misaki, T. Yamabe, H. Mori and S. Tanaka, Synth. Met., 1999, 103, 1885 CrossRef CAS
- M. Katsuhara, M. Aragaki, T. Mori, Y. Misaki and S. Tanaka, Chem. Mater., 2000, 12, 3186 CrossRef CAS
- M. Katsuhara, M. Aragaki, S. Kimura, T. Mori, Y. Misaki and S. Tanaka, J. Mater. Chem., 2001, 11, 2125 RSC
- M. Katsuhara, S. Kimura, T. Mori, Y. Misaki and S. Tanaka, Chem. Mater., 2002, 14, 458 CrossRef CAS
- M. Ashizawa, S. Kimura, T. Mori, Y. Misaki and K. Tanaka, Synth. Met., 2004, 141, 307 CrossRef CAS
- M. Adam, P. Wolf, H. J. Räder and K. Müllen, J. Chem. Soc., Chem. Commun., 1990, 1624 RSC
- K. Lahlil, A. Moradpour, C. Bowlas, C. Menou, P. Cassoux, J. Bonvoisin, J. P. Launay, G. Dive and D. Dehareng, J. Am. Chem. Soc., 1995, 117, 9995 CrossRef CAS
- M. Adam, A. Bohnen, V. Enkelmann and K. Müllen, Adv. Mater., 1991, 3, 600 CrossRef
- M. Adam, U. Scherer, Y. J. Shen and K. Müllen, Synth. Met., 1993, 55–57, 2108 CrossRef CAS
- J. Peng, E. Wang, Y. Zhou, Y. Xing, H. Jia, Y. Lin and Y. Shen, J. Chem. Soc., Dalton, 1998, 3865 RSC
- C. Boulle, O. Desmars, N. Gautier, P. Hudhomme, M. Cariou and A. Gorgues, Chem. Commun., 1998, 2197 RSC
- N. Gautier, N. Gallego Planas, N. Mercier, E. Levillain and P. Hudhomme, Org. Lett., 2002, 4, 961 CrossRef CAS
- C. A. Christensen, M. R. Bryce, A. S. Batsanov and J. Becher, Org. Biomol. Chem., 2003, 1, 511 RSC
- C. A. Christensen, M. R. Bryce, A. S. Batsanov, J. A. K. Howard, J. O. Jeppesen and J. Becher, Chem. Commun., 1999, 2433 RSC
- M. Mas-Torrent, M. Durkut, P. Hadley, X. Ribas and C. Rovira, J. Am. Chem. Soc., 2004, 126, 984 CrossRef
| This journal is © The Royal Society of Chemistry 2005 |