Isotopic induction of the excited-state single-proton transfer in 7-azaindole dimer
J.
Catalán
and
P.
Pérez
Departamento de Química Física Aplicada, Universidad Autónoma de Madrid, Cantoblanco, 28049, Madrid, Spain
First published on 25th November 2004
Abstract
The electronic spectrum in 2-methylbutane of the doubly hydrogen-bonded 7-azaindole dimer in monodeuterated form, (7AI)2-hd, is consistent with a single-proton transfer that never produces the typical emission for the double proton transfer with its centre at 480 nm and onset at 413 nm.
Introduction
Multiple H-bonded base-pairing as a fundamental element of DNA structure was first described by Watson and Crick1 in stable keto-tautomer forms which, among others, can exhibit two-proton phototautomerism and its associated mutation, through electronic excitation.2 One model base pair for such a two-proton tautomeric shift is 7-azaindole (7AI) H-bonded dimer, which has been extensively studied in this respect3 ever since it was reported by Kasha et al.47AI H-bonded dimer results from the double hydrogen-bonding interaction between the corresponding pyrrole and pyridine units in the two 7AI monomers and is usually designated the C2h dimer. The C2h structure is the most stable among the five possible 7AI dimers examined to date;5 two involve a double hydrogen bond and another a single bond on the carbon atom at position 3, whereas the other two are of the card-pack type. The C2h structure constitutes the true energy minimum for 7AI dimers in the ground electronic state.5
The electronic excitation of the C2h dimer of 7AI results in (a) the delocalization of the excitation over its two halves via an excitonic mechanism4,6–8 and (b) a simultaneous, marked increase in the acidity of its pyrrole proton and the basicity of its pyridine nitrogen.9 Consequently, the C2h dimer can follow a concerted proton phototransfer; this assumption is strongly supported by the latest two pieces of experimental evidence obtained by using femtosecond spectroscopy10 and UV–UV hole-burning spectroscopy.11 Once the debate over whether the double proton transfer in 7-azaindole dimer involves the simultaneous (concerted mechanism) or stepwise shift of the two protons (sequential mechanism) has been closed by these recent experimental contributions,10,11 the time has come to approach new experimental situations allowing us to examine schemes involving single-proton transfers in the dimers and their potential role in the formation of the tautomer produced by a simultaneous double proton transfer.
To the authors’ minds, there are a number of situations where the doubly hydrogen-bonded 7-azaindole dimer may yield single-proton transfers. One involves a change in its vibronic system altering in a differential manner its two 7-azaindole fragments without affecting the overall structure of the dimer. Also, the dimer can lose its centrosymmetry through interaction with either a polar molecule or a polar environment. As a result, something will occur that will distinguish its two halves and facilitate the localization of the electronic excitation on one of the monomer units alone. The former hypothesis is discussed in this paper and the latter is examined in other work.12
Let us analyse how these perturbed dimeric forms can be isotopically generated. In principle, a 2-methylbutane (2MB) solution containing equal amount of undeuterated 7AI (7AI-h) and of monodeuterated 7AI (7AI-d) can form three different dimers, undeuterated dimers that we shall designate (7AI)2-hh (undeuterated), (7AI)2-hd (monodeuterated) and (7AI)2-dd (dideuterated). In previous work,3 we characterized the dimerization of 7AI in its (7AI)2-hh and (7AI)2-dd forms in 2MB by electronic spectroscopy and analysed its photophysical implications. In this work, we addressed the characterization of similar 7AI solutions in order to examine the above-described situation from emission spectra with a view to deriving valuable information about the proton phototransfer in 7AI dimer: particularly in the hope of determining whether the well-known photophysics of the double proton phototransfer in the dimer3,4 could be resolved into two single-proton phototransfers.
Experimental and theoretical
7-Azaindole was obtained from Sigma in 99% purity and recrystallized twice in spectroscopic-grade cyclohexane. N1-deuterated 7-azaindole was prepared by refluxing 7-azaindole in alkaline D2O for 1 h. NMR spectroscopy revealed an isotopic purity of at least 90%. In this work we are mainly concerned with 7AI solutions in 2MB (Merck, Uvasol-grade) were 0.5 × 10−4 M concentration in both 7AI-h and 7AI-d, and also the fluorescence emission of solutions containing 0.5 × 10−5 M 7AI, and 0.25 × 10−6 M of both 7AI-h and 7AI-d are analyzed. The sample temperature ranged from 127 K to 293 K and was controlled by using an Oxford DN1704 cryostat equipped with an ITC4 controller interfaced to the spectrometers. The cryostat was purged with dried nitrogen 99.99% pure.UV-Vis spectra were recorded on a Cary-5 spectrophotometer, using a Suprasil quartz cell of 1 cm light path. All spectroscopic emission measurements, for the 10−4 M 7AI samples, were done in Suprasil cylindrical cells of 3 mm light path; as a result, the path length to the centre of the cell, which governs so-called “filtering effects” on fluorescence, a major influential factor with highly absorbing solutions, was less that 1.5 mm: in fact, the average path length ranged from 0 to 1.5 mm. Corrected fluorescence and excitation spectra were obtained by using a precalibrated Aminco–Bowman AB2 spectrofluorimeter. All 7AI samples were excited at 315 and 325 nm by using light from a continuous (CW) 150 W xenon lamp for steady-state spectra. The spectral widths used were as follows: 8 and 2 mm in the respective monochromators for the emission spectra; and 2 and 8 nm in those for the fluorescence excitation spectra obtained by monitoring light at 360 and 380 nm and 2 and 16 nm by monitoring light at 480 nm. The more diluted 7AI solutions, 10−5 and 5 × 10−6 M, have been studied in cells of 1 cm optical light path, on excitation at the same wavelength (325 nm), but increasing the emission spectral widths to 4 nm and the sensibility of the photomultiplier.
All computations were done within the framework of density functional theory (DFT), using the Gaussian 98 software package.13 Full geometry optimizations for the electronic ground state were carried out by using the hybrid functional B3LYP14,15 with the 6-31G** basis set. The optimized geometries for the ground state were used to compute the Franck–Condon (FC) excitation energies for the singlet excited state (S1(π,π*)) in light of the recently developed time-dependent density functional theory (TDDFT), which has yielded excellent results so far.16–19
Results and discussion
On the formation and photophysical characterization of the potential dimers
The formation of 7-azaindole dimers, eqn. (1), by double hydrogen bonding,| 7AI-h + 7AI-h ↔ (7AI)2-hh | (1) |
Photophysically, the (7AI)2-hh and (7AI)2-dd dimers exhibit a first UV absorption band with its onset at 320 nm and 0–0 component at 315 nm. Upon excitation of these dimeric forms at 315 nm, where 7AI monomer ceases to absorb, (7AI)2-hh only exhibits the classical emission assigned to the double proton transfer with its onset at 413 nm,3,20 the strength of which increases substantially with decreasing solution temperature, and (7AI)2-dd only the emission centred at 350 nm, the strength of which also increases significantly as the temperature is lowered, and assigned to the dimer that transfers neither proton.3,20
Near-UV absorption spectra for 0.5 × 10−4 M 7AI-h and 7AI-d in 2MB
Decreasing the temperature of a 7AI solution in a non-hydrogen-bonding interacting solvent such as 2MB over the range where its dimerization occurs inevitably displaces the monomer–dimer equilibrium and produces an isobestic point in the absorption spectrum. If the whole sample consists of pure dimers at a given temperature, then cooling it will produce no isobestic points as the curve will merely reflect cooling and an increase in solution absorbance. If, as was the case here, the solution contains equimolar amounts of 7AI-h and 7AI-d, then these monomeric forms will take part in equilibria involving three potential dimers, viz. (7AI)2-hh, (7AI)2-dd and (7AI)2-hd, so one can expect the presence of a single isobestic point in the corresponding UV–Vis spectra recorded at lower temperatures.Fig. 1 shows the absorption spectra at temperatures between 293 and 127 K for a solution containing 0.5 × 10−4 M 7AI-h and an identical concentration of 7AI-d. The situation is clearly so different from that for a 10−4 M 7AI-h solution in 2MB (see Fig. 1 in ref. 3) that it warrants careful examination. Thus, the spectrum at 293 K is typical of monomer forms as its first absorption peak lies at ca. 293 nm; its variation as the temperature is lowered suggests increasing dimerization, in any case, the isobestic point is shifted as a result of the potential occurrence of various dimerization equilibria.
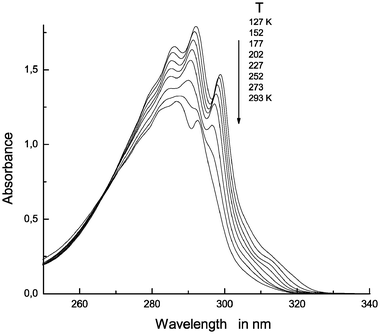 | ||
| Fig. 1 Near-UV absorption spectra for a 0.5 × 10−4 M solution of 7AI-h and 7AI-d in 2MB at 293, 277, 252, 227, 202, 177, 152 and 127 K. | ||
As with 7AI-h (see Fig. 1 in ref. 3), no monomers appear to exist below 227 K, so only the typical doubly hydrogen-bonded dimers should be present in solution. However, the spectral picture at low temperatures is unusual. Thus, unlike (7AI)2-hh and (7AI)2-dd (see Figs. 1 and 3 in ref. 3), the spectral onsets no longer fall at 320 nm; rather, they are shifted to the 330 nm region. Also, the peak at 315 nm for the 0–0 component is only, faintly, observed in the spectra recorded at temperatures between 202 and 152 K, and is completely absent from that obtained at 127 K. From the foregoing it follows that, at low temperatures, the situation is governed by the presence of a new dimer which must be the monodeuterated form, (7AI)2-hd.
The spectra in Fig. 1 clearly indicate that the photophysical of these solutions should be examined at 315 nm [where the (7AI)2-hh and (7AI)2-dd dimers potentially present will be active] and at 325 nm [where only the new monodeuterated dimer, (7AI)2-hd, will be excited].
Emission spectra for 0.5 × 10−4 M 7AI-h and 7AI-d in 2MB
Fig. 2 shows the emission spectra obtained with excitation at 315 nm. Except for the small emission traces observed by lowering the temperature from 202 K to 152 K, the 480 nm region exhibits none of the typical emission that increases significantly with decreasing solution temperature and has been assigned to the (7AI)2-hh dimer (see ref. 3). We should note that the emission ceases to be apparent as the temperature is lowered further (see the spectrum recorded at 127 K). The region from 340 nm to 420 nm exhibits some weak emission that in principle cannot be assigned to (7AI)2-hh, so it may be generated by (7AI)2-dd and (7AI)2-hd. | ||
| Fig. 2 Emission spectra (λexc = 315 nm) for a 0.5 × 10−4 M solution of 7AI-h and 7AI-d in 2MB at temperatures from 293 K to 127 K. | ||
Fig. 3 shows the emission spectra obtained with excitation at 325 nm, which should in principle be assigned to the new dimer, (7AI)2-hd, as the others [(7AI)2-hh and (7AI)2-dd] do not absorb in this region; their onsets are at 320 nm. The emission bands are centred at 390 nm and, more important, are apparent in the 480 nm region; however, their shape is inconsistent with that of a band produced by a typical double proton transfer.
 | ||
| Fig. 3 Emission spectra (λexc = 325 nm) for a 0.5 × 10−4 M solution of 7AI-h and 7AI-d in 2MB at temperatures from 293 K to 127 K. | ||
Fig. 4 shows the excitation spectra obtained by monitoring light at 360 nm, which reveal that, between 292 K and 252 K, monomeric forms prevail; however, based on the virtual absence of the peak at 315 and on the fact that the onset of these spectra is beyond 320 nm, (7AI)2-dd and, especially, (7AI)2-hd, must also be present at these and, in particularly, lower temperatures.
 | ||
| Fig. 4 Excitation spectra obtained by monitoring light emitted at 360 nm by a 0.5 × 10−4 M solution of 7AI-h and 7AI-d in 2MB at temperatures from 293 K to 127 K. | ||
Fig. 5 shows the excitation spectra obtained by monitoring light at 380 nm. From the profiles one can conclude that monomeric forms prevail between 293 K and 277 K. However, based on the virtual absence of the peak at 315 nm and on the shift of the onset beyond 320 nm, the dominant form at lower temperatures must be the new dimer: (7AI)2-hd.
 | ||
| Fig. 5 Excitation spectra obtained by monitoring light emitted at 380 nm by a 0.5 × 10−4 M solution of 7AI-h and 7AI-d in 2MB at temperatures from 293 K to 127 K. | ||
Fig. 6 shows the excitation spectra obtained by monitoring light at 480 nm. Based on them, emission in this spectral region is governed by the (7AI)2-hh dimer, which, however, is accompanied by the new dimer: (7AI)2-hd.
 | ||
| Fig. 6 Excitation spectra obtained by monitoring light emitted at 480 nm by a 0.5 × 10−4 M solution of 7AI-h and 7AI-d in 2MB at temperatures from 293 K to 127 K. | ||
With reference to what has been discussed in this manuscript, it is true that for a 10−4 M solution of 7AI in 2MB at 77 K a phosphorescence emission is recorded, which Catalán and Kasha3 have ascribed to phosphorescence from higher aggregates than the usual C2h 7AI dimer. Based on the stoichiometry of these aggregates, Bulska et al.21 proposed a structure composed of five molecules of 7AI linearly assembled, which is very improbable for a 10−4 M solution. Also, a recent proposal22 deals with a 7AI dimer complexed with another 7AI monomer molecule, thus breaking the C2h symmetry. We must check whether the fluorescence detected comes from those higher aggregates. If the fluorescence comes from higher aggregates, one must conclude that in 10−5 M concentration of 7AI in 2MB at 77 K that phosphorescence is not monitored22, and therefore, those aggregates must not exist.
Emission spectra for 0.5 × 10−5 M and 0.25 × 10−6 M 7AI-h and 7AI-d in 2MB at 127 K
For the sake of rejecting the possibility of that the fluorescence monitored in 0.5 × 10−4 M solutions of 7AI-h and 7AI-d is only owed to higher aggregates of 7AI, formed at low temperatures of the sample, lower concentrations of 7AI should be prepared that do not have to exhibit the same fluorescence band. The planning of experiments on this subject becomes difficult since Taylor23 demonstrated that a 10−5 M solution of 7AI in 3MP at room temperature does not show fluorescence emission from the 7AI dimer. Nevertheless, even at very low 7AI concentrations if the temperature decreases the 7AI will start forming the dimer. Fig. 7 exhibits the absorption for the 0.5 × 10−5 M solution of 7AI-h and 7AI-d at 127 K and clearly the sample has generated the dimer. Fig. 8 shows the fluorescence emission on excitation at 325 ± 4 nm of a 0.5 × 10−5 M solution of 7AI-h and 7AI-d at 127 K using an emission slit of 4 nm, that is, the new fluorescence already recorded in the previous and more concentrated 7AI solution at 0.5 × 10−4 M concentration. This fluorescence band is also recorded at 0.25 × 10−6 M of 7AI-h and 7AI-d at 127 K, and since it is clear-cut that the dominant 7AI species in these dilute solutions are the doubly-h-bonded 7AI dimers one can draw the conclusion that as the well-known emission generated by means of excited state double proton transfer (which possesses an onset at 413 nm and a peak maximum at 480 nm) is not observed, the new fluorescence emission at 420 nm must be ascribed to the single-proton transfer in the 7(AI)2-hd dimer. | ||
| Fig. 7 Near-UV absorption spectrum for a 0.5 × 10−5 M solution of 7AI-h and 7AI-d in 2MB at 127 K. | ||
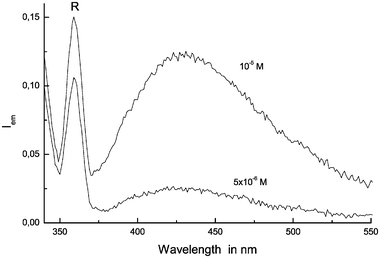 | ||
| Fig. 8 Emission spectra (λexc = 325 nm) for a 0.5 × 10−5 M and 0.25 × 10−6 M solutions of 7AI-h and 7AI-d in 2MB at 127 K. (R means Raman peak). | ||
In view of the fluorescences shown in Figs. 3 and 8, it is appropriate to point out that in a 10−4 M solution of 7AI in 2MB emits fluorescence from the single proton transfer 7AI dimer together with the 7AI oligomers. These oligomers may be those 7AI tetramers found by Dufour et al.24 in a 7AI single-crystal sample which fluoresces at 350 nm.25 The emission of tetramer species at about 350 nm would help explain that the fluorescence at 420 nm in very dilute solutions, 10−5 and 5 × 10−6 M of 7AI, is found to be shifted to 390 nm in 10−4 M solutions of 7AI, as a result of the overlap with the oligomers emission.
Scheme for the single-proton transfer in 7AI dimer
Although, structurally, the monodeuterated dimer of 7AI, (7AI)2-hd, appears to possess C2h symmetry, this is not so as, photophysically, its vibronic system departs from such a symmetry. The two vibrations that in the ground electronic state move the two protons synchronously within the C2h dimer of (7AI)2-hh are localized only on the N–H bond (at 3224 cm−1) and only N–D bond (at 2383 cm−1), respectively, in the monodeuterated dimer. Spectroscopically, such a dimer must behave as if it had Cs symmetry and this symmetry loss must involve substantial spectral changes such as a shift in the onset of its first absorption band.Evaluation of the S0 → S1 transition for the C2h molecular structure in 7-azaindole normal dimer, where both N–H distance are 1.033 Å, reveals that it is primarily a transition between the molecular orbitals (MOs) HOMO and LUMO. The analysis of these MOs (Fig. 9) reflects the excitonic delocalization and simultaneous exacerbation upon electronic excitation of the two hydrogen bonds supporting the molecular structure of 7-azaindole, the acidity of its two pyrrole sites and the basicity of its two pyridine nitrogens. The reported description for these MOs is consistent with a simultaneous double proton transfer in the C2h dimer of 7-azaindole.
 | ||
| Fig. 9 HOMO and LUMO of 7-azaindole dimer: (a)C2h; (b) Cs with N–H = 1.05 Å; and (c) Cs with N–H = 1.1 Å. | ||
On the other hand, fixing the N–H distance of one of the bonds at 1.05 Å and optimizing the molecular geometry of the dimer yields a dimer of Cs symmetry representative of the new situation in the monodeuterated dimer, (7AI)2-hd, where the other N–H distance is 1.033 Å. The S0 → S1 transition in this new molecular structure is also largely described by a HOMO → LUMO transition; also, based on the MOs of Fig. 9, the situation is rather different. Thus, the excitonic effect is now irrelevant as both the HOMO and the LUMO are preferentially localized on one dimer half; even more important, these MOs are preferentially located on a different dimer half. The shape of the HOMO and LUMO clearly indicates that, because the electronic excitation affects only one of the hydrogen bonds that support the dimer structure, its acid site (the N–H bond) becomes more acidic and, simultaneously, its basic site (the pyridine nitrogen) more basic. In conclusion, these MOs reveal that an Cs dimer of 7-azaindole produces a single-proton transfer upon excitation. The molecular orbitals corresponding to the structure with an N–H distance of 1.1 Å (Fig. 9c), exhibit with clarity the total localization of the electronic excitation in each molecular half of the 7-azaindole dimer.
The regular decrease of the energy for the S1 state of the Cs dimer of 7-azaindole as the N–H distance, in one of this bonds, increases from 1.05 Å to 1.20 Å indicates that the proton-transfer process is barrierless. The results obtained are: −759.610 043, −759.612 020, −759.616 517 and −759.622 159 Eh at an N–H distance of 1.05, 1.10, 1.15 and 1.20 Å, respectively. It is pointed out that these energy values are FC, and are evaluated by adding the corresponding energy of the ground state to the FC transition energy at the TDDFT level. This behaviour provides a plausible explanation for the significant red-shift in the onset of first absorption band for 7-azaindole dimer from a C2h symmetry to a Cs symmetry (see Fig. 10).
 | ||
| Fig. 10 Near-UV absorption spectra for (a) a 10−4 M solution of 7AI-h in 2MB at 127 K and (b) a 0.5 × 10−4 M solution of 7AI-h and 7AI-d in 2MB at 127 K. | ||
The evidence gathered about the photophysics of the (7AI)-hd dimer allows us to propose Scheme 1. If (7AI)2-hd possesses an isotopically perturbed Cs symmetry, then the phototransfer will involve a single-proton and exhibit an emission signal at ca. 420 nm. The most salient feature is that this single-proton transfer is not an intermediate step leading, via a second proton transfer in 7AI, to the tautomers reached through the double proton transfer that was reported more than 30 years ago by Kasha et al.4
 | ||
| Scheme 1 | ||
This new view on the problem raises several questions currently being addressed in our laboratory, namely: Can an electronically excited 7AI molecule in a dimer of Cs symmetry induce a double proton transfer in the dimer?
Conclusions
The loss of C2h symmetry induced by deuteration in 7-azaindole dimer was demonstrated in a solution containing 0.5 × 10−5 M 7AI-h and 0.5 × 10−5 M 7AI-d in 2MB at a low temperature. The phototransfer in the excited singlet state for 7AI dimer can theoretically take place via two different mechanisms, namely: a concerted double proton transfer if the dimer possesses C2h symmetry and a single-proton transfer if it has a Cs symmetry induced by isotopic substitution. These transfers therefore represent two different mechanisms and the single-proton one is not an intermediate step in the formation of the tautomer produced by the concerted two-proton process.Acknowledgements
We are greatly indebted to Dirección General de Investigación Cientifica y Tecnica (Spain) for support Project No. BQU2002-02106.References
- J. D. Watson and F. H. C. Crick, Nature, 1953, 171 Search PubMed , 737 and 964.
- J. D. Watson and F. H. C. Crick, Cold Spring Harbor Symp. Quant. Biol., 1953, 18, 123 Search PubMed.
- J. Catalán and M. Kasha, J. Phys. Chem. A, 2000, 104, 10812 CrossRef CAS , and references therein.
- C. A. Taylor, M. A. El-Bayoumi and M. Kasha, Proc. Acad. Natl. Sci. USA, 1969, 63, 253 Search PubMed.
- J. Catalán, P. Pérez, J. C. del Valle, J. L. G. de Paz and M. Kasha, Proc. Natl. Acad. Sci. USA, 2002, 99, 5793 CrossRef CAS.
- E. G. McRae and M. Kasha, in Physical Processes in Radiation Biology. ed. L. Augenstein, R. Mason and B. Rosenberg, Academic Press, New York, 1964, p. 23 Search PubMed.
- M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371 CrossRef CAS.
- J. Catalán, J. C. del Valle and M. Kasha, Proc. Natl. Acad. Sci. USA, 1999, 96, 8338 CrossRef CAS.
- J. Catalán, J. Am. Chem. Soc., 2001, 123, 11940 CrossRef CAS.
- S. Takeuchi and T. Tahara, Chem. Phys. Lett., 2001, 347, 108 CrossRef CAS.
- K. Sakota, A. Hara and H. Sekiya, Phys. Chem. Chem. Phys., 2004, 6, 32 RSC.
- J. Catalán, Phys. Chem. Chem. Phys., 2004, 6, 4467 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Sakrzewski, J. A. Montgomery, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millan, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, M. Barone Cossi, R. Cammi, B. Mennuci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaroni, R. Gompers, R. L. Martin, D. J. Fox, T. Keith, M. A. Ai-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challaconbe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, J. L. Andrés, M. Head-Gordon, E. S. Replogle and J. A. Pople, GAUSSIAN 98, Gaussian Inc., Pittsburgh PA, 1998 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5642 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 3, 785 CrossRef.
- K. G. Wiberg, R. E. Stratmann and M. J. Frisch, Chem. Phys. Lett., 1998, 297, 60 CrossRef CAS.
- S. Hirata, T. J. Lee and M. J. Head-Gordon, Chem. Phys., 1999, 111, 8904 CAS.
- J. Catalán, J. Chem. Phys., 2003, 119, 1373 CrossRef CAS.
- J. Catalán and J. L. G. de Paz, J. Chem. Phys., 2004, 120, 1864 CrossRef CAS.
- J. Catalán, P. Pérez, J. C. del Valle, J. L. G. de Paz and M. Kasha, Proc. Natl. Acad. Sci. USA, 2004, 101, 419 CrossRef CAS.
- H. Bulska, A. Grabowska, B. Pakula, J. Sepiol, J. Waluk and P. Wild Urs, J. Lumin., 1984, 29, 65 CrossRef CAS.
- J. Catalán, Int. J. Quantum Chem., 2005, 102 Search PubMed , in press.
- C. A. Taylor, PhD Thesis, Florida State University, Tallahassee, FL, 1968, p. 71, Fig. 23.
- P. Dufour, Y. Dartiguenave, M. Dartiguenave, N. Dufour, A. M. Lebuis, F. Belanguer-Gariepy and A. L. Beauchamp, Can. J. Chem., 190, 68–193 Search PubMed.
- P. T. Chou, J. H. Liao, C. Y. Wei, C. Y. Yang, W. S. Yu and Y. H. Chou, J. Am. Chem. Soc., 2000, 122, 986 CrossRef CAS.
| This journal is © the Owner Societies 2005 |