Laser-induced fluorescence excitation and dispersed fluorescence spectroscopy of the Ã(1B1)–![[X with combining tilde]](https://www.rsc.org/images/entities/h2_char_0058_0303.gif) (1A1) transition of dichlorocarbene
(1A1) transition of dichlorocarbene
Joseph S.
Guss
,
Craig A.
Richmond
,
Klaas
Nauta
and
Scott H.
Kable
*
School of Chemistry, University of Sydney, Sydney, 2006, NSW, Australia. E-mail: s.kable@chem.usyd.edu.au; Tel: +61 2 9351 2756
First published on 25th November 2004
Abstract
The Ã(1B1)–![[X with combining tilde]](https://www.rsc.org/images/entities/char_0058_0303.gif) (1A1) transition of jet-cooled CCl2 has been investigated using both laser-induced fluorescence (LIF) excitation and dispersed fluorescence (DF) spectroscopy. DF spectra were taken from over 90 emitting vibronic states over the breadth of the LIF spectrum. 68 ground state vibrational levels were assigned in the C35Cl2 isotopomer and 47 in C35Cl37Cl. The ground state frequencies were fit to an anharmonic potential yielding: ω″1
= 736 cm−1, ω″2
= 338 cm−1, ν″3
= 760 cm−1, x″11
=
−3.19 cm−1, x″22
=
−0.29 cm−1, x″12
=
−1.80 cm−1, x″13
=
−5.70 cm−1 and x″23
=
−3.80 cm−1. This is the first gas phase measurement of ν″3. Despite the close frequency match between ν″1 and 2ν″2 there was no evidence for Fermi resonance. The intensities of transitions in the DF spectra were analysed to identify the nature of the emitting state. Levels below T00
+ 2500 cm−1 in the à state could be assigned simply as combinations and overtones of ν′1 and ν′2. Between T00
+ 2500 and T00
+ 5300 cm−1 the emission revealed an increasing mixing between levels within the same polyad, i.e., (m,n,0), (m
+ 1,n
− 2,0), (m
+ 2,n
− 4,0)
etc, presumably due to Fermi resonance. This mixing becomes so extensive that simple assignment of the emitting states is not possible. Above T00
+ 5300 cm−1 only K′
= 0 states could be identified, indicating that the Renner–Teller intersection between the and à states had been exceeded. This intersection is therefore 22 600 cm−1 above the ground state, in excellent agreement with a previous theoretical calculation of 23 000 cm−1.
(1A1) transition of jet-cooled CCl2 has been investigated using both laser-induced fluorescence (LIF) excitation and dispersed fluorescence (DF) spectroscopy. DF spectra were taken from over 90 emitting vibronic states over the breadth of the LIF spectrum. 68 ground state vibrational levels were assigned in the C35Cl2 isotopomer and 47 in C35Cl37Cl. The ground state frequencies were fit to an anharmonic potential yielding: ω″1
= 736 cm−1, ω″2
= 338 cm−1, ν″3
= 760 cm−1, x″11
=
−3.19 cm−1, x″22
=
−0.29 cm−1, x″12
=
−1.80 cm−1, x″13
=
−5.70 cm−1 and x″23
=
−3.80 cm−1. This is the first gas phase measurement of ν″3. Despite the close frequency match between ν″1 and 2ν″2 there was no evidence for Fermi resonance. The intensities of transitions in the DF spectra were analysed to identify the nature of the emitting state. Levels below T00
+ 2500 cm−1 in the à state could be assigned simply as combinations and overtones of ν′1 and ν′2. Between T00
+ 2500 and T00
+ 5300 cm−1 the emission revealed an increasing mixing between levels within the same polyad, i.e., (m,n,0), (m
+ 1,n
− 2,0), (m
+ 2,n
− 4,0)
etc, presumably due to Fermi resonance. This mixing becomes so extensive that simple assignment of the emitting states is not possible. Above T00
+ 5300 cm−1 only K′
= 0 states could be identified, indicating that the Renner–Teller intersection between the and à states had been exceeded. This intersection is therefore 22 600 cm−1 above the ground state, in excellent agreement with a previous theoretical calculation of 23 000 cm−1.
Introduction
Carbenes have provided a fertile area of study for spectroscopists and theoreticians with many hundreds of papers published over more than four decades. One of the principle reasons for this extended interest is the extreme sensitivity of the electronic energies to substitution about the carbene site. This sensitivity is caused by the interaction of two non-bonding, carbon-centred electrons, which may be paired in the same orbital (the “carbene” configuration), or in two separate orbitals (the “biradical” configuration). The ground state configuration, and indeed the relative energy of all states involving these electrons, varies enormously with the other substituents on the central carbon atom. For example, the prototypical carbene, methylene (CH2) has a triplet biradical ground state with the singlet carbene configuration lying 3165 cm−1 higher in energy.1 The singlet biradical lies about 12 000 cm−1 above the triplet state.2 All other hydrogen or halogen containing carbenes have a singlet carbene ground state, with the possible exception of CHI.3 In CF2, this singlet carbene configuration is so stable that the triplet lies about 20 000 cm−1 higher in energy4,5 and the singlet biradical state 37 000 cm−1 above the ground state.6,7More recently, both experimentalists and theorists have turned their attention to the photochemistry and photophysics of the à state. This has been motivated, in part, by increasing recognition that carbenes are present in a wide range of complex chemical environments, for example in flames and combustion8,9 and in the atmosphere.10–14 There is also interest in carbenes because their à and states are Renner–Teller (RT) components of the same 1Δ state in the linear configuration, which often has an excitation energy similar to the bond dissociation energy of one of the C-ligand bonds. This has the potential to lead to a fascinating interplay between photochemistry (bond breaking) and photophysics (state mixing in the RT region). The RT coupling has been examined in CHF15,16 and CHBr17,18 in some detail, while photodissociation has been explored in CFBr19 and CFCl.20
The subject of this paper is CCl2. The first experimental study of CCl2 was carried out by Milligan and Jacox in 1967.21 IR-absorption spectroscopy provided values of 748 and 721 cm−1 for the symmetric and antisymmetric stretching frequencies, although no firm assignment was made as to which was which. They were also able to obtain the C35Cl37Cl isotopomer stretching frequencies of 726 and 700 cm−1. Several more matrix studies have been performed since.22–27 Andrews22 performed an experiment similar to that of Milligan and Jacox, and obtained consistent results, but was able to assign the lower frequency to the symmetric stretch (ν1). Shirk28 excited matrix isolated CCl2 with 514.5 nm radiation from an argon ion laser and recorded the dispersed fluorescence (DF) spectrum, making the first assignment of the bending frequency ν″2 = 341 cm−1. In 1975, Tevault and Andrews refined the value of ν″2 to 326.5 cm−1 and calculated x″11 = −3 cm−1 using DF spectroscopy. In 1977, Bondybey27 measured both emission and excitation spectra of CCl2 and calculated ω″1 = 726 cm−1, ω″2 = 333 cm−1, ω″3 = 745 cm−1, x″11 = −3.7 cm−1, x″22 = −2.2 cm−1, x″12 = 0.2 cm−1 and T00 = 17 092 cm−1. Bondybey also measured the laser induced fluorescence (LIF) excitation spectrum from which he was able to calculate many of the à state vibrational constants and a fluorescence lifetime of 3.6 μs. Matrix studies have provided all three ground state frequencies and all anharmonicities associated with ν1 and ν2, as well as ω1, ω2, x11, x22 and x12 in the excited state.
Since 1977, when Huie et al. recorded an LIF spectrum of CCl2 in a bulb29, there have been many gas phase studies,30–36 the most comprehensive of which was by Clouthier and Karolczak in 1991.35 They used a pyrolysis nozzle to produce jet-cooled CCl2 in the vacuum chamber, measuring the electronic transition origin at T00 = 17 255.672 cm−1 and several rotational constants. They also assigned several ground state vibrations from an analysis of hot band structure in the spectrum.
The most recent spectroscopic study of CCl2 was a DF investigation reported by Liu et al. in 2003.37 They recorded DF spectra from four vibrational bands in C35Cl2 and two vibrational bands in C35Cl37Cl. In all, 76 ground state vibrational levels were assigned for C35Cl2 and twelve for C35Cl37Cl. An anharmonic analysis was included, which reported all harmonic and anharmonic constants involving ν″1 and ν″2. At energies near 6000 cm−1, extra transitions were observed, which were interpreted as mixing with the triplet state.
Bauschlicher et al.38 carried out the first theoretical study of CCl2 in 1976. They computed the energies, bond angles and bond lengths in the lowest energy singlet and triplet states. In 1985, Nguyen et al. published theoretical values for the vibrational frequencies of CCl2 in the ground and excited singlet states.39 Since then several theoretical studies have been undertaken.5,40–45 The most recent calculations of the vibrational frequencies of CCl2 were performed by Demaison et al.45 who used the CCSD(T) method with a cc-pV5Z basis set and reported a full set of vibrational parameters including anharmonicity constants. Sendt and Bacskay5 examined the photochemically interesting region of the à state and calculated the Renner–Teller intersection to lie near 23 300 cm−1 above the state zero-point level, or 6000 cm−1 above the zero-point energy of the à state. The C–Cl dissociation energy was calculated to be a further 3000 cm−1 above the Renner–Teller intersection.
In this work we start a comprehensive analysis of the Ã- spectroscopy of CCl2. We have measured the LIF spectrum from 18 000 to 24 000 cm−1, which is much further than any previous work. To assign the multitude of vibronic states over this range, we therefore measured 90 different DF spectra. Analysis of the spectra results in new state vibrational frequencies and anharmonicity constants, identification of extensive Fermi resonance in the à state, and measurement of the energy of the Renner–Teller intersection.
Experimental
Chloroform (l, 298 K) was seeded in argon (25 bar) and the mixture flowed to a pulsed nozzle (Precision Instruments) attached to an alumina oven heated to 1500 K.46 The sample then entered the vacuum chamber (pumped to 2 × 10−6 mbar with the nozzle off and 1 × 10−3 mbar with the nozzle on) via a free-jet expansion, cooling the fragments to 5–10 K (based on rotational temperature). The vacuum system comprised a Varian VHS-6 diffusion pump backed by a mechanical pump (Alcatel 2033). The expansion was intersected 8–12 mm downstream by an Excimer-pumped dye laser (Lambda Physik Lextra-200, XeCl at 308 nm and Lambda Physik LPD-3001E). A number of laser dyes (C540A, C503, C480, C460, C450, C440 and Exalite 428, all from Exciton) were used to produce light in the range λ = 410–550 nm. The laser frequency was calibrated using a Coherent Wavemaster wavemeter. All frequencies are reported in vacuum wavenumber and should be accurate to within ±1 cm−1.LIF spectra were recorded by imaging the CCl2 emission with an f = 50 mm quartz lens into a monochromator (Spex Minimate) with no slits (band pass 60 nm). A photomultiplier (EMI 9816QB) connected to the exit slit of the monochromator, detected the fluorescence. The photomultiplier signal was sent to a preamplifier (Stanford Research, SR-240) and then passed to a gated boxcar averager (SR-250). Although CCl2 has a long fluorescence lifetime27,29,30 we were only able to observe a small part of it as the molecules drifted in and out of the viewing region while still fluorescing. For this reason our gate was typically 100–500 ns. The output was passed through an A/D converter (SR-245) before being recorded by a PC using SR-272 software. The timing was controlled a digital delay generator (Stanford Research, DG-535).
DF spectra were measured by replacing the small monochromator with a Spex 0.75 m double monochromator with slits set typically at 1 mm (band pass 18 cm−1). The excitation laser was fixed on a peak from the LIF spectrum and the monochromator was scanned to produce the DF spectra. Only one half of the double monochromator was used. The large monochromator was calibrated with the emission lines from a mercury lamp. Data collection and processing remained the same as for the LIF spectrum.
Results and analysis
Overview of the LIF spectrum
The à ← fluorescence excitation spectrum of jet-cooled CCl2, spanning 6500 cm−1, is shown in Fig. 1. The peak intensities have been normalised to laser power, and normalised between different laser dyes; however, because seven laser dyes were used, the intensities should be considered semi-quantitative. Our spectrum does not extend further to the red than 17 700 cm−1, corresponding to the region of the 220 transition, because the Franck–Condon intensity is decreasing strongly and glow from the pyrolysis nozzle interfered with our detection. The two non-contiguous parts of the excitation spectrum at the lowest frequency could only be obtained by measuring the fluorescence through the larger 0.75 m monochromator, which was set 335 cm−1
(=
ν″2) below the laser frequency and scanned at the same rate as the laser. This provided effective discrimination against the nozzle glow, but as a consequence, intensities are less reliable than the rest of the spectrum.
 | ||
| Fig. 1 LIF spectrum of CCl2. The spectrum has been divided into three Regions (see text). Region 1: vibrational assignments are straightforward, and are shown below the spectrum; Region 2: emission is characteristic of mixed vibrational states where simple assignment is not possible; Region 3: a significant change in structure indicates that the Renner–Teller (RT) energy has been exceeded. The inferred energy of the RT intersection is indicated. | ||
We have divided the spectrum into three regions for reasons that will become apparent. In essence, Region 1 comprises all of the previous assignments in the à state (except for Gao et al., which we shall comment further upon later). In all cases in Region 1 we are able to make secure rotational, vibrational and isotopic assignments through analysis of the DF spectra and modelling of the LIF contours. Vibrational assignments are shown below the spectrum in Fig. 1 and reported in Table 1.
| Frequency/cm−1 | Frequency/cm−1 | A rotational constant/cm−1 | |
|---|---|---|---|
| Assignment | C35Cl2 | C35Cl37Cl | C35Cl2 |
| a These levels show evidence of mixing, however their vibronic character is mostly as assigned. b These levels are not assigned, the levels map to a mixture of the available assignments. | |||
| (0,0,0) | |||
| (0,1,0) | |||
| (0,2,0) | 17 861.25 | 17 853.25 | 3.8 |
| (1,0,0) | |||
| (0,3,0) | 18 164.4 | 18 155.1 | 4.0 |
| (1,1,0) | 18 187.8 | 3.9 | |
| (0,4,0) | 18 467 | 18 455 | 4.1 |
| (1,2,0) | 18 488.3 | 18 478.4 | 3.95 |
| (2,0,0) | 18 510 | 18 502.2 | 3.8 |
| (0,5,0) | 18 770.1 | 18 755.7 | 4.3 |
| (1,3,0) | 18 789.2 | 18 776.3 | 4.1 |
| (2,1,0) | 18 808.7 | 18 797.8 | 3.95 |
| (0,6,0) | 19 072.6 | 19 056.1 | 4.55 |
| (1,4,0) | 19 090.9 | 19 074.8 | 4.25 |
| (2,2,0) | 19 108.1 | 19 094.1 | 4.1 |
| (3,0,0) | |||
| (0,7,0) | |||
| (1,5,0) | 19 392.5 | 19 373.7 | 4.43 |
| (2,3,0) | 19 408.0 | 19 390 | 4.2 |
| (3,1,0) | 19 422.5 | 19 403.1 | 4.1 |
| (0,8,0) | |||
| (1,6,0) | 19 693.6 | 19 672.4 | 4.65 |
| (2,4,0) | 19 708.1 | 19 687.6 | 4.35 |
| (3,2,0) | 19 720.9 | 19 696.94 | 4.2 |
| (4,0,0) | 19 750.1 | ||
| (0,9,0)a | |||
| (1,7,0)a | 19 997.8 | 19 972.7 | 4.75 |
| (2,5,0)a | 20 008.3 | 19 985 | 4.55 |
| (3,3,0)a | 20 020.1 | 19 997.1 | 4.35 |
| (4,1,0)a | 20 030.2 | 4.2 | |
| (0,10,0)a | (20 307.4) | ||
| (1,8,0)a | (20 301.0) | 5.1 | |
| (2,6,0)a | 20 282.2 | 4.5 | |
| (3,4,0)a | 20 327 | 20 293.8 | 4.32 |
| (4,2,0)a | 20 333 | 4.2 | |
| (5,0,0)a | |||
| (1,10,0)b | |||
| (2,8,0)b | |||
| (3,6,0)b | 20 599.5 | 4.5 | |
| (4,4,0)b | 20 608 | 4.3 | |
| (5,2,0)b | 20 627.5 | 4.5 | |
| (6,0,0)b | 20 635.8 | 4.3 | |
In Region 2 the DF spectra from the various à vibronic states are increasingly complex, which indicates anharmonic mixing. Throughout this region we are able to assign the isotope and rotational structure, however conventional vibrational assignments are no longer really meaningful.
The LIF spectrum qualitatively changes in Region 3. The rotational structure cannot be modelled as it was throughout Regions 1 and 2 and we believe that the energy is now above that of the Renner–Teller intersection, where the molecule now has an averaged linear geometry.
The differentiation of the LIF spectrum into these three qualitatively different regions is based on a detailed analysis of 90 DF spectra spanning the entire CCl2 LIF spectrum. We shall discuss general features of DF spectra first, and then return to the assignment of features and regions in the LIF spectrum.
Dispersed fluorescence (DF) spectra
DF spectra were recorded from rovibronic levels over the whole range of the LIF spectrum. For ease of analysis the fluorescence spectra were all calibrated relative to the excitation frequency. In this way the DF spectra provide a “map” of the ground state energy levels with the (0,0,0) level at 0 cm−1. The only exceptions are the spectra featuring peaks at negative relative wavenumbers, which are easily assigned as originating from hot bands.Each spectrum provides the same information about the state vibrational frequencies. Much of this information was published recently in a detailed analysis of the DF spectrum of CCl2 by Liu et al.37 In that work, many vibrational levels up to 6000 cm−1 were assigned, resulting in harmonic and anharmonic constants that provided an excellent model of the observed frequencies. Our analysis is quantitatively similar to this previous work and so we shall be brief in this part of our analysis, and will concentrate on features that are different.
Fig. 2 shows a DF spectrum following excitation at 20025.14 cm−1, at the top end of Region 1. This wavenumber corresponds to overlapped transitions in the rotational contours of the 130230 and 120250 bands. The resolution in the spectrum is 18 cm−1 FWHM. The peak at 0 cm−1 with the asterisk corresponds to the excitation frequency. The intensity of this peak contains a contribution due to scattered light and its intensity is not indicative of its relative transition strength. All DF spectra were corrected for the wavelength dependent sensitivity of the PMT, which was obtained from the manufacturer.
 | ||
| Fig. 2 Dispersed fluorescence spectrum of CCl2. The excitation wavenumber is 20 025.14 cm−1. Assignments for the long progressions in ν1 and ν2 are shown underneath. Weak emission assigned as due to ν3 is shown expanded in the inset. | ||
As described by several previous authors,27,37 the spectrum is dominated by long progressions in the symmetric stretch (ν1) and the bend (ν2). Assignments for almost every vibrational level up to 5000 cm−1 are shown below the spectrum and reported in Table 2 and are quantitatively consistent with assignments by Liu et al.37 The near equivalence of ν1 and 2ν2 means that the peaks in the spectrum clump together to form polyads. When assignments involving all possible combinations of ν1 and ν2 had been exhausted there were a few bands that remained unassigned. These have been assigned to transitions involving levels containing two quanta of ν3, the asymmetric stretch. (Transitions involving odd quantum changes in ν3 are symmetry-forbidden.) The inset in Fig. 2 shows two of these peaks in greater detail.
state of CCl2 for two isotopomers and comparison with model frequencies
| C35Cl2 frequency/cm−1 | C35Cl37Cl frequency/cm−1 | ||||||
|---|---|---|---|---|---|---|---|
| Assignment | Expt. | Model | Diff. | Expt. | Model | Diff. | |
| 2n | 00 | 0 | 0 | 0 | 0 | −1 | −1 |
| progression | 21 | 335 | 335 | 0 | 331 | 331 | 0 |
| 22 | 669 | 669 | 0 | 661 | 662 | 1 | |
| 23 | 1003 | 1003 | 0 | 992 | 992 | 0 | |
| 24 | 1337 | 1336 | −1 | 1322 | 1321 | −1 | |
| 25 | 1668 | 1669 | 1 | 1647 | 1650 | 3 | |
| 26 | 2002 | 2001 | −1 | 1981 | 1977 | −4 | |
| 27 | 2333 | 2332 | −1 | 2307 | 2304 | −3 | |
| 28 | 2663 | 2663 | 0 | 2629 | 2630 | 1 | |
| 29 | 2993 | 2993 | 0 | 2954 | 2954 | 0 | |
| 210 | 3323 | 3323 | 0 | 3278 | 3278 | 0 | |
| 211 | 3649 | 3652 | 3 | 3600 | 3601 | 1 | |
| 212 | 3979 | 3980 | 1 | 3920 | 3923 | 3 | |
| 213 | 4308 | 4308 | 0 | ||||
| 214 | 4633 | 4636 | 3 | ||||
| 112n | 1120 | 727 | 726 | −1 | 723 | 723 | 0 |
| progression | 1121 | 1059 | 1059 | 0 | 1053 | 1053 | 0 |
| 1122 | 1391 | 1391 | 0 | 1381 | 1382 | 1 | |
| 1123 | 1723 | 1723 | 0 | 1710 | 1710 | 0 | |
| 1124 | 2053 | 2055 | 2 | 2039 | 2037 | −2 | |
| 1125 | 2386 | 2386 | 0 | 2365 | 2364 | −1 | |
| 1126 | 2717 | 2716 | −1 | 2688 | 2689 | 1 | |
| 1127 | 3046 | 3045 | −1 | 3010 | 3014 | 4 | |
| 1128 | 3375 | 3374 | −1 | 3342 | 3337 | −5 | |
| 1129 | 3703 | 3703 | 0 | 3665 | 3660 | −5 | |
| 11210 | 4032 | 4031 | −1 | ||||
| 11211 | 4360 | 4358 | −2 | ||||
| 11212 | 4684 | 4685 | 1 | ||||
| 11213 | 5014 | 5011 | −3 | ||||
| 11214 | 5337 | 5336 | −1 | ||||
| 122n | 1220 | 1445 | 1445 | 0 | 1441 | 1440 | −1 |
| progression | 1221 | 1775 | 1777 | 2 | 1767 | 1768 | 1 |
| 1222 | 2106 | 2107 | 1 | 2095 | 2095 | 0 | |
| 1223 | 2437 | 2438 | 1 | 2417 | 2421 | 4 | |
| 1224 | 2766 | 2767 | 1 | 2743 | 2746 | 3 | |
| 1225 | 3097 | 3096 | −1 | 3065 | 3070 | 5 | |
| 1226 | 3425 | 3424 | −1 | 3395 | 3394 | −1 | |
| 1227 | 3752 | 3752 | 0 | 3716 | 3716 | 0 | |
| 132n | 1320 | 2159 | 2159 | 0 | 2153 | 2150 | −3 |
| progression | 1321 | 2489 | 2488 | −1 | 2473 | 2476 | 3 |
| 1322 | 2817 | 2817 | 0 | 2799 | 2801 | 2 | |
| 1323 | 3145 | 3145 | 0 | 3121 | 3125 | 4 | |
| 1324 | 3474 | 3473 | −1 | 3451 | 3448 | −3 | |
| 1325 | 3802 | 3800 | −2 | 3776 | 3770 | −6 | |
| 1326 | 4128 | 4127 | −1 | ||||
| 1327 | 4454 | 4453 | −1 | ||||
| 142n | 1420 | 2866 | 2865 | −1 | 2858 | 2853 | −5 |
| progression | 1421 | 3194 | 3193 | −1 | 3179 | 3177 | −2 |
| 1422 | 3521 | 3520 | −1 | 3497 | 3500 | 3 | |
| 1423 | 3849 | 3847 | −2 | 3821 | 3822 | 1 | |
| 1424 | 4171 | 4173 | 2 | ||||
| 1426 | 4826 | 4823 | −3 | ||||
| 152n | 1520 | 3567 | 3566 | −1 | 3549 | 3550 | 1 |
| progression | 1521 | 3891 | 3892 | 1 | 3870 | 3871 | 1 |
| 1522 | 4214 | 4217 | 3 | ||||
| 1523 | 4541 | 4542 | 1 | ||||
| 1524 | 4862 | 4866 | 4 | ||||
| 1525 | 5187 | 5190 | 3 | ||||
| 162n | 1620 | 4260 | 4260 | 0 | |||
| progression | 1621 | 4584 | 4584 | 0 | |||
| 1622 | 4908 | 4908 | 0 | ||||
| 1623 | 5228 | 5231 | 3 | ||||
| 172n | 1720 | 4950 | 4948 | −2 | |||
| progression | 1721 | 5272 | 5270 | −2 | |||
| 2n32 | 2032 | 1509 | 1511 | 2 | 1491 | 1494 | 3 |
| progression | 2132 | 1839 | 1838 | −1 | 1820 | 1817 | −3 |
| 112n32 | 112032 | 2227 | 2225 | −2 | 2214 | 2211 | −3 |
| progression | 112132 | 2549 | 2551 | 2 | 2530 | 2532 | 2 |
We have assigned 47 vibrational frequencies of the C35Cl37Cl isotopomer. Twelve of these were also reported by Liu et al.37 and agree quantitatively.
The DF spectra also provide a positive assignment of the Cl isotopic composition of the emitting species because the frequencies are shifted according to the isotope effect. We attributed 63 of our DF spectra to emission from C35Cl2 while the remaining 27 were attributed to C35Cl37Cl. These correspond to distinct vibrational levels; however, due to the extensive mixing in Regions 2 and 3, they could not all be uniquely assigned and not all have been included in Table 1. No fluorescence spectra from transitions involving the C37Cl2 isotopomer were recorded.
When measured at higher resolution, the Ka value of the emitting state can also be determined from the DF spectrum. The upper spectrum in Fig. 3 is an LIF spectrum of one of the polyads in Region 1. High resolution (6 cm−1 FWHM) DF spectra were measured after exciting the three transitions indicated on the LIF spectrum. A short section of each of these DF spectra is shown below. Each DF spectrum shows a single vibrational transition. The upper DF spectrum is characterised by emission from K′a = 1 and shows a splitting due to the ΔK = ±1 selection rule. This splitting is numerically approximately equal to 4A″. Emission from levels with K′a = 2 and 3, with splittings of about 8A″ and 12A″, respectively, are shown below. This information is not very useful for measuring ground state rotational parameters (these constants are already known to much higher precision32,47), but it is invaluable for identifying the emitting state. The assignment of the K-state of the emitting level is an important check for the assignment of the LIF spectrum, such as shown in the top panel. This information is even more valuable higher up the vibrational ladder where assignments become more difficult.
 | ||
| Fig. 3 High resolution dispersed fluorescence spectra taken after exciting the three features indicated in the LIF spectrum at the top. The structure in each DF spectrum arises from a single vibrational transition. The splitting arises from excitation of different K′a states with and the subsequent ΔK = ±1 selection rule in emission. | ||
 | (1) |
| Current | Previous | |||||
|---|---|---|---|---|---|---|
| C35Cl2 | Experimentalb | Theoryc | ||||
| Parameter/cm−1 | x ″33 = 0 | Theory x″33a | C35Cl37Cl | C35Cl2 | C35Cl37Cl | C35Cl2 |
| a Our data was refit with x″33 set to the value calculated by Demaison et al. (ref. 45). b These values are derived from ref. 37, refit to eqn. (1). c Ref. 45. | ||||||
| ν ″1 | 726 ± 1 | 726 ± 1 | 724 ± 2 | 726 | 722 | 727.4 |
| ν ″2 | 335 ± 1 | 335 ± 1 | 332 ± 1 | 334 | 329 | 332.6 |
| ν ″3 | 759.5 ± 1.2 | 763 ± 1 | 751.4 ± 2.5 | — | — | 748.9 |
| ω ″1 | 735.7 ± 0.8 | 736.1 ± 0.7 | 733.3 ± 2.2 | 736.45 | 730.20 | 735.3 |
| ω ″2 | 338.2 ± 0.5 | 338.5 ± 0.5 | 336.0 ± 1.2 | 337.74 | 334.40 | 335.9 |
| ω ″3 | — | 783.9 ± 1.2 | — | — | — | 769.6 |
| x ″11 | −3.18 ± 0.06 | −3.20 ± 0.06 | −3.4 ± 0.3 | −3.385 | −2.545 | −2.24 |
| x ″22 | −0.29 ± 0.02 | −0.29 ± 0.02 | −0.45 ± 0.05 | −0.3303 | −0.5066 | −0.18 |
| x ″33 | ≡0 | ≡−7.89 | ≡0 | — | — | −7.89 |
| x ″12 | −1.79 ± 0.05 | −1.81 ± 0.06 | −2.0 ± 0.2 | −1.959 | −2.095 | −1.63 |
| x ″13 | −5.15 ± 0.8 | −5.55 ± 0.7 | −3.4 ± 1.5 | — | — | −5.49 |
| x ″23 | −3.8 ± 0.7 | −4.1 ± 0.8 | −4.4 ± 1.5 | — | — | −4.36 |
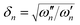 , where the ωn are the harmonic frequencies for each mode, n and Dn, which is a dimensionless parameter measuring the displacement of the normal coordinate upon electronic excitation.49 As ω″ and ω′ are known for ν1 and ν2 the δn parameter is fixed for these modes, while D1 and D2 are treated as adjustable parameters.
, where the ωn are the harmonic frequencies for each mode, n and Dn, which is a dimensionless parameter measuring the displacement of the normal coordinate upon electronic excitation.49 As ω″ and ω′ are known for ν1 and ν2 the δn parameter is fixed for these modes, while D1 and D2 are treated as adjustable parameters.
We found that the intensities of all transitions for DF spectra originating in à state levels below 19 800 cm−1
(i.e. Region 1) can be fit by just these two parameters, with optimal values being D1
= 2.4 ± 0.1 and D2
= 3.6 ± 0.1. Fig. 4 shows the experimental and model results, using these values of Dn. The error bars on the experimental intensities are 1σ from all available experimental data. Where there are no error bars, only a single measurement was available; however, errors of 10–20% are appropriate. The agreement between the model and experimental intensities is quite satisfactory. In general, values of D within ±0.1 of the values reported above also provided reasonable agreement with the experimental intensities. One important consequence of the agreement between the model and observed intensities is that although there is a near match between 2ν″2 and ν″1
(which results in polyads all the way up the state vibrational ladder), there is not any substantial anharmonic mixing between these states.
 | ||
| Fig. 4 Measured and calculated Franck–Condon (FC) factors. The measured FC factors are the average from many spectra and progressions (error = 1σ). The calculated FC factors are based on harmonic oscillator wavefunctions. | ||
A. Detailed picture of the LIF spectrum and à state vibronic Structure
Above, we presented an overview of the LIF spectrum, which was divided into three Regions. We now return to the LIF spectrum armed with detailed knowledge of the emitting state that analysis of the DF spectra provides. This information includes the Cl isotope distribution of the emitting species, the value of K′a, the vibrational character of the emitting state (leading to a unique assignment in many cases), and whether the transition in the spectrum is a hot or cold band. | ||
| Fig. 5 Expanded region of the LIF spectrum from Regions 1, 2 and 3. Simulations underneath the Region 1 spectrum show four members of a polyad, each with three isotopomers contributing to the spectrum. The simulation for a single vibronic band is shown underneath. The spectrum from Region 2 can likewise be identified and model as a set of overlapping vibronic transitions (one shown underneath), however the assignment of the constituent bands is complex. The rotational structure is completely changed in Region 3, indicating that the Renner–Teller intersection has been exceeded (see text). | ||
The variance from the harmonic FC intensities increases for DF spectra obtained from higher vibrational states. Even one rung higher in the ladder, near 20 300 cm−1, where absorption to the set of states (5,0,0) : (4,2,0) : (3,4,0) : (2,6,0) : (1,8,0) : (0,10,0) would be expected to occur, the set of DF spectra reveal extensive mixing. This is the highest that Clouthier et al., and Liu et al. have assigned. DF spectra from the next polyad are more complex again and are not readily identifiable as any simple harmonic vibrational assignment. In this region of the vibrational ladder the combination-differences no longer follow the same simple pattern as they do lower down. While the polyads appear regular throughout this region, the DF spectra demonstrate conclusively that anharmonic mixing is significant above about 20 000 cm−1, and that this mixing is what is responsible for the misbehaviour of the combination-differences above about 20 000 cm−1. We are currently evaluating the level mixing higher up the ladder in CCl2 and will report on this later.
Throughout Region 2, the rotational structure of the vibronic bands is preserved. A higher resolution scan from a polyad near 21 250 cm−1, in the middle of Region 2, is shown in Fig. 5. While the vibronic complexity increases throughout this Region, and we cannot offer a simple vibrational assignment for this polyad, or any polyad beyond 20 000 cm−1, the individual vibronic bands can be clearly identified. The DF spectra continue to indicate the value of K′a in the emitting state and the chlorine isotope distribution of the emitting species.
and à states, where the à state is strongly coupled to the state. The RT interaction depends strongly on Ka. States with Ka > 0 have increasing state character and hence a weaker transition moment in absorption and fluorescence and a much longer fluorescence lifetime. In our experiment where we integrate only the first couple of hundred nanoseconds of fluorescence, we are biased significantly against the detection of such weak, long-lived states.
The A′ rotational constants for each vibronic level of the C35Cl2 isotopomer are also listed in Table 1. The constants generally increase with increasing quanta of the bending vibration, ν2, and to a lesser extent the symmetric stretch, ν1. This is characteristic of the average bond angle increasing for higher ν2 and consistent with the presence of the RT intersection near 22 600 cm−1. This trend in A′ constants becomes obscured for the highest levels in the table, as the vibronic mixing becomes more prevalent.
Discussion
In this work we have measured the LIF spectrum and many DF spectra of jet-cooled CCl2. The DF spectra reveal a wealth of detail about the emitting state, as well as vibrational frequencies in the ground electronic state. There have been many previous experimental and theoretical investigations of this prototypical carbene, so we begin our discussion with a comparison and extension of the previous work, then consider the unanswered questions posed by this work.Comparison with previous ground (![[X with combining tilde]](https://www.rsc.org/images/entities/h3_char_0058_0303.gif) ) state studies
) state studies
In some regards the experiment and results reported here mirror those published recently by Liu et al.37 and it is appropriate to comment on the differences between our measurements. Liu et al. measured DF spectra from a small number of initial à states, with similar resolution to ours. They measured a larger number of vibrational levels, importantly many higher lying vibrational states, and identified a region where state mixing seems to indicate the presence of the triplet state near these energies. We were unable to measure this region, due to interference by the glow from the pyrolysis nozzle.
In the present study, we have excited many more initial à vibronic states with the initial aim of assigning the emitting state. The frequencies in our DF spectra agree quantitatively (typically within 1 cm−1) with those published by Liu et al. We have extended the assignments of the state vibrational levels in two important ways. First, we have assigned emission to levels involving ν″3, allowing the first measurement of the asymmetric stretch frequency in the gas phase. Second, we have measured significantly more levels in the C35Cl37Cl isotopomer than previously. The addition of ν3 to the list of assigned levels changes the vibrational constants somewhat. These constants are summarised in Table 3.
Liu et al. fit their data to a slightly different form of the anharmonic equation:
 | (2) |
 | (3) |
There is a relatively good agreement between the current spectroscopic constants and those reported previously. Because we have measured levels involving ν3 we were able to extract x13 and x23. The inclusion of these two anharmonicity constants has an effect on the rest of the constants, which accounts for the small differences between our results and other gas phase measurements.
A comparison of the current spectroscopic constants with the most recent ab initio calculations44 shows quite extraordinary agreement. There has been no experimental measurement of x33 which, according to calculation, is quite large at x33 = −7.89 cm−1. If this one theoretical value is used, and the rest of the constants re-fit according to eqn. (1), a new set of “experimental” constants are obtained as shown in Table 3. The effect on most of the other constants was negligible but it allows an estimate of the previously intractable ω″3, which becomes 784 cm−1, and is now in reasonable agreement with the calculated value of 769.9 cm−1. The agreement between the experimental and ab initio values is extraordinarily good and we believe that these constants are a good representation of the vibrational level structure of all levels in the ground state up to about 5000 cm−1.
Comparison with previous excited (Ã) state studies
There have been several previous investigations of the CCl2 absorption and fluorescence excitation spectrum.27,31–36 The most extensive report was that of Clouthier et al., who assigned almost every vibrational level up to 20 300 cm−1 (i.e. Region 1). This is the region where the DF spectra could be interpreted by harmonic Franck–Condon factors, and where analysis of the intensities provides an unambiguous assignment of the emitting state. Our DF spectra confirm all assignments of Clouthier, and have provided several new measurements, particularly for C35Cl37Cl.Gao et al. assigned the LIF spectrum to significantly higher energy than Clouthier (Region 2), assigning up to 16 quanta of bending vibration and 8 quanta of symmetric stretch. The analysis of our DF spectra does not support that assignment. Above 20 300 cm−1 assignment of the vibronic state becomes more and more complex. The vibrations no longer behave as overtones and combinations of normal modes and the DF spectra show clear character of emission from mixed states. One picture is that the vibrations are increasingly anharmonically mixed due to the fairly close resonance between 2ν′2 ≈ ν′1, which has been suggested by previous workers.33,37 We are exploring and modelling this interaction further.
There have been no assignments for the LIF spectrum throughout Region 3. Indeed the whole nature of the spectrum has changed from the Region below. We have recorded about 30 DF spectra throughout this region. Every emitting state was shown to be due to CCl2 (by the characteristic frequencies) and we have no reason to believe that any other species contributes to the spectrum in Fig. 1. The spectrum still shows what looks like polyad structure, though the polyads now have so many members that the spread of frequencies within a polyad now matches the spacing between them.
The most characteristic observation about this region of the LIF spectrum, however, is that every emitting state appears to have K′ = 0. We believe that this indicates that the Renner–Teller intersection has been exceeded. There has been an ab initio theoretical estimate of several key energy points on the à state surface.5 The energy to C–Cl bond cleavage on this surface was calculated to be 26 681 cm−1, which is above the limit of the spectrum in Fig. 1, and therefore probably not optically accessible due to poor Franck–Condon overlap. The energy of the Renner–Teller intersection, however was calculated to lie at 23 000 cm−1, in remarkable agreement with the boundary between Regions 2 and 3.
Conclusions
We have measured the Ö LIF excitation spectrum of jet-cooled CCl2 from 17700–24000 cm−1. Ninety dispersed fluorescence spectra were measured within this range. Analysis of the frequencies in the DF spectra have provided assignments of 68 vibrational levels in C35Cl2 and 47 in C35Cl37Cl, including the first gas phase measurement of ν″3
= 760 cm−1. A full set of anharmonic constants was extracted for both isotopomers.
Analysis of the intensities in the DF spectra provided identification or characterisation of the emitting à vibronic state. Levels below about 20 000 cm−1
(T00
+ 2300 cm−1) can be assigned simply as combinations of ν′1 and ν′2. Excitation of levels between 20 000 and 22 600 cm−1 result in DF spectra that cannot be assigned in the Franck–Condon limit and show evidence of increasing Fermi resonance. Levels above 22 600 cm−1 show a collapsed rotational structure in the LIF spectrum, with only transitions terminating in K′a
= 0 rotational states evident in the spectrum. This places the energy of the Renner–Teller intersection between the and à states at about 22 600 cm−1, in excellent agreement with a previous theoretical calculation of 23 000 cm−1.
Acknowledgements
This research was supported by an Australian Research Council Discovery grant. CAR and JSG gratefully acknowledge PhD stipends from an Australian Post-graduate Award and a University of Sydney, School of Chemistry Research Scholarship, respectively. KN acknowledges an ARC Post-doctoral Fellowship. All of us thank Dr George Bacskay and Dr Timothy Schmidt for stimulating discussions about various aspects of this work.References
- T. J. Sears, P. R. Bunker and A. R. W. McKellar, J. Chem. Phys., 1982, 77, 5363 CrossRef CAS.
- W. H. Green Jr., N. C. Handy, P. J. Knowles and S. Carter, J. Chem. Phys., 1991, 94, 118 CrossRef CAS.
- K. K. Murray, D. G. Leopold, T. M. Miller and W. C. Lineberger, J. Chem. Phys., 1988, 89, 5442 CrossRef CAS.
- S. Koda, Chem. Phys. Lett., 1978, 55, 353 CrossRef CAS.
- K. Sendt and G. B. Bacskay, J. Chem. Phys., 2000, 112, 2227 CrossRef CAS.
- D. S. King, P. K. Schenck and J. C. Stephenson, J. Mol. Spectrosc., 1979, 78, 1 CrossRef CAS.
- C. W. Mathews, Can. J. Phys., 1967, 45, 2355 CAS.
- K. Sendt, E. Ikeda, G. B. Bacskay and J. C. Mackie, J. Phys. Chem. A, 1999, 103, 1054 CrossRef CAS.
- M. Martoprawiro, G. B. Bacskay and J. C. Mackie, J. Phys. Chem., 1999, 103, 3923 Search PubMed.
- R. E. Rebbert and P. J. Ausloos, J. Photochem., 1975, 4, 419 CAS.
- M. J. Molina and F. S. Rowland, Nature, 1974, 249, 810 CAS.
- W. Hack, M. Wagner and K. Hoyermann, J. Phys. Chem., 1995, 99, 10847 CrossRef CAS.
- B. T. Beiderhase, K. Hoyermann and W. Hack, Z. Phys. Chem., 2000, 214, 95 Search PubMed.
- W. Hack, B. Wagner and K. Hoyermann, Z. Phys. Chem., 2000, 214, 741 Search PubMed.
- T. W. Schmidt, G. B. Bacskay and S. H. Kable, Chem. Phys. Lett., 1998, 292, 80 CrossRef CAS.
- T. W. Schmidt, G. B. Backsay and S. H. Kable, J. Chem. Phys., 1999, 110, 11277 CrossRef CAS.
- B.-C. Chang, M. L. Costen, A. J. Marr, G. Ritchie, G. E. Hall and T. J. Sears, J. Mol. Spec., 2000, 202, 131 CrossRef CAS.
- H.-G. Yu, J. T. Muckerman and T. J. Sears, J. Chem. Phys., 2002, 116, 1435 CrossRef CAS.
- P. T. Knepp and S. H. Kable, J. Chem. Phys., 1999, 110, 11789 CrossRef CAS.
- J. S. Guss, O. Votava and S. H. Kable, J. Chem. Phys., 2001, 115, 11118 CrossRef CAS.
- D. E. Milligan and M. E. Jacox, J. Chem. Phys., 1967, 47, 703 CrossRef CAS.
- L. Andrews, J. Chem. Phys., 1968, 48, 979 CrossRef CAS.
- M. E. Jacox and D. E. Milligan, J. Chem. Phys., 1970, 53, 2688 CrossRef CAS.
- H. Fan, I. Ionescu, C. Annesley, J. Cummins, M. Bowers, J. Xin and S. A. Reid, Chem. Phys. Lett., 2004, 378, 548 CrossRef.
- A. K. Maltsev, O. M. Nefedov, R. H. Hauge, J. L. Margrave and D. Seyferth, J. Phys. Chem., 1971, 75, 3984 CrossRef CAS.
- D. E. Tevault and L. Andrews, J. Mol. Spec., 1975, 54, 110 CrossRef CAS.
- V. E. Bondybey, J. Mol. Spectrosc., 1977, 64, 180 CrossRef.
- J. S. Shirk, J. Chem. Phys., 1971, 55, 3608 CrossRef CAS.
- R. E. Huie, N. J. T. Long and B. A. Thrush, Chem. Phys. Lett., 1977, 51, 197 CrossRef CAS.
- J. J. Tiee, F. B. Wampler and W. W. Rice, Chem. Phys. Lett., 1979, 65, 425 CrossRef CAS.
- D. A. Predmore, A. M. Murray and M. D. Harmony, Chem. Phys. Lett., 1984, 110, 173 CrossRef CAS.
- J.-I. Choe, S. R. Tanner and M. D. Harmony, J. Mol. Spectrosc., 1989, 138, 319 CrossRef CAS.
- D. J. Clouthier and J. Karloczak, J. Phys. Chem., 1989, 93, 7542 CrossRef CAS.
- Q. Lu, Y. Chen, D. Wang, Y. Zhang, S. Yu, C. Chen, M. Koshi, H. Matsui, S. Koda and X. Ma, Chem. Phys. Lett., 1991, 178, 517 CrossRef CAS.
- D. J. Clouthier and J. Karolczak, J. Chem. Phys., 1991, 94, 1 CrossRef CAS.
- Y.-D. Gao, C.-J. Hu, R. Qin, C. Yang and C.-X. Chen, Acta Phys.-Chim. Sin., 2002, 18, 112 Search PubMed.
- M. L. Liu, C. L. Lee, A. Bezant, G. Tarczay, R. J. Clark, T. A. Miller and B. C. Chang, Phys. Chem. Chem. Phys., 2003, 5, 1352 RSC.
- C. W. Bauschlicher Jr., H. F. Schaefer III and P. S. Bagus, J. Am. Chem. Soc., 1977, 99, 7106 CrossRef.
- M. T. Nguyen, M. C. Kerins, F. Hegarty and N. J. Fitzpatrick, Chem. Phys. Lett., 1985, 117, 295 CrossRef.
- G. L. Gutsev and T. Ziegler, J. Phys. Chem., 1991, 95, 7220 CrossRef CAS.
- K. K. Irikura, W. A. Goddard III and J. L. Beauchamp, J. Am. Chem. Soc., 1992, 114, 48 CrossRef CAS.
- N. Russo, E. Sicillia and M. Toscano, J. Chem. Phys., 1992, 97, 5031 CrossRef CAS.
- Z.-L. Cai, X.-G. Zhang and X.-Y. Way, Chem. Phys. Lett., 1993, 210, 481 CrossRef CAS.
- M. Born, S. Ingemann and N. M. M. Nibbering, J. Am. Chem. Soc., 1994, 116, 7210 CrossRef CAS.
- J. Demaison, L. Margules, J. M. L. Martin and J. E. Boggs, Phys. Chem. Chem. Phys., 2002, 4, 3282 RSC.
- M. R. Cameron and S. H. Kable, Rev. Sci. Instrum., 1996, 67, 283 CrossRef.
- H. E. Fujitake M., J. Chem. Phys., 1989, 91, 3426 CrossRef CAS.
- G. Herzberg, Molecular Spectra and Molecular Structure III. Electronic spectra of polyatomic molecules, Van Nostrand, New York, 1966, vol. 3 Search PubMed.
- J. R. Henderson, M. Muramoto and R. A. Willett, J. Chem. Phys., 1964, 41, 580 CAS.
| This journal is © the Owner Societies 2005 |