FT-IR study of CO adsorption on Pt/CeO2: characterisation and structural rearrangement of small Pt particles
P.
Bazin
*a,
O.
Saur
a,
J. C.
Lavalley
a,
M.
Daturi
a and
G.
Blanchard
b
aLaboratoire Catalyse et Spectrochimie, UMR 6506, ENSICAEN-Université, 6, Boulevard du Maréchal Juin, 14050, CAEN-Cédex, France. E-mail: philippe.bazin@ensicaen.fr; Fax: +332 31 45 28 22
bRhodia, 52 rue de La Haie-Coq, 93308, Aubervilliers, France
First published on 10th November 2004
Abstract
CO adsorption has been followed by IR spectroscopy on a Pt/CeO2 sample (Pt loading = 0.5%) treated under oxygen and vacuum or reduced by H2 and then evacuated at various temperatures. The sample contains highly dispersed Pt. Attention is paid to the presence of an unusual ν(CO) band at 1937 cm−1 on the reduced sample. Such a band is in particular not observed when the support is fully covered by CO (CO adsorption at liquid nitrogen temperature) or by methanol, allowing one to assign it to CO bridged species bound to both Pt very lowly coordinated and to the support, e.g. to sites at the periphery of very small Pt particules. Experiments performed after sample reduction at 423 K followed by increasing evacuation temperature between 423 and 673 K showed that the increase of the latter provokes a sintering of the Pt particles, due to ceria surface O2− mobility.
1 Introduction
Platinum (Pt) and cerium oxide (ceria) are two standard components in automobile three way catalysts. Ceria, one of the main components of the waste system control support, is mainly used for its oxygen storage capacity (OSC) due to the ability of CeO2 to be easily and reversibly reduced.1,2 Pt is used for the oxidation of CO and hydrocarbons3 and for NO conversion.4 Platinum supported on ceria-based oxides also presents interesting properties in hydrocarbon reforming for hydrogen production,5–8 as well as in different applications in fine chemistry.9,10 Therefore, being Pt/CeO2 a versatile and powerful system for different catalytic applications, it is necessary to get a large knowledge of its physical properties in order to understand and to improve its performances. In particular, the evaluation of the dispersion and the particle size of Pt metal are very important. These properties are mainly estimated by two ways: physical and chemical methods. The most powerful physical technique is the high resolution transmission electron microscopy (HRTEM) by which several characterisation studies of Pt/CeO2 systems have been done.10–13 They often refer to catalysts with a support of relatively low surface area or previously submitted to high temperature calcination. For high surface area samples, the HRTEM observation becomes more difficult because of the low contrast between the small metal particles and the ceria support, as well as because of the difficulty into localising the particles themselves, so that it is hard to obtain quantitative data on metal dispersion.14 Chemical methods are based on absorption or adsorption of probe molecules. In presence of supports as ceria or ceria–zirconia, probe molecules (such as hydrogen) can be adsorbed (and eventually dissociated) by both the metal and the support at the same time, depending on the conditions of the test. These spillover phenomena prevent the obtention of quantitative dispersion results. In this case, it seems necessary to follow the adsorption and the coordination state of the chemical species chosen to probe the metal phase. For this purpose we have developed a technique allowing to characterise the metal phase state by CO adsorption followed by IR spectroscopy.15 It is noteworthy to remark that our methodology can be applied also to samples containing small amounts of noble metal, as is often the case for real catalytic materials. One of our previous works16 focused on the measure of the Pt dispersion on ceria and mixed ceria–zirconia catalysts by two methods: H2 and CO chemisorption. In this study, we report the analysis (by IR spectroscopy of CO adsorption) of the dispersion and structural modifications of Pt particles on ceria (0.5 Pt wt.%) versus the temperature of the treatment under H2 and the H2 evacuation temperature.The adsorption of carbon monoxide followed by infrared spectroscopy has often been used to characterise supported or unsupported transition metals.17–20 In the case of reduced platinum, the IR spectrum obtained after CO adsorption is generally composed of two ν(CO) absorption regions: the first, between 2090 cm−1 and 2000 cm−1, is assigned to linearly adsorbed CO on a single Pt atom, while the second, situated around 1860–1780 cm−1, characterises the adsorption of CO bridged on two Pt atoms. The intensity of the bridging band is often very weak and, consequently, difficult to be observed: CO adsorption on platinum gives preferentially rise to linear species.17–20 The interaction of the CO molecule with the Pt atom is generally described using the model proposed by Blyholder,21,22 which considers molecular orbitals. The chemical bond between CO and Pt concerns the bonding 5σ and the antibonding 2π* orbitals of CO versus Pt orbitals. The 5σ–Pt interaction is electron-donating towards platinum and it strengths the Pt–C bond. The antibonding 2π*–Pt interaction corresponds to an electron transfer from platinum to an antibonding CO orbital. Therefore, an increase of backdonation (increase of electron density in the 2π* orbital) leads to a weakening of the carbon–oxygen bond in the CO molecule, i.e. to a lowering of the ν(CO) vibration. Therefore the ν(CO) vibration frequency depends on the C–O bond strength, i.e. on the backdonation phenomenon. It is sensitive to:
• The number of platinum atoms linked to CO molecules. Sheppard and Nguyen17 showed that a relationship exists between the geometry of the coordination site (which gives rise to linear, bridged species, etc.) and the vibration frequency of the CO molecule. In fact the higher the order of the site (number of platinum atoms forming the site), the greater is the back-donation phenomenon. This explains why bridged species are observed at lower wavenumbers than linear species.
• The coordinance of the platinum atom on which CO is adsorbed. In a study, Kappers and van der Mass23 have shown that a linear correlation can be found between the CO stretch and the coordinance of the platinum atom linked to CO. The greater the Pt coordination, the higher the ν(CO) vibration frequency. The adsorption sites of platinum crystallites having surface atoms with different coordinations (planes, edges, corners) are characterised by different ν(CO) wavenumbers. Greenler et al.,24 for example, reported three bands at 2081, 2070 and 2063 cm−1, assigned to a linear CO adsorption on platinum atoms constituting planes, edges and corners of Pt crystallites supported on silica. On the other side, studies carried on platinum particles supported on alumina and having different sizes have concluded to an increase of the ν(CO) vibration due to the increase of the particle size.25
• The degree of oxidation of platinum . The CO probe is very sensitive to the oxidation degree of platinum. The more the reduced character the greater is the backdonation, the lower is the ν(CO) wavenumber. Generally, CO linearly adsorbed on Pt0 shows a wavenumber ν(CO) < 2100 cm−1, whereas for Ptδ+ν(CO) > 2100 cm−1.
• CO phase coverage. When the CO coverage level increases, an increase of the ν(CO) frequency is observed. Two effects can be evoked to explain this phenomenon: the static (or chemical effect) and the dynamic effect. The first can be simply modelled considering the metal phase as an electron reservoir; it is easy to conceive that the greater the adsorbed CO molecule number, the lower is the amount of back-donation towards a single molecule, which leads to a strengthening of the CO bond, therefore to an increase of the wavenumber.21,22 The second concerns the coupling between molecular dipoles: the coupling of CO vibrators leads to the transfer of the intensity of low wavenumber vibrations to those of high wavenumber.18–20
• The presence of electrophilic or nucleophilic compounds. The interaction of the metal with a nucleophilic compound increases the electron density of the metal phase, that gives rise to an enhancing of the backdonation degree towards the 2π* CO orbital, thus to a decrease of the ν(CO) frequency. Therefore NH3 (electron-donating) adsorption on a platinum containing surface (where CO was pre-adsorbed) decreases the ν(CO) wavenumber of 25 cm−1.26 On the contrary, adsorption of electrophilic compounds such as H2S,27–31 SO42−32 or O2 (O2−) 33 leads to an increase of the ν(CO) frequency.
Therefore, the study of CO adsorption by FT-IR allows determining the supported platinum oxidation state as well as the morphology of the metal particles.
2 Experimental
2.1 Catalysts
The Pt/CeO2 oxide powder, obtained by a Rhôdia proprietary process, has a specific surface area of 147 m2 g−1. The purity of the sample used was higher than 99.5%. The Pt loading was 0.5% (wt.%) and a hexachloroplatinic solution was used as the precursor for Pt. The OH groups on the surface of cerium oxide were exchanged by PtCl62− ions in water. The material obtained was dried in an oven at 393 K and calcinated in a muffle furnace at 773 K. After several washing treatments, drying and a final calcination, elemental analysis showed that the residual chlorine content in the sample was below the detection limit.2.2 Analysis methods
The powder was pressed and used as wafers of 10 mg cm−2. The pellet was then placed in an IR cell equipped with KBr windows and connected to a vacuum system (pressure P < 10−5 Torr). A pressure gauge (Boc Edwards Barocel® pressure sensor, accuracy = 10−3 mbar) is connected to the top of the IR cell, to measure the gas pressure inside the analysis compartment. Then samples were activated at 723 K under oxygen atmosphere for 2 h in order to eliminate water and the majority of carbonate species and evacuated for 30 min at the same temperature. A static reduction under H2 (13 kPa) was then carried out at various temperatures by sample exposure to H2 for 20 min followed by an evacuation for 20 min, this being repeated three times. Pt/CeO2 samples reduced at 423, 523, 623 and 723 K and then evacuated at 673 K were respectively noted R423E673, R523E673, R623E673 and R723E673. IR spectra were recorded at room temperature with a Nicolet Magna 550 spectrometer (resolution 4 cm−1). In order to decrease the amount of water and carbon dioxide in the infrared beam, the spectrometer and the sample compartment were purged with H2O and CO2 free air produced by a Floxal purge gas generator. The reported spectra are differences between spectra recorded after and before adsorption of CO.2.3 Pt accessibility
After reduction at different temperatures CO has been introduced by small doses. We have assumed that CO was quantitatively adsorbed on metal particles, since no residual gas phase was detected in the IR cell before sample saturation (a highly sensitive pressure gauge was connected to the cell for this purpose and gas phase spectra were recorded as well), nor CO adsorbed species were measured on the support. The total band integrated area has been measured for ν(CO) stretching from 2120 to 1850 cm−1 after each introduction. The amount of Pt accessible to CO has been determined by the intersection of the straight lines joining values resulting from CO adsorption before and after the monolayer achievement, following the method described by Binet et al. on Pd supported compounds15 and recently adapted to CeO2–ZrO2 supported platinum.16 For theses works, the adsorption CO/Pt stoichiometry has been considered to be unity, particularly considering that the percentage of doubly bridged CO is very low, so that we can easily calculate the amount of surface platinum sites equal to the number of adsorbed CO at metal saturation. Results reported in Table 1 show that nearly 60% of Pt is accessible to CO for Tred. ≤ 626 K. Pt not in contact with CO must be either inside the support, either in the bulk or the crystallites. The amount of platinum atom at the surface of crystallites should be at least 60%, which confirms their small size. The decrease in accessibility observed after reduction at 623 K is due to decoration/encapsulation phenomena or to Pt sintering. Nevertheless, after reduction at 723 K, 40% of platinum is still accessible.| Reduction temperature/K | 423 | 523 | 623 | 723 |
| Pt dispersion CO/Pt (%) | 63 | 63 | 58 | 41 |
3 Results and discussion
3.1 ν(CO) band assignment
The FTIR spectrum of CO adsorbed species on platinum strongly depends on the treatment conditions of the catalyst. The Pt/CeO2 sample has been activated under different atmospheres and the results are the following.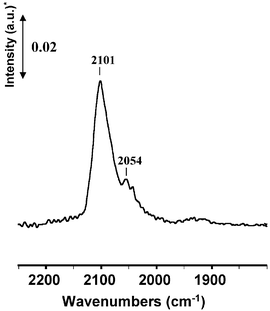 | ||
| Fig. 1 FT-IR spectra recorded after CO adsorption (0.7 kPa) at rt and evacuated at the same temperature on Pt/CeO2 oxidized by O2 at 723 K and then evacuated at 473 K * a.u.: absorbance units. | ||
 | ||
| Fig. 2 FT-IR spectra of species formed after CO adsorption (Pe ∼ 0.7 kPa) at r.t. and then evacuated at the same temperature on Pt/CeO2. Samples have been previously reduced by H2 at the indicated temperatures then evacuated at 673 K (1) sample reduced by H2 at 723 K, oxidised by O2 at 723 K and then reduced at 423 K. | ||
Band at 2096 cm−1. According to the literature results, two interpretations could be proposed for the origin of the band at 2096 cm−1. First, previous studies of CO adsorption on Pt/SiO236 or Pt/Al2O324 reduced samples, with a poor dispersion and then large particles (ca. 4 nm), revealed a ν(CO) band near 2090 cm−1. Therefore, the band located at 2096 cm−1 might be ascribed to the CO adsorption on a Pt0 atom – with an important coordination degree – of large crystallites. Second, the position of the band is similar to that assigned above for the ν(CO) of the CO–O co-adsorption (2101 cm−1), corresponding to an oxidised Ptδ+ atom.
Taking into account the high metal dispersion of the sample used in the present study (Table 1), the presence of large Pt particles is very unlikely. The band at 2096 cm−1, disappearing after reduction at increasing temperature, is therefore assigned to CO adsorption on Ptδ+ atoms resulting from local platinum oxidation at the metal-support interface.
Band at ∼2054 cm−1. The ν(CO) frequency of this band is typically assigned to linearly adsorbed CO on Pt0 atoms.16
Shoulder at ∼2010 cm−1. Since the ν(CO) frequency is sensitive to the Pt coordination and shifts to lower frequencies when the Pt coordination decreases, the shoulder at 2010 cm−1 is probably due to the linearly CO adsorption on a weakly coordinated Pt atom. Two possibilities could be proposed for the origin of such a Pt site. On one hand, this Pt coordination site might be localised onto steps or kinks of small particles (∼1.5 nm) denoted PS. In this case, CO adsorption on terrace sites would give rise to the band at ∼2054 cm−1 assigned above. On the other hand, the band at 2010 cm−1 might correspond to linearly CO species on very small Pt particles (<1.5 nm) denoted PVS, mainly populated by low coordination sites. The intensity of the 2010 cm−1 band is certainly due to the presence of these two types of sites but, according to the results detailed below, the occurrence of the second phenomenon seems to be greater. Therefore, in the present work we consider that the shoulder at 2010 cm−1 is mainly due to the CO adsorption on weakly coordinated Pt atoms of very small PVS particles.
Band at 1937 cm−1. Generally, CO adsorption on Pt does not lead to bands in this spectral region. In fact, linearly adsorption gives rise to bands in the 2100–2000 cm−1 range and the ν(CO) region of bridging CO is located near 1850 cm−1. To determine the nature of the adsorption site corresponding to this new band, we have carried out complementary experiments.
12CO/13CO isotopic exchange. We have reported in Fig. 3, respectively curve a and b, the IR spectra recorded at room temperature after 12CO and 13CO adsorption (Pe ∼ 0.7 kPa) on the Pt/CeO2–R423E673 sample. Main bands are observed at 2096, 2046 and 1937 cm−1 after 12CO introduction, whereas a shift of ca. 50 cm−1 to lower frequencies is observed for all bands using 13CO. In particular, a shift from 1937 cm−1 to 1891 cm−1 is observed for the above-mentioned 1937 cm−1 band. This result confirms that this band is effectively due to a ν(CO) vibration, and not to other features, as forbidden electronic transitions observed in the spectrum of reduced ceria.37
 | ||
| Fig. 3 FT-IR spectra recorded after carbon monoxide adsorption (0.7 kPa) at r.t. on Pt/CeO2–R423E673 followed by evacuation at the same temperature: (a) 12CO and (b) 13CO. | ||
Thermal stability of adsorbed CO species. The thermal stability of carbonyl species (especially those characterised by the band at 1937 cm−1) has been studied, in particular to determine the effect of CO coverage on the different ν(CO) frequencies. Fig. 4 shows IR spectra of Pt/CeO2–R423E673 after CO adsorption at room temperature followed by successive evacuations up to 573 K. After evacuation at 298 K, several components appear at about 2096, 2051, 2014 and 1937 cm−1 (Fig. 4a). By increasing the evacuation temperature, a shift to lower wavenumbers and an overall decrease of the intensity are observed, especially for the 2051 cm−1 band. After desorption at 498 K (spectrum i), bands are located at 2033, 2008 and 1937 cm−1. The 2033 cm−1 band is assigned to CO adsorbed on Pt0 atom belonging to small particles (PS) mainly low coordinated.16,23–25,37–39 The band at 2008 cm−1 is attributed to CO adsorbed on Pt sites of very low coordination23,39 on very small particles (PVS). It is worthy of remark that the higher the wavenumber associated to a species the lower its stability toward evacuation; in fact the carbonyl bands disappear in the order 2096 < 2051 < 2014 < 1937 cm−1. In particular, the band at 1937 cm−1, being at the lowest wavenumber, corresponds to the most stable species; moreover no modification of the frequency of the band at 1937 cm−1 is observed by increasing the evacuation temperature. To explain this fact we should remember that temperature rising leads to CO molecule desorption, which reduces the dipole–dipole coupling between CO adsorbed molecules on neighbouring sites, as observed for the other bands. If the species is isolated, on the contrary, intermolecular coupling does not take place, so no modification of the wavenumber is expected upon desorption. This result implies that the species characterized by the 1937 cm−1 band are not dipole–dipole coupled with neighbouring adsorbed CO molecules; we conclude that this band is due to the adsorption of isolated CO molecules.
 | ||
| Fig. 4 FT-IR spectra recorded after CO adsorption (0.7 kPa) at r.t. on Pt/CeO2–R423E673 followed by evacuation at (a) 298, (b) 323, (c) 348, (d) 373, (e) 398, (f) 423, (g) 448, (h) 473, (i) 498, (j) 523, (k) 548 and (l) 573 K. | ||
Gandao et al.39 have followed by IR spectroscopy the adsorption of CO on a Pt/Mg(Al)O catalyst and they have observed a band in the 1960–1930 cm−1 region. They have tentatively assigned it to CO species bridge-bonded on Pt (through the carbon atom) and Mg2+ cations (via the oxygen atom). They have also shown that these species are the most stable upon desorption at high temperature, but, contrarily to the present results, they observed a red frequency shift by heating under vacuum. We have proposed above that the band at 2010 cm−1 characterises CO molecules adsorbed on Pt atoms of very small particles (PVS), which might be in strong interaction with the support, that could be the case in particular for the molecules at the periphery of the crystallites. Therefore, we propose that the band at 1937 cm−1 is due the adsorption of CO molecules (in an isolated way) on very low coordinated Pt atoms in strong interaction with the support.
Note that an assignment of the 1937 cm−1 band to carbonyl complexes like Ptx(CO)y2− can be discarded though such species give rise to a band near 1940 cm−1.40–42 In fact formation of carbonyl complexes are mainly observed on zeolitic supports and are generally very volatile, in complete disagreement with the present results relative to the thermal stability of these species.
CO adsorption at low temperature (compacity of adsorbed CO molecules). We have performed an experiment where CO (Pe ∼ 0.26 kPa) has been introduced in the IR cell at liquid nitrogen temperature (noted TN2,liq); results are reported in Fig. 5. Spectrum a has been recorded after evacuation at TN2,liq. The intense bands at 2148 cm−1 and 2163 cm−1 are respectively due to physisorbed CO and CO adsorbed on Ce3+ sites, since CO adsorption on Ce4+ sites gives rise to a band at higher wavenumber, the site being more acidic.43 The presence of the natural isotopic molecule 13CO in 12CO (c.a. 1%) gives rise to the bands at 2107 and 2095 cm−1 corresponding to the same species. Bands in the 2100–1900 cm−1 range correspond to CO adsorption on Pt sites. The band at 1937 cm−1 is hardly detectable, while the two bands at 2006 and 1992 cm−1 are more intense than when CO adsorption is performed at r.t.
 | ||
| Fig. 5 FT-IR spectra recorded after CO adsorption (0.26 kPa) at liquid nitrogen temperature (TN2,liq) on Pt/CeO2–R423E673 followed by evacuation at (a) TN2,liq to (e) 298 K. (b, c, d: intermediate temperatures). | ||
Progressive heating under vacuum up to 298 K provokes important spectral evolutions (Fig. 5b–e):
(i) The band due to physisorbed CO progressively vanishes.
(ii) An intensity decrease and a shift is observed for bands due to CO adsorption on Ce3+ sites (2163 cm−1 at TN2,liq, to 2169 cm−1 before reaching 298 K).
(iii) The intensity of 2006 and 1992 cm−1 bands decreases.
(iv) The band at 1937 cm−1 clearly appears.
(v) The temperature does not affect the 2057 cm−1 band intensity.
The position of the band at 2169 cm−1 probes the presence of a few number Ce4+ sites,43 which should be also present at low temperature, but the band at 2169 cm−1 is probably hidden at TN2,liq by the tail of that due to CO–Ce3+ species. The existence of few unreduced sites on the support will be probed by methanol adsorption, as reported below.
The species characterised by a band at 1937 cm−1 is not formed at TN2,liq but it appears on increasing the vacuum temperature. Two hypotheses could be proposed to explain these phenomena. Firstly, activation energy is necessary to form such species. Since the appearance of the 1937 cm−1 band is accompanied by a decrease of the 2006 and 1992 band intensities due to CO adsorbed on PVS, we could propose that these species might be formed from the corrosive effect of CO on very small particles PVS to give rise to a carbonyl complex like Ptx(CO)y2− for example, but the formation of such complexes has been rejected above. The second hypothesis is to suppose that the interaction between CO molecules adsorbed on Pt atoms at the periphery of PVS and the support is inhibited at TN2,liq. For TN2,liq, the amount of CO molecules adsorbed on cationic sites of ceria is very important with respect to the intensity of the band near 2163 cm−1; this could inhibit the interaction with the support. By heating until 298 K, CO molecules desorb leading to the liberation of cationic sites Ce3+ which could therefore interact with CO molecules adsorbed on Pt sites at the periphery of PVS particles. According to this hypothesis, the band at 1937 cm−1 would be due to a strong interaction with the support, as supposed before. This latter assignment is quite reliable and in nice agreement with our previous results.
Whatever the hypothesis, a correlation seems to exist between CO species formed on PVS (band at 2010 cm−1) and the species characterised by the band at 1937 cm−1. This suggests that the species characterized by the 1937 cm−1 band give rise to that at 2010 cm−1 when the interaction of CO with the support is prevented.
CH3OH adsorption at rt. CH3OH molecules are dissociatively adsorbed on CeO2 to form methoxy species.44 Therefore, these species block the whole cationic coordinatively unsaturated sites, preventing the interaction of the support with other molecules. To ascertain our hypothesis of CO molecules both adsorbed on the support and on a peripheral Pt site of PVS particles, we have progressively introduced small methanol doses at r.t. in the IR cell after CO adsorption and evacuation at the same temperature. Results concerning the Pt/CeO2–R423E673 catalyst are reported in Fig. 6, which presents the ν(C
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O) and the ν(O–CH3) stretches. The addition of methanol leads to the formation of bands at 1102, 1060 and 1022 cm−1 characteristic of methoxy species linearly, bridged and threefold coordinated on cerium sites, respectively.44 Concomitantly, the 1937 cm−1 band intensity progressively decreases. In parallel, the intensity of that at 2011 cm−1 increases. This result confirms that (i) a relation exists between species characterised by bands at 2011 cm−1 and 1937 cm−1 and, (ii) the species characterised by this last band is partly due to an interaction with the cationic sites of the support. We can therefore explain the effect of methanol by the following Scheme 1.
O) and the ν(O–CH3) stretches. The addition of methanol leads to the formation of bands at 1102, 1060 and 1022 cm−1 characteristic of methoxy species linearly, bridged and threefold coordinated on cerium sites, respectively.44 Concomitantly, the 1937 cm−1 band intensity progressively decreases. In parallel, the intensity of that at 2011 cm−1 increases. This result confirms that (i) a relation exists between species characterised by bands at 2011 cm−1 and 1937 cm−1 and, (ii) the species characterised by this last band is partly due to an interaction with the cationic sites of the support. We can therefore explain the effect of methanol by the following Scheme 1.
 | ||
| Fig. 6 FT-IR spectra recorded after CO adsorption (0.7 kPa) at room temperature on Pt/CeO2–R423E673 followed by evacuation at rt. (a). Small doses of CH3OH introduced at rt: (b) 3.7 μmol, (c) 7.5 μmol, (d) 11.2 μmol, (e) 14.9 μmol. Part A: ν(CO) region. Part B: ν(O–C) region of methoxy species. | ||
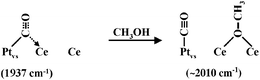 | ||
| Scheme 1 | ||
We therefore conclude that the 1937 cm−1ν(CO) band results from a bridged CO adsorption at the metal support interface on mixed Pt,Ce sites, at the periphery of PVS particles.
3.2 Structural rearrangements during redox treatment
We have reported in Fig. 7B the spectra recorded after CO introduction (Pe ∼ 0.7 kPa) and evacuation at room temperature on the Pt/CeO2 catalyst previously reduced by H2 at 423 K and evacuated at 423 K (spectrum a), 573 K (spectrum b) and 673 K (spectrum c). Although the reduction treatment has been carried out at the same temperature, recorded spectra are quite different. | ||
| Fig. 7 FT-IR spectra of: (a) Pt/CeO2–R423E423, (b) Pt/CeO2–R423E573, (c) Pt/CeO2–R423E673, (d) Pt/CeO2–R673E423. A: Hydroxyls range; B: ν(CO) stretching range after CO adsorption (Pe ∼ 0.7 kPa) followed by evacuation at rt; C: ν(CO) stretching range after CH3OH adsorption (Pe ∼ 0.13 kPa) followed by evacuation at r.t. | ||
For the sample reduced by H2 and evacuated at 423 K (spectrum Ba), bands at 2008 and 1937 cm−1 due to CO adsorption on Pt of PVS particles are mainly observed. Only shoulders located at 2047 and 2092 cm−1 (respectively assigned to CO adsorbed on Pt sites of PS particles and Ptδ+) are apparent. Evacuation at higher temperatures, 573 and 673 K (Fig. 7Bb and 7Bc), gives rise to an increase of the 2047 cm−1 band intensity at the expense of that at 2008 cm−1 suggesting the transformation of PVS particles into PS particles. This phenomenon evidences the effect of the evacuation temperature on the particle morphology. H2 desorption can be, in fact, reversible (giving back to molecular hydrogen) or irreversible, leading to water desorption, i.e. to metal and support reduction. Therefore the reduction of ceria by H2 at 423 K might lead to a metastable surface structure, subsequently destabilised by the mobility of O2− surface ions when the temperature is high enough (∼573 K).45 Consequently, the destabilization of PVS particles by evacuation at temperature near 573 K could arise from a surface reorganisation of ceria.
Two spectroscopic probes have been used in order to determine the effect of the evacuation temperature on the superficial structure of the support: (i) residual OH and (ii) OCH3 methoxy species formed by dissociative adsorption of methanol. Residual hydroxyls spectrum recorded for the sample reduced at 423 K followed by an evacuation at the same temperature (Pt/CeO2–R423E423, Fig. 7Aa) is mainly composed by two sharp bands at 3681 and 3644 cm−1 assigned to two different OH bridged species on Ce3+ atoms or on Ce4+ atoms, in proximity of oxygen vacancies, and also by a broad band near 3400 cm−1, due to associated OH species perturbed by hydrogen bond.46,47 The band at 3500 cm−1, mainly observed after evacuation at 673 K (Fig. 7Ac) is ascribed to oxyhydroxy species.46,47 Bands due to OH species linked by hydrogen bond and to bridged OH of lower wavenumber (3644 cm−1) essentially vanish by evacuation at 573 or 673 K (Fig. 7Ab and c). This transformation is reversible by H2 adsorption.46 Accordingly, evacuation at 573 or 673 K provokes (at least partially) the desorption of molecular hydrogen. The intensity of the band initially at 3681 cm−1 is not affected by the desorption but its wavenumber is shifted down to 3676 cm−1 (Fig. 7Ab), as well as the wavenumber of the other band due to bridged species, initially at 3644 cm−1 and shifted to 3634 cm−1 (residual band) after evacuation at 573 K. These lowering of wavenumbers reveal a re-oxidation of the surface by H2 desorption47,48 (reversible reduction of the support by H2).
The spectrum of methoxy species adsorbed on the reduced catalyst at 423 K and evacuated at the same temperature is shown in Fig. 7Ca. It is mainly composed by two ν(OC) bands at 1068 et 1050 cm−1 characterising bridged species on two cerium atoms, called OCH3 species (II).49 In detail, they are assigned to methoxy groups on reduced Ce3+ or mixed Ce3+-Ce4+ (ν(OC) band at 1068 cm−1), or oxidized Ce4+ (ν(OC) band at 1050 cm−1). The 1102 cm−1 band, generally absent in the case of OCH3 adsorption on surface reduced ceria, corresponds to linearly adsorbed methoxys on oxidised Ce4+ atoms [OCH3 species(I)]. The shoulder near 1020 cm−1 is attributed to OCH3 threefold coordinated on Ce4+ cations.49 The presence of oxidised Ce4+ cations at the surface could explain an oxidised Ptδ+ state of platinum at the metal–support interface (by oxygen migration phenomena, as reported by Galdikas et al.50), which gives rise to a ν(CO) band at 2092 cm−1 after CO adsorption (Fig. 7B). By evacuation at 573 and 673 K (Fig. 7C, spectra b and c), the intensity of the band initially at 1050 cm−1 (spectrum a) assigned to methoxy species of type II seems distributed in 1102 cm−1 and 1023 cm−1 bands, respectively due to type I and III species. Type III species mainly characterises (111) faces of highly crystallised ceria. After evacuation in the 573–673 K range, the surface state is partially modified showing unreduced (110) and (111) faces on which methoxy species are adsorbed to form type I (ν(OC) band at 1102 cm−1) or type III (ν(OC) band at 1023 cm−1) species. The mode initially at 1068 cm−1 (curve a) shifts to 1061 cm−1 (curves b, c). A similar behaviour is observed for the ν(OH) band at 3681 cm−1, assigned to bridged species on reduced Ce3+ cations. There is a similarity of behaviours of OH and methoxy species for sample reduced at 423 K (curve a) or at 673 K (curve d) followed by an evacuation at the same temperature 423 K (Fig. 7C). The same observation might be proposed in the range of ν(OH) (curves a and d, Fig. 7A).
From the above mentioned spectral evidence and taking into account previous studies,47,51,52 we conclude that a modification of the ceria surface oxidation state is possible by hydrogen evacuation and by the mobility of surface O2− ions. Moreover, we suggest that this process can explain a concomitant sintering of PVS particles into small PS particles.
In Fig. 8, we have schematised the morphological evolution of Pt crystallites probed by CO adsorption, depending on the temperature of the treatment (oxidized, reduced and evacuated). The Pt/CeO2 structures depicted in Fig. 8A, B, C, G, J and K are not shown but deduced from CO adsorption. On fresh Pt/CeO2, oxidised at 723 K and then evacuated at 473 K, the band at ∼2100 cm−1 is observed on the recorded spectra only (Fig. 8I and Fig. 1). This band results from the co-adsorption of CO and O on Pt atoms with an oxidised character. CO adsorption at room temperature on Pt/ceria treated under H2 at 523 K (Fig. 8B) and then evacuated at the same temperature (Fig. 8G) mainly leads to the presence of peaks at 2010 and 1937 cm−1 (Fig. 8H and Fig. 7Ba), which characterise very small Pt particles (noted PVS). In this work, we have considered that the bands at 2010 and 1937 cm−1 are due to the CO adsorption on Pt atoms weakly coordinated and to a bridged adsorption of CO at the metal-support interface on mixed Pt, Ce sites, respectively. The spectrum recorded after CO adsorption on the sample reduced at the same temperature (Tr = 523 K), followed by an evacuation at higher temperature (Te = 673 K) is quite different (Fig. 8D and Fig. 7Bc) from that obtained after evacuation at 523 K. Bands at 2010 and 1937 cm−1 are still present but their intensity is lower. The band at 2045 cm−1 is characteristic of small Pt crystallites bigger than PVS, noted PS. Adsorption of CO at low temperature (Fig. 8E) or introduction of CH3OH after CO (Fig. 8F) converts the band at 1937 cm−1 into that at 2010 cm−1. This phenomenon is explained by the saturation of the cationic sites of the support. Only the band at 2054 cm−1 is observed on spectra recorded after CO adsorption on Pt/CeO2 reduced at 723 K and evacuated at 673 K (Fig. 8M and Fig. 2d). After such a treatment only PS particles are detected. However, PVS particles are again observed by an oxidation at 723 K followed by a reduction at 423 K (Fig. 8L and Fig. 2e). This means that this kind of metal particles modification is reversible.
 | ||
| Fig. 8 Schematisation of CO adsorption on Pt/CeO2. (Tr: H2 reduction temperature, Te: H2 evacuation temperature, To: oxidation temperature). | ||
4 Conclusion
In this work we have investigated metal-support interaction in a Pt/CeO2 catalytic material, using CO probe adsorption followed by FT-IR spectroscopy. By this technique we have been able to determine the dispersion and the size modification of Pt particles, when the sample was submitted to different temperature treatments. Sintering phenomena leading to particles coalescence were observed and ascribed to the surface oxygen mobility of the ceria support phase. Moreover, we have detected and assigned an exotic ν(CO) band at 1937 cm−1 to a bridged CO adsorption at the metal support interface on mixed Pt, Ce sites, at the periphery of very small platinum particles.This study, besides its fundamental interest of associating chemical physics properties and modifications of platinum metal particles supported on an oxide to CO adsorbed probe molecule vibrations (observed by infrared), allows one to give a view inside a catalytic material submitted to different thermal treatment protocols. It is worth mentioning that the analysis method used is particularly powerful, allowing one to differentiate various structures and morphologies for supported metal particles, even when very small and highly dispersed. Moreover, the Pt loading being rather low (0.5%) and the support having a high surface area, the studied material is very close to a real catalyst.
References
- H. S. Gandhi, A. G. Piken, M. Shelef and R. G. Delesh, SAE Tech. Pap., 1976, 760201 Search PubMed.
- A. Trovarelli, Catal. Rev. – Eng., 1996, 38, 439 Search PubMed.
- Y. J. Mergler, A. van Aalst, J. van Delft and B. E. Nieuwenhuys, Appl. Catal. B: Environ., 1996, 10, 245 CrossRef CAS.
- R. Burch, J. P. Breen and F. C. Meunier, Appl. Catal. B: Environ., 2002, 39, 283 CrossRef CAS.
- C. Wheeler, A. Jhalani, E. J. Klein, S. Tummala and L. D. Schmidt, J. Catal., 2004, 223, 191 CrossRef CAS.
- G. Jacobs, L. Williams, U. I. Graham, G. A. Thomas, D. E. Sparks and B. H. Davis, Appl. Catal. A: Gen., 2003, 252, 107 CrossRef CAS.
- J. P. Breen, R. Burch and H. M. Coleman, Appl. Catal. B: Environ., 2002, 39, 65 CrossRef CAS.
- A. Ghenciu, Curr. Opin. Solid State Mater. Sci., 2002, 6, 389 CrossRef.
- A. Sepúlveda-Escribano, F. Coloma and F. Rodríguez-Reinoso, J. Catal., 1998, 178, 649 CrossRef CAS.
- M. Abid, G. Ehret and R. Touroude, Appl. Catal. A: Gen., 2001, 217, 219 CrossRef CAS.
- S. Bernal, G. Blanco, J. M. Gatica, C. Larese and H. Vidal, J. Catal., 2001, 200, 411 CrossRef CAS.
- S. Bernal, J. J. Calvino, J. M. Gatica, C. Larese, C. Lopez-Cartes and J. A. Pérez Omil, J. Catal., 1997, 169, 510 CrossRef CAS.
- S. Bernal, J. J. Calvino, M. A. Cauqui, J. M. Gatica, C. Larese, J. A. Pérez Omil and J. M. Pintado, Catal. Today, 1999, 50, 175 CrossRef CAS.
- F. Fajardie, J. F. Tempère, J. M. Manoli, O. Touret and G. Djéga-Mariadassou, Catal. Lett., 1998, 54, 187 CrossRef CAS.
- C. Binet, A. Jadi and J. C. Lavalley, J. Chim. Phys., 1989, 86, 451 CAS.
- V. Perrichon, L. Retailleau, P. Bazin, M. Daturi and J. C. Lavalley, Appl. Catal. A: Gen., 2004, 260, 1 CrossRef CAS.
- N. Sheppard and T. T. Nguyen, Adv. Infrared Raman Spectrosc., 1978, 5, 67 Search PubMed.
- P. Hollins, Surf. Sci. Rep., 1992, 16, 51 CrossRef.
- F. M. Hoffmann, Surf. Sci. Rep., 1983, 3, 107 CrossRef CAS.
- F. Maugé, C. Binet, J. C. Lavalley, in Catalysis by Metals, ed. A. J. Renouprez and H. Jobic, Springer-Verlag, 1997, 6L1 Search PubMed.
- B. J. Blyholder, J. Phys. Chem., 1964, 68, 2772 CrossRef CAS.
- B. J. Blyholder, J. Phys. Chem., 1975, 79, 756 CrossRef CAS.
- M. J. Kappers and J. H. van der Mass, Catal. Lett., 1991, 10, 365 CrossRef CAS.
- R. G. Greenler, K. D. Burch, K. Kretzchmar, R. Klauser, A. M. Bradshaw and B. E. Hayden, Surf. Sci., 1985, 152, 338 CrossRef.
- L. C. de Ménorval, A. Chaqroune and B. Coq et F. Figueras, J. Chem. Soc., Faraday Trans., 1997, 93, 3715 RSC.
- M. Primet, J. M. Basset, M. V. Mathieu and M. Prettre, J. Catal., 1973, 29, 213 CrossRef CAS.
- S. P. Mehandru and A. B. Anderson, J. Phys. Chem., 1989, 93, 2044 CrossRef CAS.
- D. K. Lambert, J. Chem. Phys., 1989, 89, 3847 CrossRef CAS.
- E. L. Garfunkel, J. E. Crowell and G. A. Somorjai, J. Phys. Chem., 1982, 86, 310 CrossRef CAS.
- J. E. Crowell, E. L. Garfunkel and G. A. Somorjai, Surf. Sci., 1982, 121, 303 CrossRef CAS.
- P. Gallezot, J. Datka, J. Massardier, M. Primet and B. Imelik, Proc. 8th Int. Congr. Catalysis, London, 1976, The Chemical Society, London, 1977, vol. 2, p. 696 Search PubMed.
- P. Bazin, PhD Thesis, University of Caen, 1998.
- D. W. Daniel, J. Phys. Chem., 1988, 92, 3891 CrossRef CAS.
- T. Jin, Y. Zhou, G. J. Mains and J. M. White, J. Phys. Chem., 1987, 91, 5931 CrossRef CAS.
- T. Jin, T. Okuhara, G. J. Mains and J. M. White, J. Phys. Chem., 1987, 91, 3310 CrossRef CAS.
- R. K. Brandt, M. R. Hughes, L. P. Bourget, K. Truszkowska and R. G. Geenler, Surf. Sci., 1993, 286, 15 CrossRef CAS.
- C. Binet, A. Badri and J. C. Lavalley, J. Phys. Chem., 1994, 98, 6392 CrossRef CAS.
- A. A. Solomennikov, Yu. A. Lokhov, A. A. Davydov and Yu. A. Ryndin, Kinet. Katal., 1979, 20, 714 CAS.
- Z. Gandao, B. Coq, L. C. de Ménorval and D. Tichit, Appl. Catal., 1996, 147, 395 Search PubMed.
- H. Bischoff, N. I. Jaeger, G. Schulz-Ekloff and L. Kubelkova, J. Mol. Catal., 1993, 80, 95 CrossRef CAS.
- J. S. Bradley, J. M. Millar, E. W. Hill and S. Behal, J. Catal., 1991, 129, 530 CAS.
- G. Longoni and P. Chini, J. Am. Chem. Soc., 1976, 98, 7225 CrossRef CAS.
- M. Daturi, C. Binet, J.-C. Lavalley, A. Galtayries and R. Sporken, Phys. Chem. Chem. Phys., 1999, 1, 5717 RSC.
- E. Finocchio, M. Daturi, C. Binet, J. C. Lavalley and G. Blanchard, Catal. Today, 1999, 52, 53 CrossRef CAS.
- D. Martin and D. Duprez, J. Phys. Chem., 1996, 100, 9429 CrossRef CAS.
- A. Badri, C. Binet and J. C. Lavalley, J. Chem. Soc., Faraday Trans., 1996, 92, 4669 RSC.
- M. Daturi, E. Finocchio, C. Binet, J. C. Lavalley, F. Fally and V. Perrichon, J. Phys. Chem., 1999, 103, 4884 Search PubMed.
- A. Galdikas, C. Descorme and D. Duprez, Solid State Ionics, 2004, 166, 147 CrossRef CAS.
- A. Badri, C. Binet and J. C. Lavalley, J. Chem. Soc., Faraday Trans., 1997, 93, 1159 RSC.
- S. Bernal, J. J. Calvino, G. A. Cifredo, J. M. Gatica, J. A. Pérez Omil and J. M. Pintado, J. Chem. Soc., Faraday Trans., 1993, 89, 3499 RSC.
- M. Daturi, E. Finocchio, C. Binet, J. C. Lavalley, F. Fally, V. Perrichon, H. Vidal, N. Hickey and J. Kaspar, J. Phys. Chem., 2000, 104, 9186 Search PubMed.
- C. Binet and M. Daturi, Catal. Today, 2001, 70, 155 CrossRef CAS.
| This journal is © the Owner Societies 2005 |