Highly dispersed magnesium oxide species on silica as photoactive sites for photoinduced direct methane coupling and photoluminescence
Leny
Yuliati
a,
Tadashi
Hattori
a and
Hisao
Yoshida
*b
aDepartment of Applied Chemistry, Graduate School of Engineering, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, 464-8603, Japan
bDivision of Environmental Research, EcoTopia Science Institute, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, 464-8603, Japan. E-mail: h-yoshida@esi.nagoya-u.ac.jp; Fax: +81-52-789-5849
First published on 15th November 2004
Abstract
Photoinduced direct methane coupling proceeded around room temperature over highly dispersed magnesium oxide species on silica, which exhibited fine structure in photoluminescence emission spectra. It was found that increasing the emission intensity tends to give an increase in the photoactivity for this reaction. The emission sites in the silica-supported magnesia have vibrational energy around 950 cm−1 and lifetime of excited state around 38 ms, which were similar properties to the previously reported other silica-based photoactive systems for this reaction, such as silica–alumina and silica-supported zirconia. These photoluminescence spectra could be similarly quenched by methane molecules. Thus, it is commonly suggested that in the systems of highly dispersed metal oxide species (MOx) on silica, the surface Si–O–M bonds are deeply related to the dominant photoactive sites for both the fine structural photoluminescence spectra and photoinduced direct methane coupling.
Introduction
Direct production of higher hydrocarbon from methane is one of the most attractive routes for the efficient use of natural gas and has been investigated over the past two decades. Especially the oxidative coupling of methane (OCM) has been extensively studied. However, it tends to produce COx, and no catalyst has reached the criteria for industrial application.1,2 Thus, the researches in the field have been gradually weakened. On the other hand, the movement of fossil fuels age to future hydrogen energy age makes the non-oxidative direct methane conversion become one of the alternative approaches that still attracts the attention of many researchers.1,3 However, the conversion of methane to higher valuable chemicals such as ethane and hydrogen under non-oxidative condition is thermodynamically unfavorable and very difficult at low temperature. In our research, this became feasible by using photoenergy even at room temperature. It was reported that photoinduced non-oxidative methane coupling proceeded at room temperature without any formation of COx over silica-based materials such as silica–alumina,4–6 silica-supported zirconia,7,8 silica–alumina–titania,9,10 and H-zeolites.11 The starting of this reaction was:| 2CH4 → C2H6 + H2, ΔG = 68.6 kJ mol−1, |
Photoluminescence spectroscopy is a good method for obtaining significant information about the structures of photoactive sites in the catalyst. Especially because of its high sensitivity, it is applicable for samples with low concentration, e.g. <1%.12 In the case of silica–alumina and silica-supported zirconia, it was found that the catalysts exhibiting the characteristic fine structure in photoluminescence spectra showed much higher activity in photoinduced direct methane coupling than the ones exhibiting no or broad emission band.4,5,7,8 Thus, the unique emission sites are suggested to be the active sites for photoinduced non-oxidative methane coupling.
Silica–magnesia is well known as acid and base catalyst for dehydration of alcohol,13 aldolic condensation of ketone,14etc. Due to the discovery of photoluminescence spectra exhibited by silica-supported magnesia,15,16 some studies in photocatalytic reactions have been examined using silica-supported magnesia as a photocatalyst. It has been reported that silica-supported magnesia showed activity in photooxidation of CO15 and photoepoxidation of propene by using molecular oxygen.17–19 In the present study, we found that silica-supported magnesia exhibited activity in the photoinduced direct methane coupling. Then, by photoluminescence spectroscopy we investigated the photoactive sites in silica-supported magnesia and discussed the photocatalytic active sites. In addition, we considered a general aspect for photoactive sites in the systems of highly dispersed metal oxide on silica.
Experimental
Materials
Amorphous silica was synthesized from Si(OEt)4 by a sol–gel method, followed by calcination in a flow of dry air (1 cm3 s−1) at 773 K for 5 h.17 MgO/SiO2 samples with different amount of MgO were prepared by impregnation of the silica (554 m2 g−1) with an aqueous solution of Mg(NO3)2·6H2O (Kishida). The water was slowly removed by stirring the mixture on a hot plate temperature of which was 393 K. Then, the sample was dried at 393 K overnight in an oven, followed by calcination in a flow of dry air (1 cm3 s−1) at 773 K for 5 h. The Mg content (x mol%) was defined as x = NMg(mol)/(NMg(mol) + NSi(mol)) × 100 and referred to as to MgO/SiO2(x). For comparison, silica–alumina (SiO2–Al2O3, Al = 9.1 mol%) and silica-supported zirconia (ZrO2/SiO2, Zr = 0.1 mol%) samples were prepared according to the methods in previous studies.4,7Photoreactions
The activities of catalysts were tested in the photoinduced direct methane coupling in a similar way to the previous studies.4–11 The sample (0.2 g) was spread on the flat bottom (14 cm2) of a closed quartz reaction vessel (30 cm3), and treated with 100 Torr (1 Torr = 133 Pa) oxygen for 1 h at 1073 K, followed by evacuation for 1 h at 1073 K as pre-treatment. After cooling to room temperature, 200 μmol of methane was introduced and the photocatalytic reaction was carried out for 3 h upon irradiation from a 300 W Xe lamp (used at 17.5 A, 15 V, intensity of light measured at 220–300 nm was ca. 10 mW cm−2). The products in the gaseous phase were separately collected and analyzed by gas chromatography. The products that were strongly adsorbed on the catalyst were collected by heating at 573 K for 15 min and analyzed by gas chromatography. Two kinds of detectors were used to analyze the products. Thermal conductivity detector with Ar carrier was applied for detection of hydrogen and the detection limit in this case was estimated as 0.04 μmol. Flame ionization detector was applied for detection of hydrocarbons and the measurements were reproducible within 96%. The experimental error on the reaction tests was estimated within 4%.Characterizations
BET specific surface area of samples was calculated from the amount of nitrogen adsorption at 77 K. Before the measurement, the sample was heated at 673 K for 30 min in a flow of He. The amount of N2 adsorbed was determined by a thermal conductivity detector.Phosphorescence spectra and decay curves were recorded at 77 K by Hitachi F-4500 fluorescence spectrophotometer, using a UV-cut filter (λtransmittance > 330 nm) to remove the scattered light from Xe lamp and an attachment for phosphorescence measurement. Before recording phosphorescence spectra and decay curves, 0.15 g of sample was treated with 100 Torr oxygen for 1 h at 1073 K in a specially designed in situ cell, followed by evacuation for 1 h at 1073 K. Then, the sample was transferred to the optical part without exposing to air.
Results
Photoactivity in direct methane coupling
The photoactivities of silica and silica-supported magnesia samples in photoinduced direct methane coupling are listed in Table 1. There were no COx molecules formed under this non-oxidative photoreaction. All silica-supported magnesia samples showed much higher activity than silica itself (Table 1, entry 1–4 and 6). On either the silica or the silica-supported magnesia samples, the main product was C2H6 (>90%) which was obtained as a gaseous product. Trace amounts of C2H4 and C2H6 were observed as adsorbed products that were detected through a thermal desorption procedure at 573 K. After a 3-h irradiation, the production of H2 could not be detected by gas chromatography in most cases (Table 1, entry 1–4) due to less sensitivity, but it was confirmed after 24 h of irradiation (Table 1, entry 5). The stoichiometric ratio of C2H6 and H2 formation is confirmed here (C2H6/H2 = 0.96) within experimental error.| Entry | Sample | BET specific surface area/m2 g−1 | Yield of gaseous phase products/10−2 C%b | Yield of thermally desorbed productsc/10−2 C%b | Total yield/10−2 C%b | Yield of H2d/10−2 μmol | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| C2H6 | C3H8 | C2H4 | C2H6 | C3H6 | Experimental | Theoreticale | ||||
| a Reaction temperature was ca. 310 K, sample was 0.2 g, initial methane was 200 μmol, irradiation time was 3 h. tr. = trace, n.d. = not detected. b Based on the initial amount of methane, analyzed by FID (reproducibility 96%). c These products were desorbed by heating at 573 K for 15 min after photoreaction. d Analyzed by TCD (limit detection 0.04 μmol). e Based on the yield of gaseous phase and thermal desorption products. f Irradiation time was 24 h. | ||||||||||
| 1 | MgO/SiO2(0.3) | 509 | 1.77 | 0.07 | 0 | tr. | 0 | 1.83 | n.d. | 1.8 |
| 2 | MgO/SiO2(0.5) | 495 | 2.62 | 0.08 | 0.08 | 0.03 | 0 | 2.73 | n.d. | 2.9 |
| 3 | MgO/SiO2(2.0) | 471 | 2.72 | 0.07 | tr. | 0.03 | 0 | 2.81 | n.d. | 2.8 |
| 4 | MgO/SiO2(5.0) | 445 | 1.32 | 0.07 | tr. | tr. | 0 | 1.39 | n.d. | 1.4 |
| 5 | MgO/SiO2(0.5)f | 495 | 8.16 | 0.73 | 0.02 | 0.02 | 0 | 8.93 | 7.8 | 9.2 |
| 6 | SiO2 | 554 | 0.42 | 0.03 | 0 | 0 | 0 | 0.45 | n.d. | 0.5 |
| 7 | SiO2-Al2O3(9.1) | 517 | 1.42 | 0.04 | tr. | 0.09 | 0.13 | 1.66 | n.d. | 1.8 |
| 8 | ZrO2/SiO2(0.1) | 500 | 2.14 | 0.11 | 0.02 | tr. | 0 | 2.25 | n.d. | 2.4 |
In order to compare the activity of the silica-supported magnesia system to other silica-based systems, we prepared SiO2-Al2O3 and ZrO2/SiO2 samples and applied them in the photoreaction under the same condition. The amount of alumina and zirconia loading on silica were chosen to exhibit the fine structural emission spectra and to show high activity in the photoreaction.4,7 The difference in optimal loading amount of metal cations on silica will be described in the Discussion section. It was demonstrated that the silica-supported magnesia system showed a similar level of activity to silica–alumina and silica-supported zirconia systems.
Structure of MgO/SiO2 samples
It was confirmed that there was no crystalline structure of MgO observed from an X-ray diffraction pattern, even in the sample with the highest Mg content (5 mol%). This indicates that all samples may contain very small particles or less crystallinity of MgO.The specific surface area of MgO/SiO2 samples are listed in Table 1. They were not drastically different from that of the pure SiO2. The surface area per unit weight of SiO2 was almost the same as the pure SiO2. These indicate that the magnesium oxide species on the samples were well dispersed on the silica surface without significantly changing the surface area of silica support. Similar results have been observed in the ZnO/SiO2 system.20
The amount of magnesium oxide species on the silica surface corresponds to 1 atom nm−2 for MgO/SiO2(2). Monolayer coverage of the highly dispersed magnesium oxide on the silica surface corresponds to ca. 36 mol% (32 wt%) of magnesium content. This means that the present samples have enough high surface area to provide highly dispersed magnesium oxide species on the silica surface. Besides, from XANES spectra it was reported that silica-supported magnesia prepared by impregnation of silica with aqueous solution of magnesium nitrate (up to 30 wt% MgO) produced highly dispersed magnesium oxide species on the silica surface.18,21 Thus, it is considered that all of the samples in the present study would have the structure of highly dispersed magnesia oxide species on the silica surface.
Photoluminescence spectra
Photoluminescence excitation and emission spectra of MgO/SiO2 samples evacuated at 1073 K are shown in Fig. 1. The intensities of both excitation and emission spectra increased with an increase of Mg loading amount, then decreased at higher loading of Mg (more than 2 mol%). The excitation spectra of MgO/SiO2 samples showed two maxima around 240 and 300 nm (Fig. 1A). The band intensity at 300 nm was lower than that at 240 nm. These two kinds of excitation maxima suggested the presence of different types of magnesium species on the silica surface or different excitation transitions. As the shape of excitation spectra did not change by changing the monitoring emission wavelength, it can be considered that there would be only one kind of photoactive sites that has at least two dominant absorption transitions from ground state to excited state.22 | ||
| Fig. 1 Photoluminescence excitation (A) and emission (B) spectra recorded at 77 K of (a) MgO/SiO2(0.1), (b) MgO/SiO2(0.3), (c) MgO/SiO2(0.5), (d) MgO/SiO2(2), and (e) MgO/SiO2(5). Monitoring emission wavelength for A was 525 nm, excitation wavelength for B was 300 nm. | ||
As shown in Fig. 1B, MgO/SiO2(0.1) sample exhibited a weak and broad emission band centered around 440 nm wich is probably due to surface silanols of silica.7,8,16,18 Other MgO/SiO2 samples exhibited emission spectra with fine structure centered around 525 nm, which were similar to each other in spectral shape. This suggests the presence of the similar emission sites in 0.3–5.0 mol% MgO/SiO2 samples. The shapes of the emission spectra are clearly different from the emission spectra of either silica or bulk MgO,23,24 but are similar to the reported one which is assigned to the highly dispersed magnesium oxide species on the silica surface.15,18 The shape of the fine structural emission spectra did not vary with the excitation wavelength, suggesting that there was probably only one kind of phosphorescence emission sites in the samples.
There are eight maxima on the emission spectra and the energy position of each maximum is listed in Table 2. The intervals between the positions of the maxima are constant, corresponding to the vibrational energy of the photoexcited sites. The vibrational energy of the photoexcited sites can be estimated as the average of the intervals at 950 ± 48 cm−1. This value is in agreement with the vibrational energy described in the previous work.15
| No. | Maximum | The interval to the next maximum/cm−1 | |
|---|---|---|---|
| nm | × 104 cm−1 | ||
| a MgO/SiO2(2), recorded at 77 K, excitation wavelength was 300 nm. | |||
| 1 | 435.8 | 2.2946 | 959 |
| 2 | 454.8 | 2.1988 | 953 |
| 3 | 475.4 | 2.1035 | 1019 |
| 4 | 499.6 | 2.0016 | 917 |
| 5 | 523.6 | 1.9098 | 864 |
| 6 | 548.4 | 1.8235 | 975 |
| 7 | 579.4 | 1.7259 | 967 |
| 8 | 613.8 | 1.6292 | |
Interaction of methane molecules with the emission sites
Quenching experiments were carried out to get information about interactions between the emission sites of silica-supported magnesia and methane molecules. Little amount of methane was introduced to the MgO/SiO2(0.5) sample step by step. The admission of the methane molecules decreased the intensity of the emission spectrum, as shown in Fig. 2. This quenching effect clearly indicates that there were interactions between the emission sites of silica-supported magnesia and the added methane molecules.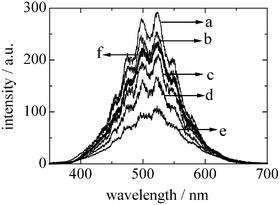 | ||
| Fig. 2 Effect of methane admission upon phosphorescence spectra of MgO/SiO2(0.5), recorded at 77 K. Excitation wavelength was 300 nm. The amount of methane was (a) 0, (b) 21, (c) 84, (d) 170 and (e) 320 Pa. The spectrum f (bold) was recorded after vacuum at room temperature for 15 min. | ||
As described elsewhere,12,23–25 two mechanisms of phosphorescence quenching are possible, dynamic quenching (collisional or weak interaction), and static quenching via formation of surface complexes. In silica-supported magnesia, the emission spectra were quenched by methane molecules, but about 80% of the initial intensity was recovered by evacuation at room temperature for 15 min. It shows that there were weak interactions between the emission sites and methane, though some small parts were not reversible at room temperature, as shown in Fig. 2. Thus, it suggests that the quenching of emission spectra in silica-supported magnesia by methane molecules follows the dynamic quenching mechanism dominantly.
Analysis of decay curve
Fig. 3 shows the decay curves of the emission light from MgO/SiO2 samples that exhibited the fine structure of emission spectra (0.3–5 mol%). The decay curves were measured at excitation wavelength 300 nm and emission wavelength 525 nm. The decay curve of MgO/SiO2 sample can not be described by one exponential function, but it can be described by two exponential functions as the following equation:| I = I0[A1 exp(−t/τ1) + A2 exp(−t/τ1)] |
 | ||
| Fig. 3 Experimental (circle) and simulation (line) decay curve of (a) MgO/SiO2(0.3), (b) MgO/SiO2(0.5), (c) MgO/SiO2(2) and (d) MgO/SiO2(5). The spectra were recorded at 77 K. Excitation and emission wavelengths were 300 nm and 525 nm, respectively. | ||
The best fitting parameters are presented in Table 3. At least two kinds of emission fractions that have different lifetime can be deduced from the decay curve. The shorter lifetime emission (SLE) was dominant for all samples (0.3–0.5 mol%) that exhibited the fine structural spectra, and the lifetime did not show remarkable changes although the Mg content is increasing. It shows that the same kind of emission sites was formed in all samples, and the fractions in the emission sites were almost the same in all of these samples. The lifetime of the SLE fraction (τ1) was estimated to be 38 ms.
| Sample | SLE | LLE | ||
|---|---|---|---|---|
| A 1 | τ 1 b/ms | A 2 | τ 2 b/ms | |
| a Recorded at 77 K, excitation wavelength was 300 nm, emission wavelength was 525 nm, measurement time was 750 ms. b The value in parentheses is the error allowed for the fitting. | ||||
| MgO/SiO2(0.3) | 0.92 | 38(5) | 0.08 | 5(1) × 102 |
| MgO/SiO2(0.5) | 0.97 | 38(5) | 0.03 | 7(1) × 102 |
| MgO/SiO2(2) | 0.95 | 38(5) | 0.05 | 5(1) × 102 |
| MgO/SiO2(5) | 0.89 | 38(5) | 0.11 | 7(1) × 102 |
The longer lifetime emission (LLE) was found as the minor component in all MgO/SiO2 samples exhibiting fine structural spectra. The lifetime (τ2) was around 500–700 ms. In the silica–alumina system, it has been suggested that the LLE fractions are due to surface OH groups and did not play an important role in photoinduced direct methane coupling.26 In other words, the SLE fraction was clarified as the fraction exhibiting the fine structure in the emission spectra and the emission sites would be the expected active sites responsible for the photoreaction.7,26
As shown in Table 3, the SLE and LLE fraction ratios were obtained as almost constant in the present samples by the present fitting procedures. Since all the samples presented in Table 3 showed clear fine structural spectra, it was predicted that the ratios would be similar to each other. The sample of MgO/SiO2(0.1) exhibiting a broad spectrum would show a different kind of emission sites with different lifetimes from those on these samples. Even though the decay curve of the MgO/SiO2(0.1) was difficult to be analyzed due to large noise and low intensity (not shown), it was confirmed that the emission sites in this sample have the character of longer lifetime emission. On the other hand, the optical cell also exhibited a very low intensity of broad emission spectra with a long lifetime (not shown), which may partly be contributed to the LLE fraction. Although the intensity was very low, the contribution of this optical cell will be higher when the emission intensity is lower. Thus, the measured decay curve of the sample exhibiting low intensity would have a lower accuracy of determining the fraction originated from the sample. At least, however, it is clear that the LLE was a minor component, i.e. the SLE was the dominant emission sites in these samples.
Discussion
The emission sites on MgO/SiO2
The relationship between the total intensity of emission spectra (excitation wavelength was 300 nm, emission wavelength was 525 nm) and Mg content is shown in Fig. 4. Fig. 4 also displays the relationship between the emission intensity of shorter lifetime (SLE) and Mg content. The SLE intensity and total intensity of emission spectra did not show much difference since the SLE is the dominant emission fraction, as shown in Table 3. The variation of Mg content resulted in the variation of emission intensity. It is clear that the Mg content affects the number of emission sites formed in silica-supported magnesia. Since the emission intensity increased with increasing Mg content up to 2 mol%, the amount of SLE species would increase up to around 2 mol%. When assuming that the emission efficiency is constant or not drastically changed, the curve in Fig. 4 means that the number of emission sites in silica-supported magnesia would initially increase with the increasing of Mg content, but too much Mg loaded on silica would cause the reduction of the emission sites in number. This phenomenon indicates that only highly dispersed magnesium species on the silica surface would exhibit the fine structure in emission spectra.18 | ||
| Fig. 4 Plot of the emission intensity of the MgO/SiO2 samples (open circle), SLE intensity (cross) and the total yield in the photoinduced direct methane coupling (closed circle, broken line) versus Mg content. | ||
As reported previously, SLE is suggested as the emission sites that play an important role in photoinduced direct methane coupling.7,26 In silica–alumina, the proposed SLE site is a Si–O–Al linkage,26 and in silica-supported zirconia the SLE site proposed is a Si–O–Zr linkage.7 These linkages have already been confirmed by Raman and infrared spectroscopy.27–31 In a separate experiment, it has been confirmed that the fine structure of emission spectra in silica-supported magnesia was not obtained when the vacuum pretreatment was done at low temperature, such as 773 K.15 We also confirmed that the fine structure of emission spectra on the present samples could not be obtained by vacuum pre-treatment at 873 K. The high temperature (1073 K) is necessary to obtain the fine structure of emission spectra in silica-supported magnesia, as also needed in silica–alumina5 and silica-supported zirconia.7,8 The evacuation at high temperature is important for desorption of hydroxyl groups on the surface, which would produce coordinatively unsaturated sites and would correspond to photoexcitation sites. In silica-supported magnesia, it has been reported that the emission site is a coordinatively unsaturated Mg (Mg–O bond) on the surface of the tetrahedral silica network which was only produced by evacuation at high temperature.15,16,18 These facts mentioned above suggest that the Si–O–Mg linkages as the emission sites would be responsible for exhibiting the fine structure in the phosphorescence emission spectra.
The active sites in photoinduced direct methane coupling
Fig. 4 also shows the photoactivity of silica-supported magnesia samples (total yield) as a function of Mg content. As shown in Fig. 4, both the emission intensity and the total yield similarly varied with Mg content. This good relationship suggests a possibility that the emission sites in silica-supported magnesia are the dominant active sites in the photoinduced direct methane coupling. This suggestion is also supported by the result of the quenching effect (see Fig. 2) that showed the interactions between methane and the emission sites of silica-supported magnesia.In Fig. 5, we can see the relationship between the SLE emission intensity and the photocatalytic activity of the samples in the photoinduced direct methane coupling. The increase of the emission intensity tended to give an increase in the photoactivity of the sample, suggesting that the emission sites would be the active sites in the reaction. However, the increase of the emission intensity did not show a proportional increase with the photoactivity of the sample. It would be reasonable to assume that in the sample with higher Mg content, the excitation state would be delocalized over Mg oligomer which might cause a decrease in the photocatalytic activity. We confirmed similar phenomena in the silica–alumina system from the reported data of intensity and photoactivity.4 The tendency of this saturation has also been reported in silica-supported zirconia.7
 | ||
| Fig. 5 Relation between SLE intensity of the MgO/SiO2 samples (closed circle), the SiO2-Al2O3(9.1) (opened circle), the ZrO2/SiO2(0.1) (triangle) and the total yield on them in the photoinduced direct methane coupling. | ||
General aspect on a silica-based photocatalyst
Fig. 5 also shows the photoactivity of silica–alumina and silica-supported zirconia in this reaction. It was elucidated that the silica-supported magnesia showed an activity at almost the same level as other reported systems; or rather, when they were compared at the same SLE intensity, the silica-supported magnesia was found to be slightly superior to others.Even though these three systems showed a similar level of activity, the optimum loading amounts of each metal oxide on the silica support were different. The difference of the cation or the sample preparation method will affect the optimum loading amount of the cation on the silica support. The ratio of the amount of the formed active species to the loading amount on silica was high in the order of silica-supported zirconia > silica-supported magnesia > silica–alumina. When the activity was normalized by the SLE intensity, these three different kinds of cations showed almost the same activity even though the apparent optimum concentrations on silica were different from each other. In addition, the lifetimes of the excitation state and vibrational energy were also similar to each other (Table 4). These facts suggest that there might be similar active sites for the photoinduced methane coupling which do not strongly depend on the kind of cations.
| Sample | Vibrational energya/cm−1 | Lifetime (SLE)b/ms |
|---|---|---|
| a Calculated from the average interval of the maximum in fine structure of emission spectra. b The dominant fraction calculated from the decay curve, see text. c The value in parentheses is the error allowed for the fitting. d Data from ref. 26. e Data from ref. 7. | ||
| MgO/SiO2 | 950(48)c | 38(5) |
| SiO2-Al2O3d | 985(54) | 33(3) |
| ZrO2/SiO2e | 955(50) | 28(5) |
These similar properties may bring a similar performance in the interaction with the methane molecules, which can be proved by studying the quenching effect by methane molecules. When the illumination intensity and concentration of the molecules are maintained to be constant, in the presence of quencher molecules the relative photoluminescence intensity will be expressed as a function of the concentration of the quencher molecules, according to the following Stern–Volmer expression:12

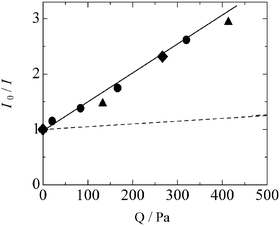 | ||
| Fig. 6 Stern–Volmer plots of relative emission intensity versus partial pressure of methane molecules on MgO/SiO2(0.5) (circle), SiO2-Al2O3 sample (triangle) and ZrO2/SiO2 sample (square). The broken line is the interaction between SiO2-Al2O3 sample and N2 molecules. The data for SiO2-Al2O3 and ZrO2/SiO2 samples were taken from refs. 5 and 7, respectively. | ||
As shown in Fig. 6, in the presence of methane molecules the relative emission intensity is a linear function of methane pressure, with an intercept equal to 1. Here, the slope corresponds to kτ. This clearly indicates that the quenching of emission intensity occurred by interactions between the added methane molecules and the emission sites of the silica-supported magnesia through non-radiative deactivation pathways. In other words, dynamic quenching mainly happened, in which the quenching depended on the amount of the quencher molecules. When we also plotted the results on the silica–alumina5 and the silica-supported zirconia7 in Fig. 6, it was clarified that these three systems have similar slope in the Stern–Volmer plot. Since the lifetimes of the emission sites in all systems are similar to each other (Table 4), the similar slope of Stern–Volmer plot indicates that the silica-supported magnesia exhibits similar methane quenching rate to the silica–alumina and the silica-supported zirconia. This result suggests that the emission sites in these systems would be concerned with activation of methane molecules in a similar way. As a comparison, introducing the same amount of an inert gas (N2) to the silica–alumina system5 instead of methane reduced much less the emission intensity, as shown in Fig. 6 as a broken line, which confirmed a certain interaction between the emission site and the methane molecules.
Since the photoreaction could not occur without irradiation, it is most likely that the emission sites give the photoexcitation energy to the methane molecules through adsorption, or the complex of the active sites and adsorbed methane might be activated by the photoenergy.7 Similar quenching efficiency of the silica-supported magnesia to the silica–alumina and the silica-supported zirconia gives us information that the performance of the photoactive sites on the silica-supported magnesia is similar to those on the silica–alumina and the silica-supported zirconia systems. It indicates that they have a similar key-step in the activation of methane; probably methane would be activated by the same mechanism in photoinduced direct methane coupling,7 which would bring similar photoactivity in the reaction as shown in Fig. 5. Thus, it can be expected that other cations than Mg, Al, and Zr will show activity toward photoinduced direct methane coupling with similar mechanism if they could be highly dispersed on a silica support.
It has been reported that silica itself also shows photocatalytic activity in photoinduced direct methane coupling4–10 and some other reactions, such as photometathesis,32,33 photoepoxidation20,34,35 and photooxidations.36 Also in the present study, the activity in photoinduced direct methane coupling was confirmed as listed in Table 1
(entry 6). The pure silica materials evacuated at higher temperature have a kind of surface quantum defect that has a function as photoactive sites for photoreactions such as photometathesis.37 It has been clarified that the radical sites ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) Si–O˙
(NBOHC, non-bridging oxygen hole center) can be photoexcited upon light at 4.8 eV (258 nm) and the charge transfer occurs from the bonding orbital of Si–O to 2p nonbonding orbital of nonbridging oxygen,32,38 as shown in Scheme 1. The stretching vibration of NBOHC on the silica has been revealed to be 891 and 910 cm−1.32
Si–O˙
(NBOHC, non-bridging oxygen hole center) can be photoexcited upon light at 4.8 eV (258 nm) and the charge transfer occurs from the bonding orbital of Si–O to 2p nonbonding orbital of nonbridging oxygen,32,38 as shown in Scheme 1. The stretching vibration of NBOHC on the silica has been revealed to be 891 and 910 cm−1.32
 | ||
| Scheme 1 NBOHC as photoactive sites on the silica surface. | ||
On the other hand, it has been reported that in highly dispersed TiO4 species on silica, the photoexcitation on Ti–O is explained as a charge transfer from ligand oxygen to titanium (LMCT, ligand-to-metal charge transfer) as shown in Scheme 2.12 The vibrational energy of the excited sites for Ti–O–Si in TS-1 was around 965 cm−1, which was estimated by the photoluminescence study.39 However, a recent study by UV resonance Raman spectroscopy revealed that the band at 965 cm−1 should be assigned to silicalite moiety that was affected by the introduced Ti cation, not to Ti–O–Si bond vibration.40 A previous study on photoluminescence of silica–alumina also revealed that the photoactive AlO4 species on silica were not acid sites at Al cations,26 implying that the Si–O bond affected by incorporated Al cation would be the active sites.
 | ||
| Scheme 2 LMCT on highly dispersed Ti–O bond on silica. | ||
Since evacuation at high temperature is necessary to generate the active sites in the pure silica,20,32–35 the silica–alumina,5 the silica-supported zirconia,7,8 and the silica-supported magnesia, we propose that the active sites in highly dispersed metal oxide in a silica system would be formed in a similar way to those in the pure silica materials. The presence of the metal cation in silica would result in an ionic character bond to some extent which has different electron density from the covalent Si–O bond and causes the easiness in vibration. From the discussion above, the difference of the element (M = Al, Zr, Mg) in silica did not show any remarkable changes in either emission properties or photoactivity in the reaction, although these three elements have many differences, e.g. group classification, valence, and ionic radius. It brings us to a consideration that the photoexcitation of the Si–O–M linkage would be strongly influenced by the character of the Si–O bond moiety, which might be the charge transfer in the Si–O bond. In our opinion, this might be expressed as shown in Scheme 3. This also gives us one possible explanation for the difference between the photoexcitation of metal oxide (via LMCT) and the photoexcitation of the highly dispersed metal oxide in silica (via CT on Si–O bond). At present, no direct and clear results have been elucidated to describe the details of the charge transfer. Further study on this idea is required to get more understanding of the electron charge transfer mechanism for the photocatalytic field.
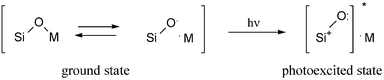 | ||
| Scheme 3 Plausible model of photoactive sites on a silica-based photocatalyst. | ||
Conclusion
Silica-supported magnesia promoted photoinduced direct methane coupling. The highly dispersed magnesium oxide species on silica would be the active sites responsible for both exhibiting the fine structural photoemission spectra and the photoactivity in the reaction.The similar photoactivity and properties of the emission sites in the silica-supported magnesia, the silica–alumina, and the silica-supported zirconia suggest the general aspect on a silica-based photocatalyst, as follows:
(i) The highly dispersed metal oxide species on silica having Si–O–M linkages (M = metal cation) that are generated by desorption of hydroxyl groups on the silica surface at high temperature would become the photoemission sites and the active sites for the photoinduced direct methane coupling.
(ii) These emission sites would be concerned with the activation of methane molecules in a similar way. Probably the reactions would proceed with similar mechanisms.
(iii) The photoexcitation of the species would be deeply related to the Si–O bond moiety.
Acknowledgements
This work was partially supported by a Grant-in-Aid for Scientific Research on Priority Areas (417) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of the Japanese Government. We acknowledge the invaluable contribution by the reviewers.References
- T. V. Choudhary, E. Aksoylu and D. W. Goodman, Catal. Rev., 2003, 45, 151 CrossRef CAS.
- Y. Xu and L. Lin, Appl. Catal. A, 1999, 188, 53 CrossRef CAS.
- Y. Xu, X. Bao and L. Lin, J. Catal., 2003, 216, 386 CrossRef CAS.
- H. Yoshida, N. Matsushita, Y. Kato and T. Hattori, Phys. Chem. Chem. Phys., 2002, 4, 2459 RSC.
- H. Yoshida, Y. Kato and T. Hattori, Stud. Surf. Sci. Catal., 2000, 130, 659.
- Y. Kato, H. Yoshida and T. Hattori, Chem. Commun., 1998, 2389 RSC.
- H. Yoshida, M. G. Chaskar, Y. Kato and T. Hattori, J. Photochem. Photobiol. A, Chem., 2003, 160, 47 CrossRef CAS.
- H. Yoshida, M. G. Chaskar, Y. Kato and T. Hattori, Chem. Commun., 2002, 2014 RSC.
- Y. Kato, N. Matsushita, H. Yoshida and T. Hattori, Catal. Commun., 2002, 3, 99 CrossRef CAS.
- H. Yoshida, N. Matsuhita, Y. Kato and T. Hattori, J. Phys. Chem. B, 2003, 107, 8355 CrossRef CAS.
- Y. Kato, H. Yoshida, A. Satsuma and T. Hattori, Microporous Mesoporous Mater., 2002, 51, 223 CrossRef CAS.
- M. Anpo and M. Che, Adv. Catal., 1999, 44, 119 CAS.
- T. López, R. Gómez, M. E. Llanos and E. López-Salinas, Mater. Lett., 1999, 38, 283 CrossRef CAS.
- T. López, R. Gómez, M. E. Llanos, E. García-Figueroa, J. Navarrete and E. López-Salinas, Mater. Lett., 1999, 39, 51 CrossRef CAS.
- T. Tanaka, H. Yoshida, K. Nakatsuka, T. Funabiki and S. Yoshida, J. Chem. Soc., Faraday Trans., 1992, 88, 2297 RSC.
- H. Yoshida, T. Tanaka, T. Funabiki and S. Yoshida, J. Chem. Soc., Faraday Trans., 1994, 90, 2107 RSC.
- H. Yoshida, C. Murata and T. Hattori, J. Catal., 2000, 194, 364 CrossRef CAS.
- H. Yoshida, T. Tanaka, M. Yamamoto, T. Yoshida, T. Funabiki and S. Yoshida, J. Catal., 1997, 171, 351 CrossRef CAS.
- H. Yoshida, T. Tanaka, M. Yamamoto, T. Funabiki and S. Yoshida, Chem. Commun., 1996, 2125 RSC.
- H. Yoshida, T. Shimizu, C. Murata and T. Hattori, J. Catal., 2003, 220, 226 CrossRef CAS.
- H. Yoshida, T. Yoshida, T. Tanaka, T. Funabiki, S. Yoshida, T. Abe, K. Kimura and T. Hattori, J. Phys. IV, 1997, 7, C2–911.
- B. Shelimov, V. Dellarocca, G. Martra, S. Coluccia and M. Che, Catal. Lett., 2003, 87, 73 CrossRef CAS.
- M. Anpo, Y. Yamada, Y. Kubokawa, S. Coluccia, A. Zecchina and M. Che, J. Chem. Soc., Faraday Trans., 1988, 84, 751 Search PubMed.
- M. Anpo and Y. Yamada, Mater. Chem. Phys., 1988, 18, 465 CrossRef CAS.
- M. Anpo, M. Kondo, S. Coluccia, C. Louis and M. Che, J. Am. Chem. Soc., 1989, 111, 8791 CrossRef CAS.
- Y. Kato, H. Yoshida and T. Hattori, Phys. Chem. Chem. Phys., 2000, 2, 4231 RSC.
- H. Yoshida, T. Tanaka, A. Satsuma, T. Hattori, T. Funabiki and S. Yoshida, Chem. Commun., 1996, 1153 RSC.
- G. Mestl and H. Knözinger, Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger and J. Weitkamp, VCH, Weinheim, 1997, vol. 2, p. 539 Search PubMed.
- S. W. Lee and R. A. Condrate Sr, J. Mater. Sci., 1988, 23, 2951 CrossRef CAS.
- Z. Dang, B. G. Anderson, Y. Amenomiya and B. A. Morrow, J. Phys. Chem., 1995, 99, 14437 CrossRef CAS.
- B. Rakshe, V. Ramaswamy and A. V. Ramaswamy, J. Catal., 1996, 163, 501 CrossRef CAS.
- Y. Inaki, H. Yoshida, T. Yoshida and T. Hattori, J. Phys. Chem. B, 2002, 106, 9098 CrossRef CAS.
- T. Tanaka, S. Matsuo, T. Maeda, H. Yoshida, T. Funabiki and S. Yoshida, Appl. Surf. Sci., 1997, 121–122, 296 CrossRef.
- H. Yoshida, C. Murata and T. Hattori, Chem. Commun., 1999, 1551 RSC.
- C. Murata, H. Yoshida and T. Hattori, Chem. Commun., 2001, 2412 RSC.
- A. Ogata, A. Kazusaka and M. Enyo, J. Phys. Chem., 1986, 90, 5201 CrossRef CAS.
- H. Yoshida, Curr. Opin. Solid Mater. Sci., 2003, 7, 435 CrossRef CAS.
- L. Skuja, Solid State Commun., 1992, 84, 613 CrossRef CAS.
- Allison S. Soult, Duke D. Pooré, Elizabeth I. Mayo and A. E. Stiegman, J. Phys. Chem. B, 2001, 105, 2687 CrossRef CAS.
- C. Li, G. Xiong, J. Liu, P. Ying, Q. Xin and Z. Feng, J. Phys. Chem. B, 2001, 105, 2993 CrossRef CAS.
| This journal is © the Owner Societies 2005 |