Photoinduced oxidation of [Mn(L)3]2+ and [Mn2O2(L)4]3+ (L = 2,2′-bipyridine and 4,4′-dimethyl-2,2′-bipyridine) with the [Ru(bpy)3]2+/-aryl diazonium salt system
Carole
Baffert
,
Stéphane
Dumas
,
Jérôme
Chauvin
,
Jean-Claude
Leprêtre
*,
Marie-Noëlle
Collomb
* and
Alain
Deronzier
*
Laboratoire d’Electrochimie Organique et de Photochimie Rédox, Université Joseph Fourier, UMR CNRS 5630, Institut de Chimie Moléculaire de Grenoble, FR CNRS 2607, BP 53, 38041, Grenoble, Cedex 9, France. E-mail: marie-noelle.collomb@ujf-grenoble.fr; alain.deronzier@ujf-grenoble.fr; jean-claude.lepretre@ujf-grenoble.fr; Fax: (33)(0)4 76 51 42 67
First published on 25th November 2004
Abstract
The photophysical, photochemical and electrochemical studies of a mixture of bipyridinyl ruthenium ([RuII(bpy)3]2+) and manganese ([MnII(L)3]2+ or [Mn2III,IVO2(L)4]3+, L = bpy (2,2′-bipyridine) or dmbpy (4,4′-dimethyl-2,2′-bipyridine)) complexes have been investigated in CH3CN. Electrochemical oxidations of [MnII(L)3]2+ are irreversible and lead, by subsequent chemical reactions, to the corresponding di-μ-oxo complex [Mn2III,IVO2(L)4]3+ with a good yield. These latter complexes can be then reversibly oxidized into the stable [Mn2IV,IVO2(L)4]4+ species. The luminescence lifetime of the excited state of the photosensitizer [RuII(bpy)3]2+* in the presence of variable concentration of manganese complexes has been determined. Using the Stern–Volmer equation, the quenching constant rate kq have been estimated. It appears that the [MnII(L)3]2+ mononuclear complexes quench only very weakly the excited state [RuII(bpy)3]2+* since the magnitude of kq determined for the bpy and dmbpy complexes is about 107 M−1 s−1. In contrast, a strong decrease of the luminescence lifetime is observed by addition of an increasing concentration of [Mn2III,IVO2(L)4]3+. The kq values obtained for the bpy and dmbpy complexes are, respectively, 2.3 × 109 and 2.5 × 109 M−1 s−1. The major quenching pathways of [RuII(bpy)3]2+* by those binuclear complexes of manganese are presumably energy transfer processes. Finally, the possibility of photocatalytic oxidation of [MnII(L)3]2+ and [Mn2III,IVO2(L)4]3+ has been evaluated by continuous irradiation in the presence of the photosensitizer [RuII(bpy)3]2+ and an aryl diazonium salt, ArN2+, playing the role of an irreversible electron acceptor. The photooxidation process transforming [MnII(L)3]2+ into [Mn2III,IVO2(L)4]3+ by intermolecular electron transfers between photogenerated [RuIII(bpy)3]3+ and [MnII(L)3]2+ occurs for the bpy and dmbpy complexes with a high efficiency. The subsequent photooxidation leading to [Mn2IV,IVO2(L)4]4+ is efficient only in the case of the dmbpy complex. The formation of those different species by electrochemical or photochemical ways has been demonstrated and quantified by coupled UV-visible absorption and EPR spectroscopy experiments. The efficiencies of the photoinduced oxidative processes have been correlated to the electrochemical data.
Introduction
In recent years, important efforts have been made to mimic the function of the donor-side of the photosystem II (PS II) using superstructured heterometallic complexes.1–6 In the natural PS II system, under illumination, the photoactive P680 chlorophylls center oxidizes water into dioxygen through a catalytic system containing an oxo cluster of four Mn and one Ca ion and a tyrosine residue; the detailed mechanism remaining not entirely elucidated, as is the case for the structure of the Mn cluster.7–9 The main strategy used in literature to elaborate models of PS II involves the covalent coupling of a photoactive [RuII(bpy)3]2+ (bpy = 2,2′-bipyridine) moiety, playing the role of the P680 chlorophylls, to some mono or binuclear manganese complexes. In Scheme 1 are shown some relevant superstructured bimetallic complexes developed by Styring and co-workers.3–6,10–15 Laser flash photolysis experiments on the two first bimetallic complexes (Scheme 1A, B) have shown that an intramolecular electron transfer can occur from the MnII site to the photogenerated [RuIII(bpy)3]3+ species, using an external electron acceptor-like viologen in acetonitrile. However, it was found that the electron transfer (ET) rate constant is fairly slow (kET in the range 105–2 × 107 s−1) and depends on the distance between the two metallic centers.6 These authors have also developed some [RuII(bpy)3]2+ complexes covalently linked to a tyrosine residue.16,17 In the presence of an external electron acceptor, an intramolecular electron transfer between the photogenerated [RuIII(bpy)3]3+ species and the tyrosine has been characterized by nanosecond laser flash photolysis experiments. The oxidized tyrosyl radical can then oxidize an oxo Mn2III,III complex added to the solution into a Mn2III,IV one.18 Finally, following these results, a superstructured heterobimetallic complexes has been proposed where the [RuII(bpy)3]2+ center and a binuclear Mn2II,II complex are covalently linked by a tyrosine bridge (Scheme 1C);3,13–15 the tyrosyl moiety playing the role of an electron relay between the photogenerated [RuIII(bpy)3]3+ species and the Mn2II,II complex. A relative faster electron transfer rate constant (kET > 107 s−1) has been determined, leading to the Mn2II,III species or to the Mn2III,IV analogue if experiments are carried out in the presence of water.14 All these studies have been conducted by flash photolysis, but the different oxidation states of the Mn center were not quantitatively generated by continuous photoirradiation of the solution.![Structure of model complexes developed by Styring et al. [ref. 3, and refs. therein].](/image/article/2005/CP/b411365a/b411365a-s1.gif) | ||
| Scheme 1 Structure of model complexes developed by Styring et al. [ref. 3, and refs. therein]. | ||
Paradoxically, photophysical and photochemical properties of a simple mixture of the [RuII(bpy)3]2+ and [MnII(bpy)3]2+ or [Mn2III,IVO2(bpy)4]3+ complexes have never been examined to our knowledge. In order to perform such photoinduced processes at the quantitative scale, one needs the use of sacrificial oxidant that is irreversibly reduced allowing the net conversion of RuII into RuIII. Recently we demonstrated that the 4-bromophenyl diazonium salt (ArN2+) played the role of efficient sacrificial oxidant towards a simple mixture of [RuII(bpy)3]2+ and [FeII(bpy)3]2+ complexes as well as some heterobinuclear polypyridinique complexes of RuII and FeII (RuII–FeII) in organic solvent-like CH3CN.19 In these experimental conditions, the effective energy transfer from the 3MLCT excited state of the RuII(bpy)32+ center to the FeII(bpy)32+ unit is short-circuited allowing the photoinduced formation of RuIII(bpy)33+ and the oxidation of the FeII(bpy)32+ subunit with an overall high efficiency. The oxidation of [FeII(bpy)3]2+ occurs at a potential close to that of [MnII(bpy)3]2+. However, in contrast to the FeII/FeIII redox couple which is perfectly reversible in CH3CN, the MnII/MnIII one is irreversible, [MnIII(bpy)3]3+ reacting with the residual water to form the [Mn2III,IVO2(bpy)4]3+ complex.20–22
Thus, we investigated the photophysical and photochemical properties of a mixture of [RuII(bpy)3]2+ with [MnII(L)3]2+ (L = bpy or dmbpy (4,4′-dimethyl-2,2′-bipyridine)) and with corresponding binuclear complexes [Mn2III,IVO2(L)4]3+ in CH3CN in the presence of ArN2+. In these experimental conditions, the successive photoinduced intermolecular electron transfers shown in Scheme 2 would be expected. The formation of the oxidized forms under continuous irradiation has been demonstrated by coupled UV-visible absorption and EPR spectroscopy experiments. Finally, we show that the efficiencies of the photoredox processes of the different systems can be correlated to the electrochemical data, especially for the second photooxidation.
![Schematic presentation for the photooxidation mechanism of [MnII(L)3]2+ by [RuII(bpy)3]2+ in the presence of an external electron acceptor ArN2+.](/image/article/2005/CP/b411365a/b411365a-s2.gif) | ||
| Scheme 2 Schematic presentation for the photooxidation mechanism of [MnII(L)3]2+ by [RuII(bpy)3]2+ in the presence of an external electron acceptor ArN2+. | ||
Experimental
Materials and general methods
Acetonitrile (CH3CN, Rathburn, HPLC grade) and tetra-n-butylammonium perchlorate (Bu4NClO4, Fluka) were used as received and stored under an argon atmosphere in a dry glove box. X-band electron paramagnetic resonance (EPR) spectra were recorded with a Bruker ESP 300 E spectrometer at 100 K, with the following parameters: microwave 1 mW, modulation amplitude 0.197 G, time constant 327.68 ms, scan rate 1342 s, scan width 8 G, modulation frequency 100 kHz.General synthesis
Electrochemistry
All electrochemical measurements were run under an argon atmosphere in a dry glove box at room temperature. Cyclic voltammetry (CV) and controlled potential electrolysis experiments were performed using an EG&G model 173 potentiostat/galvanostat equipped with a PAR model universal programmer and a PAR model 179 digital coulometer. The standard three-electrodes electrochemical cell was used. Potentials were referred to an Ag/0.01 M AgNO3 reference electrode in CH3CN + 0.1 M Bu4NClO4. Potentials referred to that system can be converted to the ferrocene/ferrocenium couple by subtracting 87 mV, to SCE by adding 298 mV or to ENH reference electrode by adding 0.548 V. The working electrodes were platinum disks polished with 2 μm diamond paste (Mecaprex Presi) that were 5 mm in diameter for cyclic voltammetry (Epa, anodic peak potential; Epc, cathodic peak potential; E1/2 = (Epa + Epc)/2; ΔEp = Epa − Epc) and 2 mm in diameter for rotating disk electrode experiments (RDE). Exhaustive electrolyses were carried out on a platinum plate (5 cm2) or on a carbon felt electrode (RCV 2000, 65 mg cm−3). The auxiliary electrode was a Pt wire in CH3CN + 0.1 M Bu4NClO4. For electrochemical experiments, electronic absorption spectra were recorded on a Hewlett-Packard 8452 A diode array spectrophotometer. Initial and electrolyzed solutions were transferred to a conventional 0.1 or 1 cm path length quartz cells in the glove box.Spectroscopy
The luminescence lifetime of [RuII(bpy)3]2+* without or with a manganese complex was performed after irradiation at λ = 337 nm with a 4 ns pulsed N2 laser (optilas VSL-337ND-S) and recorded at λ = 650 nm using filter and a photomultiplier tube (Hamamatsu E2380-01) coupled with an ultra-fast oscilloscope (LeCroy 9310 AM).
Results and discussion
Electrochemistry and spectroscopy
Since the photocatalytical oxidation experiments have been carried out in CH3CN + 0.1 M Bu4NClO4 solutions containing a mixture of [RuII(bpy)3]2+ and manganese complexes ([MnII(L)3]2+ or [Mn2III,IVO2(L)4]3+) in the concentration ratio of about 1/10, it was important to known precisely the spectroscopic (UV-visible and EPR) and electrochemical properties of such solutions. We firstly reinvestigated in details the properties of each complex and their corresponding oxidized forms in this solvent and secondly those of a mixture of manganese and ruthenium complexes. Table 1 and Table 2 summarize, respectively, the electrochemical and UV-visible data of the complexes.| Complexes | Oxidation processesa | Reduction processesa | ||
|---|---|---|---|---|
| a Versus Ag/Ag+ (0.01 M AgNO3 in CH3CN + 0.1 M Bu4NClO4). b Irreversible processes. | ||||
| E 1/2/V (ΔEp/mV) | E 1/2/V (ΔEp/mV) | |||
| RuII ⇄ RuIII | 1st | 2nd | 3rd | |
| [RuII(bpy)3]2+ | 0.98 (80) | −1.64 (60) | −1.83 (60) | −2.08 (70) |
| E 1/2/V (ΔEp/mV) | E pc/V | |||
| Mn2III,IVO2 ⇄ Mn2IV,IVO2 | Mn2III,IVO2 ⇄ Mn2III,IIIO2 → MnII | |||
| [Mn2III,IVO2(bpy)4]3+ | 1.02 (90) | 0.04 | ||
| [Mn2III,IVO2(dmbpy)4]3+ | 0.86 (80) | −0.10 | ||
| E pa/Vb | E 1/2/V (ΔEp/mV) | |||
| MnII → MnIII | 1st | 2nd | 3rd | |
| [MnII(bpy)3]2+ | 0.84 | −1.66 (60) | −1.83 (60) | −2.07 (80) |
| [MnII(dmbpy)3]2+ | — | −1.77 (80) | −1.95 (70) | −2.16 (80) |
| Complexes | λ abs/nm (ε/M−1 cm−1) |
|---|---|
| [RuII(bpy)3]2+ | 432 (sh. 13030) 452 (15000) |
| [RuIII(bpy)3]3+ | 416 (4700) 668 (460) |
| [Mn2III,IVO2(bpy)4]3+ | 406 (sh. 1950) 470 (sh. 1100) 525 (556) 555 (460) 684 (560) |
| [Mn2III,IVO2(dmbpy)4]4+ | 406 (sh. 2060) 470 (sh. 1250) 528 (550) 559 (450) 688 (560) |
| [MnII(dmbpy)3]2+ | 249 (22900) 295 (32800) 304 (sh. 25000) |
| [MnII(bpy)3]2+ | 243 (25300) 295 (31300) 307 (sh. 23000) |
| [Mn2III,IVO2(L)4]3+ ⇄ [Mn2IV,IVO2(L)4]4++ e− | (1) |
| [Mn2III,IVO2(L)4]3+ + e− → [MnII(L)3]2+ + (MnIVO2)x + L | (2) |
These oxidation processes are associated by significant changes in UV-visible absorption (Table 2). The visible absorption spectra of the initial green MnIII–MnIV solutions show three bands at 525, 555 and 684 nm for L = bpy (528, 559 and 688 for L = dmbpy) (Fig. 1). Similar bands have been detected for other μ-oxo binuclear complexes28 allowing us to assign them as MnIV d–d transitions for those at 525 and 555 nm and ligand to metal charge transfer (LMCT) from oxo ligands to MnIV for the band at 684 nm. In the fully oxidized brown MnIV–MnIV solutions, new shoulders appear at 565 and 638 nm for L = bpy (570 and 640 for L = dmbpy). In addition, the intensity of the absorbance between 400 and 500 nm increases strongly and is associated with the appearance of two clear shoulders at 420 and 490 nm (420 and 485 nm for L = dmbpy). These latter shoulders were assigned to the contribution of the MnIV d–d transitions and to the LMCT of oxo ligands to MnIV ion.28
![Visible absorption spectra in CH3CN + 0.1 M Bu4NClO4, of (a) 2 mM [MnII(dmbpy)3]2+; (b) 0.9 mM [Mn2III,IVO2(dmbpy)4]3+ and (c) solution (b) after exhaustive oxidation at 1.10 V (formation of [Mn2IV,IVO2(dmbpy)4]4+), l = 1 cm. Insert, l = 1 mm.](/image/article/2005/CP/b411365a/b411365a-f1.gif) | ||
Fig. 1 Visible absorption spectra in CH3CN + 0.1 M Bu4NClO4, of (a) 2 mM [MnII(dmbpy)3]2+; (b) 0.9 mM [Mn2III,IVO2(dmbpy)4]3+ and (c) solution (b) after exhaustive oxidation at 1.10 V (formation of [Mn2IV,IVO2(dmbpy)4]4+), l![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) =1 cm. Insert, l=1 mm. =1 cm. Insert, l=1 mm. | ||
The oxidation processes are also evidenced by EPR spectroscopy. Indeed, X-band EPR spectroscopy allows to easily distinguish the MnIV–MnIV and MnIII–MnIV species since MnIV–MnIV is silent whereas the mixed-valence species presents the regular 16 line spectrum centred at g = 2.29 Experimentally, exhaustive oxidations lead to the quasi-total disappearance of the initial 16 line signal of the starting complexes on the X-band EPR spectra.
In the case of the dmbpy complex, the markedly lower oxidation potential of [Mn2III,IVO2(dmbpy)4]3+ (E1/2 = 0.86 V) compared to [RuII(bpy)3]2+ (E1/2 = 0.98 V), ΔE1/2 = 120 mV, leads to the clear observation of both of these successive oxidation systems in the cyclic voltammogram. This demonstrates that the oxidized RuIII form could act as an efficient oxidant towards [Mn2III,IVO2(dmbpy)4]3+. An electrolysis performed at E = 1.10 V and stopped after one electron has been consumed allows the selective oxidation of the manganese complex into [Mn2IV,IVO2(dmbpy)4]4+ with a 95% yield. At this electrolysis potential, both [Mn2III,IVO2(dmbpy)4]3+ and [RuII(bpy)3]2+ can be oxidized. The electrogenerated [RuIII(bpy)3]3+ oxidizes in turn the manganese complex. That is the reason why, when electrolysis is stopped after one electron has been consumed, only the manganese complex is oxidized. The pursuit of the electrolysis at this potential induced the quantitative oxidation of [RuII(bpy)3]2+ into [RuIII(bpy)3]3+.
The [Mn2III,IVO2(bpy)4]3+ (E1/2 = 1.02 V) complex is slightly more difficult to oxidize than [RuII(bpy)3]2+. The very small difference of potential between RuII/RuIII and MnIIIMnIV/MnIVMnIV redox systems (ΔE1/2 = 40 mV) implies that these two oxidation processes cannot be clearly distinguished, the cyclic voltammogram of the mixture exhibiting only one single reversible system at E1/2 = 1.00 V. Consequently, an electrolysis of the solution at 1.20 V yields to the concomitant formation of [RuIII(bpy)3]3+ and [Mn2IV,IVO2(bpy)4]4+ and does not allow the selective formation of [RuIII(bpy)3]3+. This result shows that [RuIII(bpy)3]3+ should not be a good candidate for an efficient oxidation of [Mn2III,IVO2(bpy)4]3+ in contrast to the case of [Mn2III,IVO2(dmbpy)4]3+ (see the ‘Continuous photolysis’ section).
The UV-visible spectra of the initial and oxidized solutions are the results of the superimposition of the absorption bands of the ruthenium and binuclear manganese complexes in their initial and oxidized forms.
The EPR spectra of the initial solutions present the typical 16 line spectra of the [Mn2III,IVO2(L)4]3+ species centred at g = 2 (Fig. 2A for L = dmbpy). In the fully oxidized solutions, the EPR spectra show now only a low intensity broad signal at g = 2.7 corresponding to [RuIII(bpy)3]3+ (Fig. 2B for L = dmbpy).18
![EPR spectra of a mixture of 1.2 mM [Mn2III,IVO2(dmbpy)4]3+ and 0.1 mM of [RuII(bpy)3]2+ in CH3CN + 0.1 M Bu4NClO4. (A) Initial solution, (B) after exhaustive electrolyses at 1.10 V: formation of [Mn2IV,IVO2(dmbpy)4]4+ and [RuIII(bpy)3]3+.](/image/article/2005/CP/b411365a/b411365a-f2.gif) | ||
| Fig. 2 EPR spectra of a mixture of 1.2 mM [Mn2III,IVO2(dmbpy)4]3+ and 0.1 mM of [RuII(bpy)3]2+ in CH3CN + 0.1 M Bu4NClO4. (A) Initial solution, (B) after exhaustive electrolyses at 1.10 V: formation of [Mn2IV,IVO2(dmbpy)4]4+ and [RuIII(bpy)3]3+. | ||
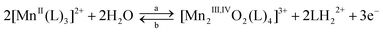 | (3) |
![Cyclic voltammograms at a Pt electrode in CH3CN + 0.1 M Bu4NClO4 of (A) 1.2 mM [MnII(bpy)3]2+ with sweep rate ν = 100 mV s−1; (B) ν = 20 mV s−1; (C) after addition of 0.1 mM of [RuII(bpy)3]2+, ν = 100 mV s−1. (D) 1.2 mM [MnII(dmbpy)3]2+, ν = 100 mV s−1; (E) ν = 20 mV s−1; (F) after addition of 0.1 mM of [RuII(bpy)3]2+, ν = 100 mV s−1.](/image/article/2005/CP/b411365a/b411365a-f3.gif) | ||
| Fig. 3 Cyclic voltammograms at a Pt electrode in CH3CN + 0.1 M Bu4NClO4 of (A) 1.2 mM [MnII(bpy)3]2+ with sweep rate ν=100 mV s−1; (B) ν=20 mV s−1; (C) after addition of 0.1 mM of [RuII(bpy)3]2+, ν=100 mV s−1. (D) 1.2 mM [MnII(dmbpy)3]2+, ν=100 mV s−1; (E) ν=20 mV s−1; (F) after addition of 0.1 mM of [RuII(bpy)3]2+, ν=100 mV s−1. | ||
An exhaustive potential-controlled oxidation at E = 0.84 V consumes 1.5 electrons per manganese atom and furnishes a green solution that exhibits the spectroscopic and electrochemical features of the [Mn2III,IVO2(bpy)4]3+ species (yield: 75%). In addition to the typical electroactivity of the binuclear complex, the cyclic voltammogram shows an additional irreversible peak at Epc = −0.70 V typical of the reduction of the released protonated bipyridine ligand (eqn. (3)). This transformation is reversible as confirmed by a controlled-potential reduction at −0.1 V which consumes 3 electrons per molecule of binuclear and regenerates a solution of [MnII(bpy)3]2+ with a 90% yield.
In the case of [MnII(dmbpy)3]2+, the rate of formation of the corresponding di-μ-oxo binuclear complex is markedly slower than that of [MnII(bpy)3]2+ as displayed on the cyclic voltammograms recorded at scan rate of 20 and 100 mV s−1 (Fig. 3D, E). At 20 mV s−1 the shape of the cyclic voltammogram is closed to that of the bpy complex recorded at 100 mV s−1 with a shift towards the lower potential values of about 140 mV, due to the electronic donating effect of the methyl substitution. However, no clear shoulder related to the MnII/MnIII oxidation process is observed. At 100 mV s−1, the formation of the binuclear di-μ-oxo is less visible and the oxidation and reduction irreversible peaks (Epa = 1.10 V and Epc = 0.61 V, respectively) can be attributed to intermediate species produced during the electrochemical transformation of [MnII(dmbpy)3]2+ into [Mn2III,IVO2(dmbpy)4]3+.22 This latter complex can be obtained with a 75% yield by an exhaustive oxidation performed at the foot of the anodic peak (E = 0.72 V) indicating that the oxidation of MnII to MnIII occurs at this potential even if no clear shoulder is observed.
The UV-visible absorption spectra of the [MnII(L)3]2+ complexes present absorption bands only in the near UV region corresponding to π → π* bands of the ligands (Table 2, Fig. 1), and exhaustive oxidations lead to the appearance of the typical visible absorption bands of the [Mn2III,IVO2(L)4]3+ complexes (Table 2).
The X-band EPR spectra of the initial solutions of [MnII(L)3]2+ exhibit 6 line signals centred at g = 2 in accordance with a high-spin MnII (3d5) ion, the hyperfine splitting resulting from the interaction between the electronic (S = 5/2) and the nuclear spin (I = 5/2). After electrochemical oxidations into [Mn2III,IVO2(L)4]3+ complexes, the 6 line spectra are replaced by the typical 16 lines of these latter. However, these 16 line signal is slightly distorted by the presence of a weak 6 line signal of Mn(CH3CN)62+ indicating a partial decoordination of manganese during the oxidation processes (see the next section).
![Cyclic voltammograms at a Pt electrode in CH3CN + 0.1 M Bu4NClO4 obtained after oxidation at 1.10 V (1.5 electron consumed) of a mixture of 1.2 mM [MnII(L)3]2+ and 0.1 mM of [RuII(bpy)3]2+, (A) L = bpy, (B) L = dmbpy; ν = 100 mV s−1.](/image/article/2005/CP/b411365a/b411365a-f4.gif) | ||
| Fig. 4 Cyclic voltammograms at a Pt electrode in CH3CN + 0.1 M Bu4NClO4 obtained after oxidation at 1.10 V (1.5 electron consumed) of a mixture of 1.2 mM [MnII(L)3]2+ and 0.1 mM of [RuII(bpy)3]2+, (A) L=bpy, (B) L=dmbpy; ν=100 mV s−1. | ||
Concerning the EPR spectra (Fig. 5, for L = dmbpy), after oxidation into [Mn2III,IVO2(L)4]3+, the 16 line signals, characteristic of these complexes, replaced the initial 6 lines of the starting MnII species. However, it should be noted that the signal is distorted by the presence of a 6 line signal centred at g = 2 corresponding to a new mononuclear MnII species. After oxidation into [Mn2IV,IVO2(L)4]4+, only this signal is present (Fig. 5C). This spectrum is identical to that of Mn(ClO4)2·6H2O dissolved in CH3CN, allowing one to assign it, without ambiguity, to MnII(CH3CN)62+. Indeed, it has been demonstrated by EXAFS spectroscopy that in CH3CN medium, Mn2+ is coordinated by six nitrogens of the solvent.30 In the fully oxidized solutions, the additional signal centred at g = 2.7 of [RuIII(bpy)3]3+ is also observed (Fig. 5D).18
![Evolution of the EPR spectrum during electrolyses at 1.10 V of the mixture of 1.2 mM [MnII(dmbpy)3]2+ and 0.1 mM of [RuII(bpy)3]2+ in CH3CN + 0.1 M Bu4NClO4. (A) Initial solution, (B) after formation of [Mn2III,IVO2(dmbpy)4]3+, (C) after formation of [Mn2IV,IVO2(dmbpy)4]4+, (D) after formation of [RuIII(bpy)3]3+.](/image/article/2005/CP/b411365a/b411365a-f5.gif) | ||
| Fig. 5 Evolution of the EPR spectrum during electrolyses at 1.10 V of the mixture of 1.2 mM [MnII(dmbpy)3]2+ and 0.1 mM of [RuII(bpy)3]2+ in CH3CN + 0.1 M Bu4NClO4. (A) Initial solution, (B) after formation of [Mn2III,IVO2(dmbpy)4]3+, (C) after formation of [Mn2IV,IVO2(dmbpy)4]4+, (D) after formation of [RuIII(bpy)3]3+. | ||
Photophysics
Any molecule presenting an absorption in the visible region, even with a weak intensity, is likely to react by an electronic energy transfer (EET) process with the excited state of the photosensitizer, [RuII(bpy)3]2+*. This is the case for the [Mn2III,IVO2(L4)]3+ complexes and at a markedly less extent, for the [MnII(L)3]2+ ones (absorption in the near UV region; see Table 2). Since an energy transfer process between [RuII(bpy)3]2+* and the different Mn complexes cannot be ruled out, and may limit the photooxidation rate constant of [RuII(bpy)3]2+* by the irreversible electron acceptor used (ArN2+) it was important to investigate the effect of the presence of the manganese complexes on the luminescence lifetime of [RuII(bpy)3]2+* (τ) before studying the electron transfer (ET) processes between [RuIII(bpy)3]3+ and the manganese complexes.| τ0/τ = 1 + kqτ0[Q] | (4) |
![Stern–Volmer plots in deoxygenated CH3CN + 0.1 M Bu4NClO4 of [RuII(bpy)3]2+* (0.013 mM) in the presence of variable concentration of quencher Q, Q = (a) [MnII(dmbpy)3]2+, (b) [Mn2III,IVO2(dmbpy)4]3+ and (c) ArN2+.](/image/article/2005/CP/b411365a/b411365a-f6.gif) | ||
| Fig. 6 Stern–Volmer plots in deoxygenated CH3CN + 0.1 M Bu4NClO4 of [RuII(bpy)3]2+* (0.013 mM) in the presence of variable concentration of quencher Q, Q=(a) [MnII(dmbpy)3]2+, (b) [Mn2III,IVO2(dmbpy)4]3+ and (c) ArN2+. | ||
| [RuII(bpy)3]2+* + [Mn2III,IVO2(L)4]3+ → [RuI(bpy)3]+ + [Mn2IV,IVO2(L)4]4+ | (5) |
| [RuII(bpy)3]2+* + [Mn2III,IVO2(L)4]3+ → [RuIII(bpy)3]3+ + [Mn2III,IIIO2(L)4]2+ (→MnII) | (6) |
| [RuII(bpy)3]2+* + [Mn2III,IVO2(L)4]3+ → [RuII(bpy)3]2+ + [Mn2III,IVO2(L)4]3+* | (7) |
The reductive process following eqn. (5) has to be ruled out since in the most favourable case, this process would be endergonic of 400 mV (E1/2 MnIV–MnIV/MnIII–MnIV = 0.86 V for L = dmbpy and E1/2 RuII*/RuI = 0.46 V). For the oxidative one, (eqn. (6)), although it is presumably strongly exoergonic, its contribution to the quenching process is rather difficult to evaluate. Indeed the [Mn2III,IVO2(L)4]3+ complexes are irreversibly reduced and the potential measured (Epc = 0.04 and −0.10 V for bpy and dmbpy, respectively) does not represent the thermodynamic E1/2 value of the couple [Mn2III,IVO2(L)4]3+/[Mn2III,IIIO2(L)4]2+. Moreover, after a continuous visible irradiation (3 h), no net changes of the absorption spectra of the solutions are detected. Meanwhile, this observation is not a conclusive proof that the quenching following eqn. (6) does not occur. Indeed, as observed from the electrochemical study, these complexes are irreversibly reduced in mononuclear [MnII(L)3]2+ species. These latter complexes are then easily reoxidized via the transient photogenerated [RuIII(bpy)3]3+ one, restoring the initial [Mn2III,IVO2(L)4]3+ complexes, explaining the absence of spectroscopic change during continuous irradiation experiments.
So for thermodynamic and kinetic point of view, the energy transfer (eqn. (7)) represents presumably the main contribution to the quenching process of [RuII(bpy)3]2+*. This process is likely owing to the large overlap of emission band centred at 600 nm of [RuII(bpy)3]2+* with the visible absorption bands of the [Mn2III,IVO2(L)4]3+ complexes (Table 2).
The origin of the energy transfer process is rather difficult to determined since it might be due to a coulombic interaction (Förster) or a short range electron exchange (Dexter).33,34 In our case, due to the important likeness of the UV-visible spectra of the binuclear bpy and dmbpy, the values of kq are similar for both complexes studied and smaller than the diffusion rate constant of the molecules. That could suggest that the interaction is due to a short-range electron exchange, since this process involves the interchange of electrons between [RuII(bpy)3]2+* and the [Mn2III,IVO2(L)4]3+ complexes and thus these complexes must diffuse in order to permit the overlap of the orbitals of the two components.35 Moreover, the energy transfer processes allowed by the coulombic interaction are those in which there is no change in spin multiplicity in either component. In the case of [RuII(bpy)3]2+*-like complexes, the MLCT excited states are a mixture of three states in rapid Boltzmann equilibrium but largely triplet in character (more than 90%).36 The ground state of [RuII(bpy)3]2+ is, on contrary, a pure singlet one which makes the Förster interaction unlikely. Finally we must notified that the Dexter mechanism, which is not favourable if we consider the strong electronic repulsion between the complexes, has already been suspected in the case of similar complexes of RuII* and FeII.37,38 After the quenching process, the populated excited state of the [Mn2III,IVO2(L)4]3+ complexes quickly relax to the ground state via a radiationless deactivation pathway.
Continuous photolysis
| [RuII(bpy)3]2+* + ArN2+ → [RuIII(bpy)3]3+ + ArN2˙ | (8) |
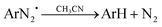 | (9) |
In addition, it should be notified that no interactions have been identified between the mono or binuclear manganese complexes and the diazonium salt.
![Evolution of the visible absorption spectra of a CH3CN + 0.1 M Bu4NClO4 solution containing a mixture of [Mn2III,IVO2(dmbpy)4]3+ (0.7 mM), [RuII(bpy)3]2+ (0.05 mM) and ArN2+ (15 mM) under visible irradiation. (A): (a) initial solution, (b) after 140 s, (c) 270 s, (d) 480 s, (e) 680 s, (f) 900 s, (g) 1160 s, (h) 1650 s. (B): (h) 1650 s, (i) 2300 s, (j) 3500 s; l = 1 cm.](/image/article/2005/CP/b411365a/b411365a-f7.gif) | ||
| Fig. 7 Evolution of the visible absorption spectra of a CH3CN + 0.1 M Bu4NClO4 solution containing a mixture of [Mn2III,IVO2(dmbpy)4]3+ (0.7 mM), [RuII(bpy)3]2+ (0.05 mM) and ArN2+ (15 mM) under visible irradiation. (A): (a) initial solution, (b) after 140 s, (c) 270 s, (d) 480 s, (e) 680 s, (f) 900 s, (g) 1160 s, (h) 1650 s. (B): (h) 1650 s, (i) 2300 s, (j) 3500 s; l=1 cm. | ||
In the case of the dmbpy complex, two successive steps are observed on the absorption spectra during irradiation (Fig. 7). In the first step (Fig. 7A), the visible absorption bands at 528, 559 and 688 nm of [Mn2III,IVO2(dmbpy)4]3+ progressively decrease in favour of the emergence of those at 570 and 640 nm, characteristics of the [Mn2IV,IVO2(dmbpy)4]4+ species. In addition, the intensity of the band at 432 nm increases due to the more intense absorption in this area (between 400 and 500 nm) of the MnIV–MnIV species than that of the MnIII–MnIV one (Table 2). All these spectral changes are in accordance with the photooxidation of the [Mn2III,IVO2(dmbpy)4]3+ into [Mn2IV,IVO2(dmbpy)4]4+ by the photogenerated RuIII species. The maximum yield of formation of [Mn2IV,IVO2(dmbpy)4]4+ (80%; estimated by spectroscopic titration) is reached after 1650 s of irradiation (Fig. 7, spectrum (h)). This yield is only slightly lower to that obtained by an electrochemical oxidation of [Mn2III,IVO2(dmbpy)4]3+ with or without [RuII(bpy)3]2+, demonstrating the efficiency of the photooxidation process. A more prolonged irradiation (up to 3500 s) produces quantitatively [RuIII(bpy)3]3+. This second step is illustrated by the decrease of the absorbance at 432 nm (Fig. 7B).
These two successive photoredox processes are also confirmed by following irradiation by EPR spectroscopy, the EPR spectra being almost superimposable on those obtained by electrochemical oxidation (Fig. 2). Initially, since [Mn2IV,IVO2(dmbpy)4]4+ is EPR silent, the light exposure leads to the progressive fading out of the 16 line EPR spectrum of [Mn2III,IVO2(dmbpy)4]3+ until it almost disappears. Then, in a second step, oxidation of [RuII(bpy)3]2+ is verified by the observation of a new weak signal at g = 2.7 typical of the RuIII form of the photosensitizer. These results are consistent with the fact that [RuIII(bpy)3]3+ is an efficient oxidant toward [Mn2III,IVO2(dmbpy)4]3+, as expected by comparison of the E1/2 values of the redox couples RuIII/RuII and MnIV–MnIV/ MnIII–MnIV (Table 1).
In contrast with [Mn2III,IVO2(bpy)4]3+, the photooxidation process is poorly efficient. A continuous irradiation of a solution containing this complex, [RuII(bpy)3]2+ and ArN2+ allows the formation of only 20% of [Mn2IV,IVO2(bpy)4]4+ and at the same time of [RuIII(bpy)3]3+. The yield of the oxidized manganese complex has been determined by electrochemistry by comparison of the height of the MnIII–MnIV/MnIV–MnIV waves before and after irradiation at a rotating disk electrode.
EPR analysis during irradiation unambiguously confirms the poor efficiency of the photoredox process. Indeed, contrary to the case of the dmbpy derivative, a continuous irradiation leads to a large degradation of the manganese complex into a diamagnetic, unidentified species; this is attested by the progressive decrease of the typical 16 line spectrum of [Mn2III,IVO2(bpy)4]3+ associated with the appearance of a weak 6 line signal due to the formation of a small amount of Mn(CH3CN)62+. These results are in good accordance with the electrochemical data, since the E1/2 value of the RuIII/RuII couple is 40 mV lower than that of the [Mn2III,IVO2(bpy)4]3+/[Mn2IV,IVO2(bpy)4]4+. The comparison of the efficiency of the photocatalytic process starting from [Mn2III,IVO2(L)4]3+ demonstrated that the formation of the [Mn2IV,IVO2(L)4]4+ oxidized species is thermally governed and very sensitive to the substitution of the bipyridyl ligands.
![Evolution of the visible absorption spectra of a CH3CN + 0.1 M Bu4NClO4 solution containing a mixture of [MnII(dmbpy)3]2+ (1 mM), [RuII(bpy)3]2+ (0.05 mM) and ArN2+ (15 mM) under visible irradiation. (A): (a) after 60, (b) 180, (c) 360, (d) 540, (e) 900, (f) 1260, (g) 1620 s; l = 1 cm.](/image/article/2005/CP/b411365a/b411365a-f8.gif) | ||
| Fig. 8 Evolution of the visible absorption spectra of a CH3CN + 0.1 M Bu4NClO4 solution containing a mixture of [MnII(dmbpy)3]2+ (1 mM), [RuII(bpy)3]2+ (0.05 mM) and ArN2+ (15 mM) under visible irradiation. (A): (a) after 60, (b) 180, (c) 360, (d) 540, (e) 900, (f) 1260, (g) 1620 s; l=1 cm. | ||
The photochemical induced oxidations of [MnII(L)3]2+ by [RuIII(bpy)3]3+ is efficient for L = bpy and dmbpy, as predicted by the less anodic potential values of MnII/MnIII compared to that of RuII/RuIII (Table 1). EPR spectroscopy attests also the formation of [Mn2III,IVO2(L)4]3+ by the progressive conversion of the initial 6 line signal of the MnII complexes into the typical 16 line spectra of the MnIII–MnIV ones.
A prolonged irradiation of the solutions yield similar results to those observed during irradiation of solutions containing [RuII(bpy)3]2+, [Mn2III,IVO2(L)4]3+ and ArN2+. As expected, the MnIV–MnIV species is photogenerated with a good yield only in the case of the dmbpy complex.
Conclusion
This study demonstrates that di-μ-oxo complexes [Mn2III,IVO2(L)4]3+; L = 2,2′-bipyridine and 4,4′-dimethyl-2,2′-bipyridine can be generated with high efficiency by photoinduced oxidation in CH3CN of the corresponding mononuclear [MnII(L)3]2+ using [RuII(bpy)3]2+ as photosensitizer in the presence of an aryl diazonium salt acting as an irreversible electron acceptor. A further photoinduced oxidation with the same system into the corresponding [Mn2IV,IVO2(L)4]4+ species can be achieved for the complex by having the bipyridine ligands substituted by an electron donating group (L = dmbpy) in agreement with the thermodynamic features of the reaction determined from electrochemical data. It is noteworthy that the efficiency of this photoinduced oxidation is also quite high (only 15% lower than that obtained by electrochemical oxidation) in spite of the strong quenching, presumably by an electronic energy transfer mechanism, of the 3MLCT excited state of [RuII(bpy)3]2+ by [Mn2III,IVO2(L)4]3+.The extension of this work is currently under way using a unimolecular polymetallic complex combining the properties of [RuII(bpy)3]2+ and [MnII(L)3]2+moieties, a better efficiency in terms of kinetic of the electron transfer between the ground state [RuIII(bpy)3]3+ and [MnII(L)3]2+ through an intramolecular process should be expected.
Acknowledgements
We thank A. Jeunet from Laboratoire d’Etude Dynamiques et Structurales de la Sélectivité, UMR CNRS 5616, Université Joseph Fourier Grenoble France, Institut de Chimie Moléculaire de Grenoble, FR CNRS 2607, for EPR experimental assistance.References
- D. Burdinski, K. Wieghardt and S. Steenken, J. Am. Chem. Soc., 1999, 121, 10781 CrossRef CAS.
- D. Burdinski, E. Bothe and K. Wieghardt, Inorg. Chem., 2000, 39, 105 CrossRef.
- L. Sun, M. Burkitt, M. Tamm, M. K. Raymond, M. Abrahamsson, D. le Gourriérec, Y. Frapart, A. Magnuson, P. H. Kenéz, P. Brandt, A. Tran, L. Hammarström, S. Styring and B. Akermark, J. Am. Chem. Soc., 1999, 121, 6834 CrossRef CAS.
- L. Sun, L. Hammarström, B. Akermark and S. Styring, Chem. Soc. Rev., 2001, 30, 36 RSC.
- L. Hammarström, L. Sun, B. Akermark and S. Styring, Spectrochim. Acta, 2001, 37A, 2145 CrossRef.
- M. L. A. Abrahamsson, H. B. Baudin, A. Tran, C. Philouze, K. E. Berg, M. K. Raymond-Johansson, L. Sun, B. Akermark, S. Styring and L. Hammarström, Inorg. Chem., 2002, 41, 1534 CrossRef CAS.
- A. Zouni, H.-T. Witt, J. Kern, P. Fromme, N. Krauss, W. Saenger and P. Orth, Nature, 2001, 409, 739 CrossRef CAS.
- N. Kamiya and J.-R. Shen, Proc. Natl. Acad. Sci. USA, 2003, 100, 98 CrossRef CAS.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831 CrossRef CAS.
- L. Sun, L. Hammarström, T. Norrby, H. Berglund, R. Davydov, M. Andersson, A. Börje, P. Korall, C. Philouze, M. Almgren, S. Styring and B. Akermark, Chem. Commun., 1997, 607 RSC.
- H. Berglund-Baudin, L. Sun, R. Davidov, M. Sundahl, S. Styring, B. Akermark, M. Almgren and L. Hammarström, J. Phys. Chem. A, 1998, 102, 2512 CrossRef CAS.
- L. Sun, H. Berglund, R. Davydov, T. Norrby, L. Hammarström, P. Korall, A. Börje, C. Philouze, K. Berg, A. Tran, M. Andersson, G. Stenhagen, J. Martensson, M. Almgren, S. Styring and B. Akermark, J. Am. Chem. Soc., 1997, 119, 6996 CrossRef CAS.
- L. Sun, M. K. Raymond, A. Magnuson, D. LeGourriérec, M. Tamm, M. Abrahamsson, P. H. Kenéz, J. Martensson, G. Stenhagen, L. Hammarström, S. Styring and B. Akermark, J. Inorg. Biochem., 2000, 78, 15 CrossRef CAS.
- P. Huang, A. Magnuson, R. Lomoth, M. Abrahamsson, M. Tamm, L. Sun, B. van Rotterdam, J. Park, L. Hammarström, B. Akermark and S. Styring, J. Inorg. Biochem., 2002, 91, 159 CrossRef CAS.
- A. Johansson, M. Abrahamsson, A. Magnuson, P. Huang, J. Martensson, S. Styring, L. Hammarström, L. Sun and B. Akermark, Inorg. Chem., 2003, 42, 7502 CrossRef CAS.
- A. Magnuson, H. Berglund, P. Korall, L. Hammarström, B. Akermark, S. Styring and L. Sun, J. Am. Chem. Soc., 1997, 119, 10720 CrossRef CAS.
- M. Sjödin, S. Styring, B. Akermark, L. Sun and L. Hammarström, J. Am. Chem. Soc., 2000, 122, 3932 CrossRef.
- A. Magnuson, Y. Frapart, M. Abrahamsson, O. Horner, B. Akermark, L. Sun, J.-J. Girerd, L. Hammarström and S. Styring, J. Am. Chem. Soc., 1999, 121, 89 CrossRef CAS.
- F. Lafolet, J. Chauvin, M.-N. Collomb, A. Deronzier, H. Laguitton-Pasquier, J.-C. Leprêtre, J.-C. Vial and B. Brasme, Phys. Chem. Chem. Phys., 2003, 5, 2520 RSC.
- S. A. Richer, P. K. S. Tsang and D. T. Sawyer, Inorg. Chem., 1989, 28, 2471 CrossRef.
- M. M. Morrison and D. T. Sawyer, Inorg. Chem., 1978, 17, 333 CrossRef CAS.
- M. N. Collomb Dunand-Sautier and A. Deronzier, J. Electroanal. Chem., 1997, 428, 65 CrossRef.
- H. Cano-Yelo and A. Deronzier, J. Chem. Soc., Faraday Trans. 1, 1984, 80, 3011 RSC.
- M.-N. Collomb Dunand Sauthier, A. Deronzier and X. Pradon, J. Am. Chem. Soc., 1997, 119, 3173 CrossRef.
- M.-N. Collomb Dunand Sauthier, A. Deronzier, A. Piron, X. Pradon and S. Ménage, J. Am. Chem. Soc., 1998, 120, 5373 CrossRef CAS.
- S. R. Cooper and M. Calvin, J. Am. Chem. Soc., 1977, 99, 6623 CrossRef CAS.
- M. M. Morrison and D. T. Sawyer, J. Am. Chem. Soc., 1977, 99, 257 CrossRef CAS.
- D. R Gamelin, M. L. Kirk, T. L. Stemmler, S. Pal, W. H. Armstrong, J. E. Penner Hahn and E. I. Solomon, J. Am. Chem. Soc., 1994, 116, 2392 CrossRef CAS.
- C. Hureau, G. Blondin, M. Cesario and S. Un, J. Am. Chem. Soc., 2003, 125, 11637 CrossRef CAS.
- Y. Inada, T. Sugata, K. Ozutsumi and S. Funahashi, Inorg. Chem., 1998, 37, 1886 CrossRef CAS.
- K. E. Berg, A. Tran, M. K. Raymond, M. Abrahamsson, J. Wolny, S. Redon, M. Andersson, L. Sun, L. Hammarström, H. Toftlund and B. Akermark, Eur. J. Inorg. Chem., 2001, 1019 CrossRef CAS.
- (a) C. P. Anderson, D. J. Salmon, T. J. Meyer and R. C. Young, J. Am. Chem. Soc., 1977, 99, 1980 CrossRef CAS . The potentials given in this reference vs. SCE have been converted into Ag/Ag+ (0.01 M AgNO3 in CH3CN + 0.1 M Bu4NClO4) by subtracting 298 mV following (b); (b) V. V. Pavlishchuk and A. W. Addison, Inorg. Chim. Acta, 2000, 298, 97 CrossRef.
- T. H. Förster, Discuss. Faraday Soc., 1959, 27, 7 RSC.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 838 CrossRef.
- J. A. Barltrop and J. D. Coyle, in Excited States in Organic Chemistry, John Wiley & Sons Ltd, New York, 1975, ch. 4 Search PubMed.
- E. M. Kober and T. J. Meyer, Inorg. Chem., 1982, 21, 3967 CrossRef CAS.
- C. Crentz, M. Chou, T. L. Netzel, M. Okumura and N. Sutin, J. Am. Chem. Soc., 1980, 102, 1309 CrossRef CAS.
- R. H. Schmehl, R. A. Auerbach, W. F. Wacholtz, C. M. Elliott, R. A. Freitag and J. W. Merkert, Inorg. Chem., 1986, 25, 2440 CrossRef CAS.
| This journal is © the Owner Societies 2005 |