Dynamic field sampling of airborne organophosphate triesters using solid-phase microextraction under equilibrium and non-equilibrium conditions
Sindra
Isetun
and
Ulrika
Nilsson
*
Department of Analytical Chemistry, Stockholm University, S-106 91 Stockholm, Sweden. E-mail: ulrika.nilsson@anchem.su.se
First published on 11th November 2004
Abstract
A simple setup for dynamic air sampling using a solid-phase microextraction (SPME) device designed for use in the field was evaluated for organophosphate triester vapour under both equilibrium and non-equilibrium conditions. The effects of varying the applied airflows in the sampling device were evaluated in order to optimise the system with respect to the Reynolds number and magnitude of the boundary layer that developed near the surface. Further, the storage stability of the analytes was studied for both capped and uncapped 100-µm PDMS fibres. Organophosphate triesters are utilized on large scales as flame-retardants and/or plasticizers, for instance in upholstered furniture. In indoor working environments these compounds have become common components in the surrounding air. Measurements were performed in a recently furnished working environment and the concentration of tris(2-choropropyl) phosphate was found to be 7 µg m−3.
Introduction
Solid-phase microextraction (SPME) is principally associated with sampling under equilibrium conditions using the specific fibre/sample partition coefficient, Kfs, for quantitative determinations. When the sample volume is considerably larger than the volume of the fibre coating, Vf, eqn. 1 can be used to calculate the concentration, C0, from the mass, m, extracted from the sample.1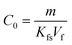 | (1) |
Parameters that affect Kfs are temperature and humidity. By decreasing the temperature of the fibre or heating up the sample Kfs can be increased, and further, as humidity in the sample is increased the coefficient is decreased.1–3 However, variations in temperature in the ‘room temperature’ range of 20–25 °C and in relative humidity up to 75% have no large effects on the partition coefficient.4–6 Agitation of the sample has proved to shorten the time required to reach equilibrium,7–10 and the mass extracted by the fibre is proportional to the concentration in the sample if the extraction time and agitation are held constant.11,12 The agitation conditions directly affect the boundary layer, δ, surrounding the fibre. In air sampling the flow velocity can be increased in order to minimize δ and consequently shorten the equilibration time. Under non-equilibrium conditions the extraction is interrupted and the concentration in the sample can be determined by using eqn. 2,13,14 where Dg is the analyte's coefficient of diffusion, L is the length of the fibre, b is the thickness of the fibre and t is the time of extraction.
 | (2) |
The magnitude of the boundary layer is given by δ = 9.52b/(Re0.62Sc0.38),1 where Re = the Reynolds number and Sc = the Schmidt number, although when the linear flow over the fibre exceeds approximately 10 cm s−1 the mass uptake rate and the size of the boundary layer are not further affected.13–15
Field sampling of volatile airborne compounds has been performed using SPME under both equilibrium5,6,16,17 and non-equilibrium18,19 conditions in various environments, where no additional flow over the fibres was applied. However, for compounds with very high partition coefficients and extended equilibration times, such as organophosphate triesters, dynamic sampling with a controlled linear airflow is more effective. Air sampling of organophosphate esters using SPME has recently been carefully evaluated and has proved to be a reliable technique for these compounds.15,20 In this work, a field sampler device was constructed and evaluated that simplifies dynamic field sampling using SPME under both equilibrium and non-equilibrium conditions. The principle of the sampler setup is similar to the design of a device evaluated in an earlier work by Pawliszyn and co-workers.14 The device enables air sampling where the linear airflow is controlled during the entire sampling period.
The air sampler device, shown in Fig. 1, is rectangular in cross-section and the SPME fibres are placed in series rather than side-by-side. It was therefore necessary to consider the effect of these novel features on the applied air flow. The parameters flow velocity (u), fluid density (ρ), fluid viscosity (μ) and the boundary layer (δ) surrounding the SPME fibre can be used to calculate a dimensionless expression, uδρ/μ, called the Reynolds number21 (Reδ), which provides useful indications about the nature of the airflow. When Reδ ≪ 1 the flow is symmetrical around the SPME fibre and no form drag occurs. As the flow is increased and Reδ > 10 the boundary layer is separated, eddies are formed, and the flow changes from laminar to turbulent. It was also important to consider the extent to which a laminar flow was maintained throughout the path length. The distance from the opening of the device to the location of the fibre is here expressed as x. At a certain critical point, xc, a transition from laminar to turbulent flow occurs. The point at which the flow changes is not sharply defined, but occurs gradually as the flow velocity increases in a given system. When the value of Rex (uxρ/μ) is less than approximately 2000 the flow is considered to be laminar and if the value exceeds approximately 4000 it is usually turbulent.21
 | ||
| Fig. 1 Schematic diagram of the SPME field sampler device showing: (1) the orifice of 3 × 60 mm; (2) the three holes to house the SPME holders; and (3) the outflow. | ||
When SPME is utilised in the field the storage stability of the analyte is a significant factor. Field sampling is often performed at some distance from the laboratory and several hours may pass before the SPME fibre is desorbed in the gas chromatograph (GC). Previous studies have evaluated storage stability for volatile organic compounds (VOCs) under various conditions.5 Therefore, this work includes a simplified study of the storage stability of organophosphate esters at room temperature.
Experimental
Chemicals and materials
Triethyl phosphate (99%), tri-n-propyl phosphate (99%), tri-n-butyl phosphate, triisobutyl (99%) and tris(2-chloroethyl) phosphate (97%) were purchased from Sigma–Aldrich, (Milwaukee, WI). Tris(chloropropyl) phosphate (100%) containing three isomers was provided by Akzo Nobel (Herkenbosch, the Netherlands). Methanol was purchased from Riedel-deHaen (Seelze, Germany) and dichloromethane from Merck (Darmstadt, Germany). The SPME sampler holders, SPME portable field sampler and polydimethylsiloxane (PDMS) SPME fibers (100 µm and 30 µm) were obtained from Supelco (Bellafonte, PA). Before use, all glassware was soaked in ethanol saturated with sodium hydroxide for >24 hours and then rinsed with water followed by ethanol and finally acetone. This was performed in order to avoid organophosphate contamination.Instrumentation
Gas chromatographic analyses of the air samples were carried out using an Agilent 6890 GC equipped with a 30 m × 0.25 mm × 0.25 µm DB-5 column (J & W Scientific, Folsom, CA), a split/splitless injector and a nitrogen–phosphorus detector (NPD). The injector temperature was set to 280 °C with the split closed for 2 min. The oven temperature was programmed to maintain a temperature of 50 °C for 2 min, then increase to 300 °C at 40° min−1. The temperature was maintained at 300 °C for 5 min before cooling. The temperature of the NPD was set to 300 °C. Agilent's Chemstation data system for GC was used for registering, storing and processing the detector signals.Gas-generating device
Triethyl phosphate was generated in an emission chamber from a permeation tube (Kin-Tek Laboratories, LaMarque, TX, USA), calibrated to emit 65 ng min−1 at 80 °C. The volumetric airflow in the chamber was adjusted to 32 L min−1. Thus, the concentration of triethyl phosphate in the chamber was 2.03 µg m−3. The emission chamber is described in detail in previous work.15 An atmosphere containing steady-state concentrations of gaseous tri-n-propyl, triisobutyl, tri-n-butyl, tris(2-chloroethyl) phosphate and three isomers of tris(2-chloropropyl) phosphate was generated from a standard mixture containing approximately 2 ng µl−1 of each of the analytes, as follows. First, the compounds were dissolved in methanol and diluted with distilled water. The solution was then placed in the chamber and agitated with a Teflon covered magnetic bar.Field sampler device
To simplify field sampling under a constant linear flow a sampling device adapted for SPME holders (Fig. 1) was constructed. A pair of plain anodized aluminium plates were fixed to opposite sides of a pair of Teflon plates, leaving a rectangular slit of 3 × 60 mm between them. Three holes in the Teflon plate were made to house the SPME needles, located 25, 60 and 95 mm from the opening of the device. The SPME fibres were placed in series and were exposed to a laminar stream of air passing through the sampler. Adjustable racks were used to set the SPME holders at an appropriate height in order to minimise disturbance of the laminar flow. The SPME fibres were extruded by at least 1 cm, since this was the length of the coated part of the fibre. A battery-operated personal sampler pump (224-PCXR7, SKC Inc) was connected to the outflow of the sampler. At the outflow, where the pump was connected, a sample holder for the filters could be connected. This setup enabled air to be sampled using SPME fibres and filters simultaneously.Air sampling
The field sampler was evaluated in an emission chamber generating a known concentration of triethyl phosphate. The sampler, equipped with the SPME fibres, was connected to the pump and placed in the chamber, whereupon air sampling commenced. The linear flow was adjusted to pass at between 10 and 35 cm s−1 over the SPME fibres, corresponding to volumetric flows in the range 1.1–3.8 L min−1.For non-equilibrium measurements the 100-µm PDMS fibres were used and extractions were performed over periods of 40–90 min. For equilibrium measurements using 30-µm PDMS fibres, the sampling duration was >18 hours. The amount of analyte desorbed from the SPME fibres was estimated from the detector response factors obtained from injections of a standard solution. The solution contained approximately 0.5 ng µl−1 of each compound dissolved in dichloromethane.
Active air sampling, using a 25-mm binder-free A/E borosilicate glass fibre filter (Gelman Sciences Inc., Ann Arbour, MI), was also performed in the chamber for comparative purposes. The pump flow was set to 3 L min−1 and the sampling periods were 2–8 hours. An internal surrogate standard, 100 ng of diphenylmethyl phosphate, was added to all the adsorbents prior to extraction. The filters were extracted in dichloromethane for 2 × 20 min in an ultrasonic bath (Bransonic 221). The extract was then gently evaporated under a stream of nitrogen before GC analysis.
Storage stability
Two types of SPME holder devices were evaluated and compared for their capacity to store the analyte over time; the ordinary uncapped fibre holder for manual sampling and the capped portable field sampler, both equipped with the 100-µm PDMS fibres. In the capped fibre holder the coated fibre is sealed behind a septum to prevent analyte losses. Extractions were performed in the emission chamber, where a stable concentration of triethyl phosphate was generated. The extraction duration was 60 min and storage intervals investigated were in the range 1–15 hours at 21–23 °C.Results and discussion
Evaluation of the field sampler
Active air sampling in the emission chamber was performed in duplicate on several occasions in order to verify the set concentration of triethyl phosphate, 2.03 µg m−3. Estimated concentrations from the filters were 1.98 µg m−3 (n = 10, RSD = 9%) as shown in Table 1.| SPMEac | Filtersbc | |
|---|---|---|
| a Average of six measurements. (*Average of eighteen measurements.) b Average of four measurements. (**Average of ten measurements.) c Relative standard deviations in percentage (% RSD) are presented in brackets. (Temp: 21–23 °C, RH: 3.5–5.5%). | ||
| Triethyl phosphate | 1995(13)* | 1980(9)** |
| Tri-n-propyl phosphate | 391(18) | 393(18) |
| Triisobutyl phosphate | 723(13) | 921(15) |
| Tri-n-butyl phosphate | 1107(13) | 860(13) |
| Tris(2-chloroethyl) phosphate | 356(27) | 386(7) |
| Tris(2-chloroisopropyl) phosphate | 696(13) | 870(13) |
The total path length of the flow in the field sampler was 150 mm, and the fibres were located 25, 60 and 95 mm from the opening. At the highest flow rate investigated the linear flow was set at 35 cm s−1 and the values of the Reynolds number (Rex) at the fibres were 486, 1167 and 1847, respectively. Consequently, even the maximal linear flows of 35 cm s−1 did not exceed the criteria for a laminar flow (Re < 2000) throughout the whole path. According to the values of the Reynolds number no turbulence was caused by the inner surfaces of the sampler. To obtain a fixed value for the boundary layer around the fibres a uniform laminar flow must be applied. In earlier work,15 linear airflows in the range 3–38 cm s−1 were tested in order to estimate the magnitude of the boundary layer as the flow was varied. The cited study showed that stable values were obtained when the fibres were placed in parallel and the flow rate exceeded 7 cm s−1. An applied flow rate above this value was considered to be sufficient to minimize the boundary layer around the SPME fibres. The flow past the fibres in our sampler device is in a perpendicular direction to its axis and the fibres are located in series. This difference from configurations used in previous studies makes it necessary to consider the type of flow created around the fibres. Predicting where turbulence occurs in the sampler could, however, be difficult. Instead, the masses extracted by each fibre at different applied flow rates were observed and compared. Flow rates investigated were 10, 15, 20, 25, 30 and 35 cm s−1, giving Reδ values of 4.9, 7.3, 9.8, 12.2, 14.7 and 17.1, respectively, at each fibre. The mass of triethyl phosphate extracted by the individual fibres at the various flow rates was then evaluated. In the range 10–35 cm s−1 the mass extracted by the three fibres showed equivalent values, 2.00 µg m−3, as shown in Table 1. Estimated concentrations obtained using SPME with the field sampler were compared with estimates obtained using filters, and no significant differences were found between the results at the 10% probability level.
A generated vapour of triisopropyl, tri-n-propyl, triisobutyl, tri-n-butyl, tris(2-chloroethyl) and tris(chloropropyl) phosphate was sampled using the field sampler in the chamber. The SPME was performed in triplicate (u = 30 cm s−1) and the active sampling on filters in duplicate, arranged such that one of the filters was connected to the field sampler device for the duration of the sampling. The procedure was repeated twice and the concentrations found are presented in Table 1. For short-term sampling the 100-µm PDMS fibre was used due to the larger volume of its coating. The difference between the concentrations yielded in the two sampling methods was not significant at the 10% probability level for tri-n-propyl and tris(2-chloroethyl) phosphate, nor at the 1% probability level for triisobutyl, tri-n-butyl and tris(2-chloropropyl) phosphate, according to Student's t-tests. The temperature in the emission chamber ranged between 21.4–22.8 °C, and the relative humidity was found to be between 3.5–5.5%.
Storage stability of the analyte
The 100-µm PDMS fibres, both capped and uncapped, were evaluated for their capacity to retain the analyte. The storage periods tested were 1, 3, 6 and 15 hours (at 22 °C). Both capped and uncapped fibres maintained 100% of the extracted mass for up to 3 hours, but after 6 hours the preserved mass on the compared fibres started to differ between the two types of fibre. After 15 hours of storage 100 percent was retained on the capped fibre and 87 percent on the uncapped. Therefore, when field sampling is performed using the uncapped fibre, analyses should be carried out during the few hours after the extraction. If a longer period of storage is necessary, a lower temperature should be used to ensure accurate results.Field measurements in a lecture room and an office
Air was sampled in a lecture room on two occasions and under different conditions. Both sampling under non-equilibrium conditions, using the 100-µm PDMS fibre, and long-term sampling under equilibrium conditions with the 30-µm PDMS fibre were carried out. When sampling under equilibrium conditions the 30-µm PDMS fibres were used because of their shorter equilibration time. In a previous work, the PDMS fibres were calibrated at different temperatures.20 It was shown that the fibre/organophosphate distribution coefficients were approximately twice as high at 22 °C compared with 27 °C. Estimated values of the coefficients at 22–23 °C were used in the present measurements. Measurements were made when the lecture room had been recently furnished and equipped with numerous brand-new computers, and compared with measurements taken six months later in the same room. Table 2 shows the concentrations of compounds obtained. A substantial concentration of tris(2-chloropropyl) phosphate was detected and was found to be 7 µg m−3 on the first sampling occasion. After six months the concentration was still high, even though it had fallen considerably. Two possible reasons for the detected concentration being lower on the second sampling occasion are that the ventilation in the room may have been stronger then or, alternatively, emissions of the compounds from the source materials may have fallen between the first and the second occasions.| SPMEad | Filterd | SPMEbd | Filtersd | |
|---|---|---|---|---|
| a Non-equilibrium sampling in June 2003. b Sampling under equilibrium condition in November 2003. c Detected but not quantified (below quantification limit S/N = 10). d Each value is an average of three measurements. Relative standard deviations in percentage (%RSD) are presented in brackets. (Temp: 21–23 °C, RH: 19–22%.) | ||||
| Tris(2-chloroethyl) phosphate | Detc | 293(17) | 12(10) | 15(6) |
| Tris(2-chloroisopropyl) phosphate | 7146(6) | 6783(11) | 1350(11) | 1493(13) |
Field sampling was also performed in a recently furnished office room. Fig. 2 shows a chromatogram from an air sample obtained using SPME under non-equilibrium conditions in the room. A number of the investigated organophosphate esters were identified in this environment, and the concentrations were determined for tri-n-butyl, tris(2-chloroethyl) and tris(2-chloropropyl) phosphate, as shown in Table 3. Triisobutyl phosphate was detected but not quantified because of coelution with an unknown compound. Here, active sampling on filters was performed in triplicate, where one of the filters was connected directly to the field sampler. The SPME was carried out under short-term conditions in triplicate and repeated twice.
 | ||
| Fig. 2 SPME chromatogram of an air sample taken in an office room. The peaks correspond to: 1, triisobutyl phosphate; 2, tri-n-butyl phosphate; 3, tris(2-chloroethyl) phosphate; and 4, three isomers of tris(chloropropyl) phosphate. The earliest eluting isomer is tris(2-chloroisopropyl) phosphate. The unnumbered peaks have not been identified. | ||
| SPMEad | Filterbd | |
|---|---|---|
| a Each value is an average of six measurements. b Each value is an average of three measurements. c Detected but not quantified due to co-elution with an interfering compound. d Relative standard deviations in percentage (% RSD) are presented in brackets. (Temp: 22 °C, RH: 16%.) | ||
| Triisobutyl phosphate | Det.c | Det.c |
| Tri-n-butyl phosphate | 18(34) | 23(9) |
| Tris(2-chloroethyl) phosphate | 37(10) | 30(19) |
| Tris(2-chloroisopropyl) phosphate | 432(19) | 426(20) |
Conclusions
Air sampling of airborne compounds with relatively low volatility, such as organophosphate triesters, requires dynamic conditions in order to reduce the sampling time. The simple, novel setup presented here for SPME under both equilibrium and non-equilibrium conditions facilitates field sampling with controlled and adjustable parameters. Even though turbulent flow is more likely to occur when the flow velocity is increased, if the fibres are placed in different vertical positions this phenomenon has less influence on the adjacent fibres.The sampler device evaluated in this work, together with a battery-operated pump, is both flexible to use and convenient to transport whenever field sampling is carried out. After sampling is concluded the capped 100-µm PDMS fibres can be stored for at least 15 h at room temperature and still yield accurate quantitative results.
Coelution, as for triisobutyl phosphate in the present work, may be a problem when using GC/NPD for complex samples such as indoor air. A sensitive GC–MS method would therefore be valuable for identification and quantification of the organophosphate triesters.
Acknowledgements
Rune Jansson is gratefully acknowledged for constructing the field sampling device.References
- J. Pawliszyn, Solid-phase Microextraction: Theory and Practice, John Wiley & Sons Inc., New York, 1997 Search PubMed.
- Z. Zhang and J. Pawliszyn, Anal. Chem., 1995, 67, 34 CrossRef CAS.
- M. Chai, C. Arthur, J. Pawliszyn, R. P. Belardi and K. Pratt, Analyst, 1993, 118, 1501 RSC.
- C. L. Arthur, L. M. Killam, K. D. Buchholz and J. Pawliszyn, Anal. Chem., 1992, 64, 1960 CrossRef CAS.
- M. Chai and J. Pawliszyn, Environ. Sci. Technol., 1995, 29, 693 CAS.
- P. A. Martos and J. Pawliszyn, Anal. Chem., 1997, 69, 206 CrossRef CAS.
- C. L. Arthur and J. Pawliszyn, Anal. Chem., 1990, 62, 2145 CrossRef CAS.
- D. Louch, S. Motlagh and J. Pawliszyn, Anal. Chem., 1992, 64, 1187 CrossRef CAS.
- Z. Zhang and J. Pawliszyn, Anal. Chem., 1993, 65, 1843 CrossRef CAS.
- A. J. Matich, D. D. Rowan and N. H. Banks, Anal. Chem., 1996, 68, 4114 CrossRef CAS.
- J. Ai, Anal. Chem., 1997, 69, 1230 CrossRef CAS.
- J. Ai, Anal. Chem., 1997, 69, 3260 CrossRef CAS.
- J. Koziel, M. Jia and J. Pawliszyn, Anal. Chem., 2000, 72, 5178 CrossRef CAS.
- F. Augusto, J. Koziel and J. Pawliszyn, Anal. Chem., 2001, 73, 481 CrossRef CAS.
- S. Isetun, A. Colmsjo, R. Johansson and U. Nilsson, Anal. Bioanal. Chem., 2004, 378, 1847 CrossRef CAS.
- J. Czerwinski, B. Zygmunt and J. Namiesnik, FEB, 1996, 5(1/2), 55 Search PubMed.
- D. Gorlo, B. Zygmunt, M. Dudek, A. Jaszek, M. Pilarczyk and J. Namiesnik, Fresenius’ J. Anal. Chem., 1999, 363, 696 CrossRef CAS.
- M. Jia, J. Koziel and J. Pawliszyn, Field Anal. Chem. Technol., 2000, 4(2–3), 73 Search PubMed.
- J. Koziel, M. Jia, A. Khaled, J. Noah and J. Pawliszyn, Anal. Chim. Acta, 1999, 400, 153 CrossRef CAS.
- S. Isetun, A. Colmsjo and U. Nilsson, Anal. Bioanal. Chem., 2004, 380, 319 CrossRef.
- J. M. Coulson and J. F. Richardsson, Chemical Engineering: Fluid flow, Heat transfer and Mass transfer, Butterworth-Heinemann Ltd., Oxford, 1996, vol. 1, pp. 52–55, 548–552 Search PubMed.
| This journal is © The Royal Society of Chemistry 2005 |