Stereoselectivity in the triplet decay of chiral benzophenone–naphthalene bichromophoric systems
M.
Consuelo Jiménez
a,
Salah-Eddine
Stiriba
b,
Rosa
Tormos
a,
Julia
Pérez-Prieto
*b and
Miguel A.
Miranda
*a
aInstituto de Tecnología Química UPV-CSIC, Departamento de Química, Universidad Politécnica de Valencia, Camino de Vera s/n, 46071 Valencia, Spain. E-mail: mmiranda@qim.upv.es; Fax: +34 963877809; Tel: +34 963877807
bDepartamento de Química Orgánica/Instituto de Ciencia Molecular, Facultad de Farmacia, Universidad de Valencia, Vicént Andrés Estellés s/n, Burjassot, 46100 Valencia, Spain. E-mail: julia.perez@uv.es; Fax: +34 963544939; Tel: +34 963543050
First published on 5th September 2003
Abstract
Chiral recognition in the intramolecular induced quenching of the methoxynaphthalene triplet by benzophenone has been observed by using diastereomeric bichromophores
Chiral recognition is a crucial phenomenon in biochemical systems as well as in technological applications. It enables the rational design of pharmaceuticals, chiral sensors and molecular devices.1 In asymmetric organic photochemistry, chiral recognition in the excited state is important to achieve enantioselectivity during photosensitization and quenching processes.2 Our group has recently succeeded in proving the existence of enantioselective discrimination in the intramolecular quenching of benzoylthiophene-derived triplets by phenols and indoles.3 In view of the current interest on stereoselectivity in photophysical/photochemical processes we decided to look for a possible chiral discrimination in systems where triplet decay is controlled by induced quenching (IQ). This has been defined as a type of quenching different from energy or electron transfer and does not lead to any photochemical product;4 it is inherent to photoreactions via triplet exciplexes4–9 They can compete with other events such as electron transfer (ET); actually, using benzophenone–anisole systems it has been demonstrated that the relationship between IQ rate constants and oxidation potentials follows Rehm–Weller behavior. This implies involvement of the same intermediate for IQ and ET processes.4
Bichromophoric compounds containing benzophenone and naphthalene units constitute classical textbook systems, that have been used to study singlet–singlet and triplet–triplet energy transfer processes.6 In this respect, Shizuka and co-workers have studied benzophenone–methoxynaphthalene bichromophores BP–(CH2)3–NPOMe. They suggested that deactivation of the methoxynaphthalene triplet, generated by intramolecular benzophenone photosensitization, occurs by interaction with the ground-state BP-moiety through triplet exciplexes with loose sandwich-like structures and weak charge transfer character.7–10
This appeared to be an adequate model system in order to investigate the possible stereoselectivity in the process by introducing chirality in the bichromophores. To this end, enantiomerically pure compounds were obtained by condensation of (R)- and (S)-naproxol† [NPX, 2-(6-methoxy-2-naphthyl)propan-1-ol] with (S)-ketoprofen [KP, (2S)-2-(3-benzoylphenyl)propanoic acid] in the presence of dicyclohexylcarbodiimide and 4-(dimethylamino)pyridine, using dry methylene chloride as solvent (Scheme 1).
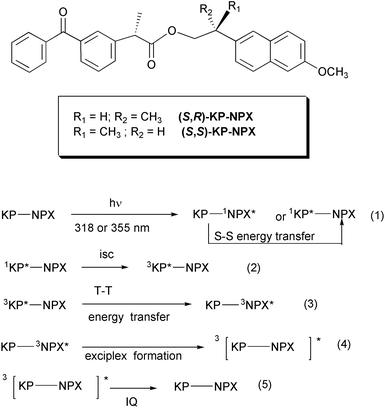 | ||
| Scheme 1 Processes involved in the photoexcitation-deactivation of KP-NPX bichromophores. | ||
The UV spectra of the bichromophoric compounds were merely the sum of the spectra of the individual chromophores, indicating little electronic interaction between the chromophores in the ground state (Fig. 1).
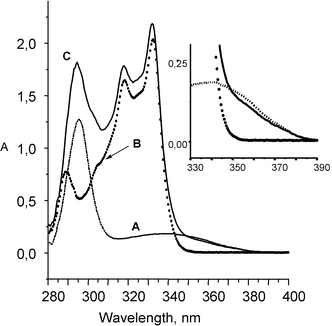 | ||
| Fig. 1 Absorption spectra of ketoprofen (A), naproxol (B) and (S,S)-KP-NPX (C). Inset: Magnification of the spectra above 340 nm. | ||
All the processes involved in the photoexcitation–deactivation of the obtained KP-NPX bichromophores are summarized in Scheme 1. Fluorescence spectra were obtained at λexc = 318 nm for (S,R)- and (S,S)-KP-NPX (2.5 × 10−4 M) in argon-saturated acetonitrile solutions and compared to those of naproxen [2-(6-methoxy-2-naphthyl)propanoic acid] under the same conditions (Fig. 2). As expected, the shapes of the three emision spectra were identical due to the nonemissive nature of the benzophenone singlet state. However, the emission intensity was considerably lower for the bichromophoric compounds, although the concentrations were adjusted to match the same absorption by the methoxynaphthalene chromophore. Thus, in the bichromophoric compounds the naphthalene singlet state is quenched by the benzophenone group, due to singlet–singlet energy transfer.11
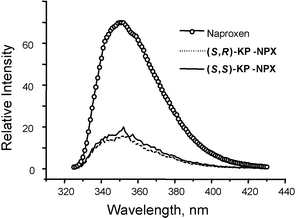 | ||
| Fig. 2 Fluorescence emission spectra of naproxen, (S,R)-KP-NPX and (S,S)-KP-NPX, in nitrogen-saturated acetonitrile at λexc = 318 nm. The intensities of the KP-NPX bichromophores have been multiplied by ten. | ||
Laser excitation‡ at 355 nm of (S,R)- and (S,S)-KP-NPX (3.0 × 10−3 M) in N2-saturated acetonitrile solutions led to the excited KP singlet state which undergoes fast intersystem crossing (isc) to the KP triplet. Intramolecular4,7,10,11 triplet–triplet energy transfer from 3KP (ET = 69 kcal mol−1)12 to the NPX moiety (ET = 62 kcal mol−1)9 results in the NPX triplet (λabs = 430 nm). This must occur within the laser pulse duration, as evidenced by the transient absorption spectrum that corresponds to the triplet naphthalene-like chromophore (Fig. 3). No absorption for triplet benzophenone was observed at 525 nm, even at short delays. The resulting triplets, KP-3NPX*, decay to the ground state; the triplet decay traces measured at 430 nm showed that the (R) and (S) NPX triplets were quenched differently by the (S)-KP moiety (Fig. 4). Thus, the 430 nm band decayed with first-order rate constants kd of 8.5 × 105 and 5.3 × 105 s−1 for (S,R)- and (S,S)-KP-NPX, respectively.13
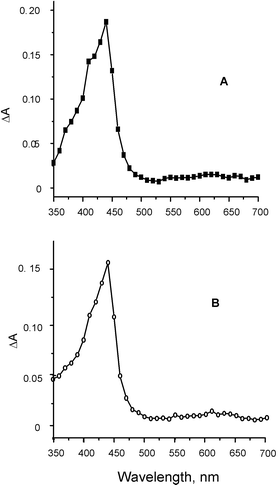 | ||
| Fig. 3 (A) Transient absorption spectrum recorded 20 ns after laser excitation (355 nm) of (S,S)-KP-NPX (3 × 10−3 M) in deaerated acetonitrile. (B) Transient absorption spectrum recorded 120 ns after laser excitation (355 nm) of (S,R)-KP-NPX (3 × 10−3 in deaerated acetonitrile. | ||
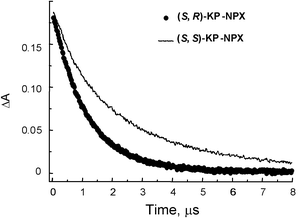 | ||
| Fig. 4 Triplet decay traces (λexc = 355 nm, λobs = 430 nm) of (S,R)-KP-NPX and (S,S)-KP-NPX in acetonitrile | ||
The stereoselectivity, as shown by the ratio between the decay rate constants, was 1.6. The sensitivity to pair configuration is a strong evidence for the involvement of intermediate (KP-NPX)* exciplexes in the NPX deactivation (steps (4) and (5), Scheme 1). Furthermore, it is indicative of specific structural requirements in the formation of the sandwich-like intermediates involved in the induced quenching processes. Diastereomeric discrimination, though associated with charge transfer quenching (rather than exciplex formation) has been previously reported for 1,2,3,4-tetraphenyl-1,4-diketones.14
In summary, we have synthesized two new chiral bichromophoric diastereomers, which are good models to demonstrate the involvement of through-space methoxynaphthalene–benzophenone interactions in the decay of the NPX triplet. This process has been found to be stereoselective.
Acknowledgements
We thank Generalitat Valenciana and the Spanish Government (Projects GV01-272, BQU2001-2725, BQU2002-00377) for generous support of this work. S. E. S. is indebted to “Ramón y Cajal” program (MCyT, Spain) for financial support.Notes and references
- (a) X. X. Zhang and J. S. Bradshaw and R. M. Izatt, Enantiomeric recognition of amine compounds by chiral macrocyclic receptors, Chem. Rev., 1997, 97, 3313–3361 CrossRef CAS; (b) C. R. Beddell, The Design of Drugs to Macromolecular Targets, ed. C. R. Beddell, Wiley, Chichester, 1992 Search PubMed.
- (a) H. Rau, Asymmetric photochemistry in solution, Chem. Rev., 1983, 83, 535–547 CrossRef CAS; (b) Y. Inoue, Asymmetric photochemical reactions in solution, Chem. Rev., 1992, 92, 741–770 CrossRef CAS; (c) N. J. Turro, Modern Molecular Photochemistry, University Science Books, CA, 1991 Search PubMed.
- (a) M. A. Miranda, A. Lahoz, R. Martinez-Máñez, F. Boscá, J. V. Castell and J. Pérez-Prieto, Enantioselective discrimination in the intramolecular quenching of an excited aromatic ketone by a ground state phenol, J. Am. Chem. Soc., 1999, 121, 11569–11570 CrossRef CAS; (b) M. A. Miranda, A. Lahoz, F. Boscá, M. R. Metni, F. B. Abdelouahab, V. Castell and J. Pérez-Prieto, Regio- and stereo-selectivity in the intramolecular quenching of the excited benzoylthiophene chromophore by tryptophan, Chem. Commun., 2000, 2257–2258 RSC.
- K. Okada, M. Yamaji and H. Shizuka, Laser photolysis investigation of induced quenching in photoreduction of benzophenone by alkylbenzenes and anisoles, J. Chem. Soc., Faraday Trans., 1998, 94, 861–866 RSC.
- (a) K. Kawaoka, A. U. Khan and D. R. Kearns, Role of singlet states of molecular oxygen in the quenching of organic triplet states, J. Chem. Phys., 1967, 46, 1842–1853 CAS; (b) J-P. Blanchi and A. R. Watkins, Quenching of triplet benzophenone by electron donors, J. Chem. Soc., Chem. Commun., 1974, 265–266 RSC; (c) C. Schweitzer, Z. Mehrdad, F. Shafii and R. Schmidt, Common Marcus type dependence of the charge transfer induced processes in the sensitization and quenching of singlet oxygen by naphthalene derivatives, J. Phys. Chem. A, 2001, 105, 5309–5316 CrossRef CAS; (d) C. Schweitzer, Z. Mehrdad, F. Shafii and R. Schmidt, Charge transfer induced quenching of triplet sensitizers by ground state oxygen and of singlet oxygen by ground state sensitizers: a common deactivation channel, Phys. Chem. Chem. Phys., 2001, 3, 3095–3101 RSC; (e) M. W. Wolf, R. E. Brown and L. A. Singer, Deactivation of benzophenone triplets via exciplex formation. Evidence for dual reaction pathways, J. Am. Chem. Soc., 1977, 99, 526–531 CrossRef CAS.
- (a) A. A. Lamola, P. A. Leermakers, G. W. Byers and G. S. Hammond, Intramolecular electronic energy transfer between nonconjugated chromophores in some model compounds, J. Am. Chem. Soc., 1965, 87, 2322–2332 CrossRef CAS; (b) R. W. Anderson Jr., R. M. Hochstrasse, H. Lutz and G. W. Scott, Direct measurements of internal conversion between excited electronic states of a molecule in the condensated phase, Chem. Phys. Lett., 1975, 32, 204–209 CrossRef; (c) M. E. Sigman and G. L. Closs, Free energy and structure dependence of intramolecular triplet energy transfer in organic model compounds, J. Phys. Chem., 1991, 95, 5012–5017 CrossRef CAS; (d) P. S. Engel, D. W. Horsey, J. N. Scholz, T. Karatsu and A. Kitamura, Intramolecular triplet energy transfer in ester-linked bichromophoric azoalkanes and naphthalenes, J. Phys. Chem., 1992, 96, 7524–7535 CrossRef CAS; (e) R. D. Fossum and M. A. Fox, Intramolecular complex formation and triplet energy transfer in polynorbornenes incorporating benzophenone, J. Am. Chem. Soc., 1997, 119, 1197–1207 CrossRef CAS.
- T. Sekiguchi, M. Yamaji, H. Tatemitsu, Y. Sakata and H. Shizuka, Laser flash photolysis studies on intramolecular hydrogen atom transfer of [(hydroxynaphthyl)propyl]benzophenone and intramolecular proton-induced electron transfer of [(methoxynaphthyl)propyl]benzophenone via triplet exciplexes, J. Phys. Chem., 1993, 97, 7003–7010 CrossRef CAS.
- M. Yamaji, Y. Aihara, T. Itoh, S. Tobita and H. Shizuka, Thermochemical profiles on hydrogen atom transfer from triplet naphthol and proton-induced electron transfer from triplet methoxynaphthalene to benzophenone via triplet exciplexes studied by laser flash photolysis, J. Phys. Chem., 1994, 98, 7014–7021 CrossRef CAS.
- T. Tanaka, M. Yamaji and H. Shizuka, Solvent dependence of triplet energy transfer from triplet benzophenone to naphtols and methoxynaphthalenes, J. Chem. Soc., Faraday Trans, 1998, 94, 1179–1187 RSC.
- For an excellent up-to-date review, see: H. Shizuka and M. Yamaji, Triplet energy transfer and triplet exciplex formation of benzophenone, Bull. Chem. Soc. Jpn., 2000, 73, 267–280 Search PubMed , and references therein.
- W. G. McGimpsey, L. Chen, R. Carraway and W. N. Samaniego, Singlet-singlet and triplet-triplet energy transfer in bichromophoric peptides, J. Phys. Chem. A, 1999, 103, 6082–6090 CrossRef CAS.
- V. Lhiaubet, F. Gutierrez, E. Penaud-Berruyer, E. Amouyal, J. P. Daudey, R. Poteau, N. Chouini-Lalanne and N. Paillous, Spectroscopic and theoretical studies of the excited states of fenofibric acid and ketoprofen in relation with their photosensitizing properties, New J. Chem., 2000, 24, 403 RSC.
- Shizuka et al., have reported a decay rate constant of 1.8 × 105 s−1 for a BP–(CH2)3–NPOMe system in acetonitrile–water (4 ∶ 1 v/v) solution (see ref. 7).
- (a) J. N. Moorthy, W. S. Patterson and C. Bohne, Remarkable discrimination in the triplet lifetimes of the diastereomers of 1,4-bis(p-methoxyphenyl)-2,3-diphenylbutan-1,4-dione, J. Am. Chem. Soc., 1997, 119, 11094–11095 CrossRef CAS; (b) J. M. Moorthy, S. L. Monahan, R. B. Sunoj, J. Chandrasekhar and C. Bohne, Modulation of lifetimes and diastereomeric discrimination in triplet-excited substituted butane-1,4-diones through intramolecular charge-transfer quenching, J. Am. Chem. Soc., 1999, 121, 3093–3103 CrossRef CAS.
Footnotes |
† Naproxol enantiomers were obtained from (R)-(+)- or (S)-(−)-naproxen by reduction with LiAlH4 in tetrahydrofuran. 1H and 13C NMR spectra were recorded in a 300 MHz spectrometer; chemical shifts (δ) are reported in ppm relative to TMS. The coupling constants (J) are in hertz (Hz).(S,R)-KP-NPX: 1H NMR (300 MHz, CDCl3): δ
(ppm) 1.26 (3H, d, J
= 7.1 Hz, CH3 NPX), 1.45 (3H, d, J
= 7.2 Hz, CH3 KP), 3.15 (1H, m, CH NPX), 3.75 (1H, q, J
= 7.2 Hz, CH KP), 3.85 (3H, s, OCH3), 4.20 (1H, dd, J1
= 10.9, J2
= 7.1 Hz, CH2 NPX), 4.30 (1H, dd, J1
= 10.9, J2
= 7.1 Hz, CH2 NPX), 7.05 (2H, m, arom. CH), 7.22 (2H, m, arom. CH), 7.30–7.71 (8H, m, arom. CH), 7.72–7.75 (3H, m, arom. CH). 13C NMR (75 MHz, CDCl3): δ 18.3 (CH3), 18.6 (CH3), 39.2 (CH NPX), 45.8 (CH KP), 55.7 (OCH3), 70.1 (CH2 NPX), 106.0 (arom. CH. NPX), 119.2 (arom. CH NPX), 125.9 (arom. CH), 126.6 (arom. CH), 127.3 (arom. CH), 128.7 (arom. CH), 128.8 (arom. CH), 129.3 (arom. CH), 129.5 (arom. CH), 130.4 (arom. CH), 131.9 (arom. CH), 132.8 (arom. CH), 133.9 (arom. C), 137.9 (arom. C), 138.1 (arom. C), 138.4 (arom. C), 141.1 (arom. C), 157.8 (arom. C NPX), 174.3 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O ester), 196.8 (CO ketone). IR (NaCl)
ν/cm−1: 1732 (CO, ester).(S,S)-KP-NPX: 1H NMR (300 MHz, CDCl3): δ 1.29 (3H, d, J
= 7.1 Hz, CH3 NPX), 1.45 (3H, d, J
= 7.2 Hz, CH3 KP), 3.15 (1H, m, CH NPX), 3.74 (1H, q, J
= 7.2 Hz, CH KP), 3.89 (3H, s, OCH3), 4.25 (2H, d, J
= 7.1 Hz, CH2 NPX), 6.95 (2H, m, arom. CH), 7.10–7.60 (10H, m, arom CH), 7.70 (3H, m, arom. CH). 13C NMR (75 MHz, CDCl3): δ 18.4 (CH3), 18.6 (CH3
), 39.2 (CH NPX), 45.8 (CH KP), 55.7 (OCH3), 70.2 (CH2 NPX), 106.0 (arom. CH NPX), 119.2 (arom. CH NPX), 125.9 (arom. CH), 126.6 (arom. CH), 127.3 (arom. CH), 128.7 (arom. CH), 128.8 (arom. CH), 129.4 (arom. CH), 129.5 (arom. CH), 129.7 (arom. CH), 130.4 (arom. CH), 131.9 (arom. CH), 132.9 (arom. CH), 133.9 (arom. C), 137.9 (arom. C), 138.2 (arom. C), 138.5 (arom. C), 141.2 (arom. C), 157.8 (arom. C. NPX), 174.3 (CO ester), 196.8 (CO ketone). IR (NaCl)
ν/cm−1: 1731 (CO, ester). O ester), 196.8 (CO ketone). IR (NaCl)
ν/cm−1: 1732 (CO, ester).(S,S)-KP-NPX: 1H NMR (300 MHz, CDCl3): δ 1.29 (3H, d, J
= 7.1 Hz, CH3 NPX), 1.45 (3H, d, J
= 7.2 Hz, CH3 KP), 3.15 (1H, m, CH NPX), 3.74 (1H, q, J
= 7.2 Hz, CH KP), 3.89 (3H, s, OCH3), 4.25 (2H, d, J
= 7.1 Hz, CH2 NPX), 6.95 (2H, m, arom. CH), 7.10–7.60 (10H, m, arom CH), 7.70 (3H, m, arom. CH). 13C NMR (75 MHz, CDCl3): δ 18.4 (CH3), 18.6 (CH3
), 39.2 (CH NPX), 45.8 (CH KP), 55.7 (OCH3), 70.2 (CH2 NPX), 106.0 (arom. CH NPX), 119.2 (arom. CH NPX), 125.9 (arom. CH), 126.6 (arom. CH), 127.3 (arom. CH), 128.7 (arom. CH), 128.8 (arom. CH), 129.4 (arom. CH), 129.5 (arom. CH), 129.7 (arom. CH), 130.4 (arom. CH), 131.9 (arom. CH), 132.9 (arom. CH), 133.9 (arom. C), 137.9 (arom. C), 138.2 (arom. C), 138.5 (arom. C), 141.2 (arom. C), 157.8 (arom. C. NPX), 174.3 (CO ester), 196.8 (CO ketone). IR (NaCl)
ν/cm−1: 1731 (CO, ester). |
| ‡ Laser Flash Photolysis experiments. Laser flash photolysis experiments were carried out by using the third harmonics (355 nm) of a pulsed Nd:YAG laser. The pulse duration was 10 ns and the energy of the laser beam was 10 mJ pulse−1. A Lo255 Oriel xenon lamp was employed as the detecting light source. The laser flash photolysis apparatus consisted of the pulsed laser, the Xe lamp, a 77200 Oriel monochromator, an Oriel photomultiplier (PMT) system made up of 77348 side-on PMT tube, 70680 PMT housing and a 70705 PMT power supply. The oscilloscope was a TDS-640A Tektronix. The output signal was transferred to a personal computer for data analysis. |
| This journal is © The Royal Society of Chemistry and Owner Societies 2004 |