Solvent effects on the photophysics of 3-(benzoxazol-2-yl)-7-(N,N-diethylamino)chromen-2-one
Antonio Eduardo da Hora
Machado
a,
Divinomar
Severino
a,
Juliana
Ribeiro
a,
Rodrigo
De Paula
a,
Marcelo Henrique
Gehlen
b,
Hueder Paulo Moisés
de Oliveira
b,
Mauricio dos Santos
Matos
c and
Jacques Antonio
de Miranda
a
aUniversidade Federal de Uberlândia, Instituto de Química/Laboratório de Fotoquímica, P.O. Box 593, CEP 38400-089 Uberlândia, MG, Brazil. E-mail: aeduardo@ufu.br
bUniversidade de São Paulo, IQSC/Laboratório de Fotoquímica, CEP 13560-970 São Carlos, SP, Brazil
cUniversidade de São Paulo, FFCLRP, Departamento de Psicologia e Educação, CEP 14040-901 Ribeirão Preto, SP, Brazil
First published on 22nd October 2003
Abstract
The photophysics of 3-(benzoxazol-2-yl)-7-(N,N-diethylamino)chromen-2-one was studied in different solvents and in SDS micelles. This compound presents characteristics which include an S0 → S11(π,π*) transition with a 1(n,π*) perturbative component, due to the electronic coupling between the diethylamino group and the coumarin ring, considerable solvatochromism, dual fluorescence and high fluorescence quantum yields in almost all solvents studied. The electronic structure of the S1 and S2 excited states permits vibronic coupling between them, making configurational changes of the S2 excited state possible, leading to the formation of an S2(TICT) state. Analysis of the TCSPC data indicates an equilibrium between the S2(TICT) and S1(LE) states in favour of the former. In protic solvents, the hydrogen bonding between the solvent and the diethylamino moiety results in the formation of an S2(HICT) state, making internal conversion an important deactivation process. Quantum mechanical calculations for the isolated molecule show that the diethylamino group in the S2(TICT) state is twisted at least 56° from the plane of the coumarin ring, with partial electronic decoupling between –NEt2 and the coumarin ring. This twisting angle must be positively influenced by solute–solvent interactions. ΦST is found to be small, but not negligible. However, ΦΔ can be considered negligible, an indication that T1 is a short-lived state. Based on the experimental data and theoretical calculations, the most probable sequence for the first excited states, including the TICT state, is T1(n,π*) < S2(TICT) < S1(π,π*) ≈ S2(n,π*).
1 Introduction
Coumarins have been extensively studied due to their practical applications,1–6 which include uses as biological and chemical sensors, fluorescent probes and laser dyes.7–10 Additionally, coumarins containing an electron-releasing group in the 7-position and a heterocyclic electron-acceptor residue in the 3-position are recognised as fluorescent dyes suitable for application to synthetic fibres.11 In sensor applications, fluorescence is frequently the observable phenomenon of choice. Sensors have been used for remote sensing of many different kinds of phenomena, all related to their solvatochromic properties.Electron-donor groups in the 4- and 7-positions of the coumarin ring cause bathochromic and bathofluoric shifts, which are become larger as the electron-donor behaviour of the substituents becomes more pronounced.4,12,13 The combination of electron-withdrawing groups in the 3-position and electron-donor groups in the 7-position intensifies electron delocalisation, with favourable implications for the fluorescence properties of these molecules. Alkylaminocoumarins, in particular, show solvatochromic behaviour. However, interactions with the solvent and the formation of hydrogen bonds involving the amino group tend to reduce the fluorescence quantum yield. As the interaction becomes more intense, internal conversion (IC) increasingly becomes the deactivation route for the excited state due to the increase in vibronic coupling.
In the present work, the photophysics and spectral properties of 3-(benzoxazol-2-yl)-7-(N,N-diethylamino)chromen-2-one (Scheme 1) were investigated in different solvents. The ET(30) solvatochromic scale was used to quantify the observed changes. Experimental and theoretical evidence on the coexistence of S1(LE), S2(TICT) and S2(HICT) states is also presented.
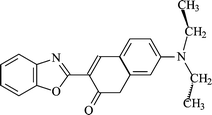 | ||
| Scheme 1 Structure of 3-(benzoxazol-2-yl)-7-(N,N-diethylamino)chromen-2-one (I). | ||
2 Experimental
3-(Benzoxazol-2-yl)-7-(N,N-diethylamino)chromen-2-one (I) was synthesised by Dr A. M. F. Oliveira-Campos and co-workers.14All solvents were of spectroscopic grade and were used without further purification. The concentration of I in the solutions was around 10−6 mol L−1. Solutions containing sodium dodecyl sulfate (SDS; 30 mmol L−1) were prepared in phosphate buffer solution (PBS; pH 7.4).
UV-Vis absorption and emission spectra were recorded using a Hach 4000U spectrophotometer and a Hitachi F-4500 spectrofluorimeter, respectively, equipped with accessories for low temperature measurements. The fluorescence spectra were obtained, using the right-angle configuration, by exciting the samples at the wavelength of maximum absorption. The fluorescence quantum yields were estimated from the corrected fluorescence spectra, using methodology proposed by Eaton.15 The fluorescence standard was 9,10-diphenylanthracene in cyclohexane (ΦF = 0.90 at 293 K). The solutions were prepared in such a way as to keep the absorbance below 0.100 at the excitation wavelength in order to avoid light reabsorption effects. Low temperature measurements were carried out at 77 K (liquid nitrogen) using methylcyclohexane as the solvent. Solutions were deoxygenated by bubbling argon through them prior to making the measurements.
Fluorescence decays were measured by the single-photon counting technique using an Edinburgh CD-900 spectrometer operating with a hydrogen-filled nanosecond flash lamp at 40 kHz pulse frequency. All lifetime values reported reflect data taken with pulses between 3000 and 5000 counts in the maximum channel and 0.900 < χ2 < 1.100.
The quantum yield of singlet oxygen generation (ΦΔ) was measured in chloroform, based on methodology proposed by Schmidt et al.,16 using solutions with an absorbance of 0.300 at 355 nm. The measurement was carried out with an Edinburgh LP 900 time-resolved system, using 355 nm laser pulses (5 ns) from a Continuum Surelite II Nd:YAG laser (Q-switched delay 200 µs). The laser power was varied from 0 to 8 mJ. A North Coast EO-817 detector was used for the detection of singlet oxygen phosphorescence at 1270 nm. The compound 1-perinaphthenone (ϕΔ = 0.97 at 293 K)16 was used as the standard in these measurements.
Quantum mechanical calculations were performed for the isolated molecule in the ground and excited states using the Gaussian 98W suite of programs,17 after optimisation of the ground-state geometry using the PM3 semi-empirical method (UHF calculation, gradient 0.1000 kcal Å−1 mol−1, Polak–Ribiere optimisation algorithm).18 The ground-state geometry was refined using a density functional theory (DFT) method (B3LYP) with a 6-31G* basis function. The Berny analytical gradient was used in this optimisation. The requested convergence limit on the RMS density matrix was 1 × 10−8 and the threshold values for the maximum force and the maximum displacement were 0.000450 and 0.001800 a.u., respectively. The ab initio RCIS method at the 3-21G** level was used to optimise the geometry of the S1, S2 and S2(ICT) states. The geometry of the S2(ICT) state was initially obtained by evaluating the changes in the state energy of the S2 state on varying the dihedral angle C(31)–N(27)–C(20)–C(15) and the distance N(27)–C(20) (single point calculations, RCIS, 3-21G basis function) to obtain the minimum energy structure, which was then optimised (Fig. 1). The state energies of I were evaluated using PM3-CI (4 occupied MOs and 4 unoccupied MOs, 4900 configurations) for the isolated molecule.18 The calculations were conducted using the optimised ground-state structure.
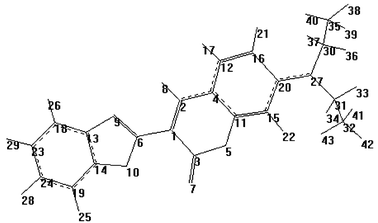 | ||
| Fig. 1 Optimised structure (DFT, B3LYP/6-31G*) of the S0 state of I, with atom numbering scheme. | ||
3 Results and discussion
3.1 Absorption spectra
Table 1 presents spectroscopic and photophysical data related to the compound under study, in different media. The absorption maximum corresponding to the S0 → S1 transition shows high molar absorptivity, between 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 54000 L mol−1 cm−1, which is also observed for analogous compounds19,20 and is attributed to a 1(π,π*) transition. In fact, the relationship between the frequency, in wavenumbers, of the absorption maximum and the solvent polarity, expressed in terms of the ET(30) scale (Fig. 2), is typical of a 1(π,π*) transition.21–23
000 and 54000 L mol−1 cm−1, which is also observed for analogous compounds19,20 and is attributed to a 1(π,π*) transition. In fact, the relationship between the frequency, in wavenumbers, of the absorption maximum and the solvent polarity, expressed in terms of the ET(30) scale (Fig. 2), is typical of a 1(π,π*) transition.21–23
| Solvent | λ abs a/nm | λ em b/nm | Δ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) b/cm−1 b/cm−1 |
log ε | ϕ F | τ 1 c/ns | E T(30)/kcal mol−1 | k nr/kF |
|---|---|---|---|---|---|---|---|---|
| a λ abs = λexc. b The first value corresponds to the LE state and the second to the TICT state. c Mean values. d Value from ref. 19. | ||||||||
| Cyclohexane | 423 | 473, 490 | 2499, 3233 | 4.76 | 0.66 | — | 30.9 | — |
| Methylcyclohexane | 426 | 478, 490 | 2553, 3066 | 4.64 | 0.64 | 2.23 | — | 0.562 |
| n-Hexane | 421 | 474, 491 | 2656, 3386 | 4.74 | 0.83 | 2.37 | 31.0 | 0.205 |
| n-Decane | 423 | 473, 491 | 2499, 3274 | 4.68 | 0.95 | 2.26 | 31.0 | 0.053 |
| n-Decane–nujol (2 ∶ 8) | 423 | 458, 489 | 1807, 3191 | — | — | 2.36 | — | — |
| Nujol | 425 | 458, 489 | 1695, 3079 | — | — | — | — | — |
| Carbon tetrachloride | 428 | 474, 493 | 2267, 3080 | 4.66 | 0.84 | 2.28 | 32.4 | 0.190 |
| Toluene | 431 | 476, 498 | 2194, 3122 | 4.62 | 0.79 | 2.14 | 33.9 | 0.266 |
| 1,4-Dioxane | 428 | 476, 495 | 2356, 3162 | 4.64 | 0.79 | 2.26 | 36.0 | 0.266 |
| Tetrahydrofuran | 432 | 482 | 2401 | 4.66 | 0.89 | — | 37.4 | — |
| Ethyl acetate | 430 | 480, 495 | 2423, 3054 | 4.62 | 0.81 | — | 38.1 | — |
| Diisopropylamine | 426 | 474, 495 | 2377, 3272 | 4.72 | 0.52 | — | — | — |
| Chloroform | 440 | 478, 498 | 1806, 2647 | 4.71 | 0.90 | 2.28 | 39.1 | 0.111 |
| Dichloromethane | 439 | 482 | 2032 | 4.78 | 0.86 | — | 40.7 | — |
| Acetone | 434 | 487 | 2507 | 4.66 | 0.76 | — | 42.2 | — |
| N,N-Dimethylformamide | 441 | 493 | 2392 | 4.67 | 0.45 | 1.93 | 43.1 | 1.222 |
| Dimethyl sulfoxide | 444 | 497 | 2402 | 4.66 | 0.60 | 1.76 | 45.1 | 0.667 |
| Acetonitrile | 438 | 490 | 2423 | 4.66 | 0.80 | 1.87 | 45.6 | 0.250 |
| 2-Butanol | 438 | 487 | 2297 | 4.65 | 0.78 | 2.37 | 47.1 | 0.282 |
| 2-Propanol | 438 | 488 | 2339 | 4.68 | 0.76 | 2.33 | 48.4 | 0.316 |
| Ethanol | 441 | 489 | 2226 | 4.70 | 0.57 | 1.95 | 51.9 | 0.754 |
| Ethylene glycol | 448 | 497 | 2200 | 4.69 | 0.46 | 1.75 | 53.8 | 1.174 |
| Methanol | 442 | 490 | 2216 | 4.70 | 0.42 | 1.43 | 55.4 | 1.381 |
| SDS micelles/PBS | 463 | 495 | 1396 | 4.83 | 0.33 | 1.16 | 61.3d | 2.030 |
| Water | 452 | 497 | 2003 | 4.61 | 0.08 | 0.66 | 63.1 | 11.11 |
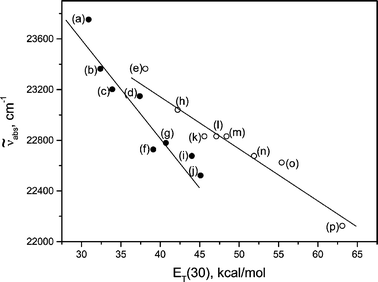 | ||
| Fig. 2 Frequency of absorption maximum () for I
(S0
→ S1)
vs. solvent ET(30): (a) n-hexane; (b) carbon tetrachloride; (c) toluene; (d) tetrahydrofuran; (e) 1,4-dioxane; (f) chloroform; (g) dichloromethane; (h) acetone; (i)
N,N-dimethylformamide; (j) dimethyl sulfoxide; (k) acetonitrile; (l) 2-butanol; (m) 2-propanol; (n) ethanol; (o) methanol; (p) water. | ||
A bathochromic shift occurs as the solvent polarity increases. This trend is approximately the same for protic and aprotic solvents, being the result of a diminishing energy gap between the ground and excited states due to the increasing strength of the polar interactions between I and the solvent.21
As a result of the anticipated electronic coupling between the diethylamino group and the coumarin ring,24,25 it seems likely that the S0 → S1 transition possesses a 1(n,π*) perturbative component, similar to that proposed for the deprotonated form of the analogue containing a hydroxyl group in the 7-position.20 This could be evidence that S2 is a 1(n,π*) state and that the energy gap between S1 and S2 is sufficiently small to make mixing of the states viable. Theoretical estimates show that the absorption band related to the S0 → S2 transition [maximum at 333 nm; oscillator strength f = 0.07, typical of a 1(n,π*) transition] must be overlapped by the band corresponding to the S0 → S1 transition.26 Despite the fact that these estimates refer to an isolated molecule, the calculated absorption maximum corresponding to the S0 → S1 transition (431 nm, f = 1.08)26 shows agreement with the experimental values observed for aprotic (around 430 nm) and protic (440 nm) solvents. The estimate of ΔE(S2,S1) based on these two theoretical absorption maxima (0.8 kJ mol−1) suggests that these two states must be very close.
Fig. 3 shows the absorption spectrum of I in cyclohexane and methanol. Seixas de Melo et al., from studies on the photophysics of coumarins,27 concluded that, for this class of compounds, the S1 state is typically 1(n,π*) and that the potential energy surfaces corresponding to S1 and S2 states are close. Thus, substitutions or changes in the solvent polarity may reduce the energy gap between these states, promoting the mixing or the inversion of states, which can result in an S1(π,π*) state.
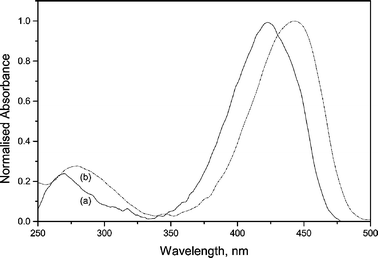 | ||
| Fig. 3 UV-Vis absorption spectra of I in cyclohexane (a) and methanol (b). | ||
For this compound, the nature of the S0 → S1 transition can be attributed to the combination of the electronic effects caused by the diethylamino and benzoxazole groups on the coumarin ring. A comparison of the geometrical data related to the S0 and S1 states shows that the bond lengths along the long axis of the molecule are shortened for S1 (Table 2), indicating an increase in the electronic conjugation over the coumarin ring in the excited state.
| Bond | S0 | S1 |
|---|---|---|
| R(1–6) | 1.457 | 1.391 |
| R(2–4) | 1.418 | 1.383 |
| R(3–5) | 1.406 | 1.402 |
| R(9–13) | 1.389 | 1.374 |
| R(10–14) | 1.370 | 1.378 |
| R(11–15) | 1.381 | 1.359 |
| R(12–16) | 1.378 | 1.357 |
| R(13–18) | 1.399 | 1.392 |
| R(14–19) | 1.386 | 1.368 |
| R(18–23) | 1.394 | 1.378 |
| R(19–24) | 1.397 | 1.388 |
| R(20–27) | 1.376 | 1.358 |
In particular, the shortening of the C(20)–N(27) bond shows the electronic coupling between the diethylamino group and the coumarin π system with electronic excitation, confirming the participation of a 1(n,π*) perturbative component in the S1 state. The bond and dihedral angles indicate that the diethylamino group nitrogen has sp2 character. For S0 and S1, this group is slightly out of the plane of the coumarin ring, being more planar in the S1 state than in the S0 (Table 3).
| State | Bond angle | Dihedral angle | ||
|---|---|---|---|---|
| C(30)–N(27)–C(20) | C(31)–N(27)–C(20) | C(30)–N(27)–C(20)–C(15) | C(31)–N(27)–C(20)–C(15) | |
| S0 | 122.118 | 121.736 | −173.595 | +5.727 |
| S1 | 122.443 | 121.997 | 184.995 | +4.716 |
This lower dihedral angle must imply an increase in the state energy of S1, due to the steric effects, and can be seen as one of the causes of the small energy gap between the S1 and S2 states. In addition, the vibronic coupling between S1 and S2, favoured by the interactions with the solvent, creates the conditions for configurational changes of the S2 state, which leads to the formation of an S2(TICT) state.
An estimate of the geometrical structure of I in the S2(TICT) state reveals that the dihedral angle C(31)–N(27)–C(20)–C(15) (Fig. 1) must be twisted at least 56° from the plane of the coumarin ring. The expected C(30)–N(27)–C(20) and C(31)–N(27)–C(20) bond angles for this state are 121.826 and 119.761°, respectively, indicating that, despite the torsion, the sp2 character of the nitrogen is not lost at all. This suggests only a partial electronic decoupling between the diethylamino group and the coumarin ring for the isolated molecule.26 However, in principle, interactions between the S2(TICT) state and the solvent might increase the electronic decoupling, with a consequent increase in the twisting angle.
3.2 Fluorescence and photophysics
Fig. 4 and Table 1 show that I exhibits considerable solvatochromism. As solvent polarity increases, the fluorescence maximum wavelength undergoes a bathofluoric shift, estimated from the Stokes' shift, which is more intense than the bathochromic shift observed for the absorption spectra. Plotting solvent ET(30) against Stokes’ shift gives two distinct graphs, one for non-protic and one for protic solvents, with good linear correlations (R < −0.97). Since the ET(30) scale is essentially a polarity scale based on the static dielectric constant, f(D), with the addition of a protic hydrogen-bonding effect, this behaviour is expected.21,28,29 The plots reveal compound I to be more sensitive to polarity changes in aprotic solvents than in protic ones. However, specific interactions induced by protic solvents seems to induce an efficient vibronic coupling with the excited states of I, reducing the fluorescence quantum yield and increasing the rate of internal conversion. Fig. 5 shows the exponential growth in the knr/kF ratio with increasing solvent polarity. The trend is more evident for protic solvents.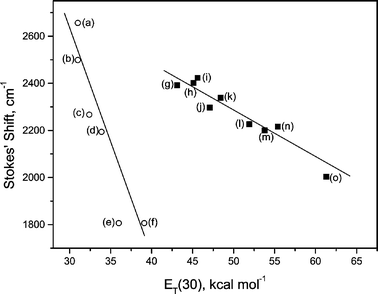 | ||
| Fig. 4 Stokes' shift for Ivs. solvent ET(30): (a) n-hexane; (b) n-decane; (c) carbon tetrachloride; (d) toluene; (e) 1,4-dioxane; (f) chloroform; (g) N,N-dimethylformamide; (h) dimethyl sulfoxide; (i) acetonitrile; (j) 2-butanol; (k) 2-propanol; (l) ethanol; (m) ethylene glycol; (n) methanol; (o) SDS micelles/PBS. | ||
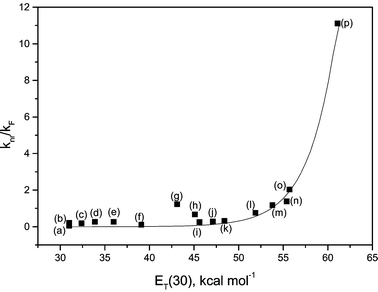 | ||
| Fig. 5 k nr/kF ratio for Ivs. solvent ET(30): (a) n-decane; (b) n-hexane; (c) carbon tetrachloride; (d) toluene; (e) 1,4-dioxane; (f) chloroform; (g) N,N-dimethylformamide; (h) dimethyl sulfoxide; (i) acetonitrile; (j) 2-butanol; (k) 2-propanol; (l) ethanol; (m) ethylene glycol; (n) methanol; (o) SDS micelles/PBS; (p) water. | ||
In contrast to the situation with 4-dimethylaminobenzonitrile, I exhibits two fluorescence bands in aprotic solvents, independent of their polarity (Table 1 and Fig. 6), attributed to the dynamic equilibrium between the S1(LE) and S2(TICT) states.21,24,25,30–32
| S1(LE) ⇄ S2(TICT) |
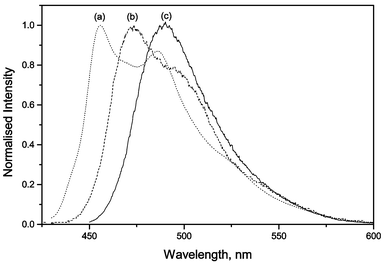 | ||
| Fig. 6 Normalised fluorescence spectra of I in n-decane (a), diisopropylamine (b) and methanol (c). | ||
In protic solvents, a red-shifted band, attributed to the S2(TICT) state, is more evident. The rate constants (Table 4) show that formation of the S2(TICT) state is favoured as solvent polarity increases. If the polar interactions are due to protic solvents, mainly alcohols capable of hydrogen bonding with the amino group, only one emission band is observed. This band, red-shifted toward the S1(LE) state, is due to the emission of the S2(TICT) state. Despite this, for all media, analysis of the TCSPC data (Table 4) indicates that the equilibrium between these species is favourable to the formation of the S2(TICT) state.
| Solvent | τ 1/ns |
|
τ 2/ns |
|
χ 2 |
|
k TICT/s−1 | k LE/s−1 |
|---|---|---|---|---|---|---|---|---|
| n-Decane | 2.197 ± 0.035 | 0.976 | 5.266 ± 1.605 | 0.024 | 1.108 | 40.67 | 4.44 × 108 | 1.09 × 107 |
| n-Hexane | 2.356 ± 0.031 | 0.976 | 5.013 ± 0.945 | 0.024 | 1.126 | 40.67 | 4.16 × 108 | 1.02 × 107 |
| 1,4-Dioxane | 2.280 ± 0.014 | 0.986 | 28.74 ± 9.78 | 0.014 | 1.157 | 70.43 | 4.33 × 108 | 6.15 × 106 |
| Chloroform | 2.277 ± 0.021 | 0.976 | 5.944 ± 1.590 | 0.024 | 1.132 | 40.67 | 4.38 × 108 | 1.05 × 107 |
| N,N-Dimethylformamide | 1.917 ± 0.009 | 0.991 | 22.83 ± 10.02 | 0.009 | 1.036 | — | — | — |
| Dimethyl sulfoxide | 1.734 ± 0.009 | 0.994 | 11.29 ± 3.27 | 0.006 | 1.138 | — | — | — |
| 2-Propanol | 2.334 ± 0.006 | 1.000 | — | — | 0.965 | — | — | — |
| Ethanol | 2.037 ± 0.006 | 1.000 | — | — | 1.095 | — | — | — |
| Ethylene glycol | 1.859 ± 0.006 | 1.000 | — | — | 1.091 | — | — | — |
| Methanol | 1.449 ± 0.005 | 1.000 | — | — | 1.089 | — | — | — |
| SDS micelles/PBS | 1.203 ± 0.006 | 0.994 | 15.41 ± 2.87 | 0.006 | 1.099 | — | — | — |
TCSPC measurements show a biexponential profile for the fluorescence decay of I in aprotic solvents,
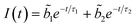
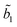 predominates, the participation of the second term being less than 3% in the best cases. For the majority of the protic solvents,
predominates, the participation of the second term being less than 3% in the best cases. For the majority of the protic solvents, 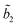 is very small, and the fluorescence decay was considered, in principle, as almost completely due to the deactivation of the S2(TICT) state.
is very small, and the fluorescence decay was considered, in principle, as almost completely due to the deactivation of the S2(TICT) state.
Since 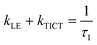 and
and 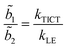 ,28 the rate constants for the forward (kTICT) and backward (kLE) ICT reactions can be estimated. The calculated constants (Table 4) confirm the favourability of populating the S2(TICT) state, even in aprotic solvents. The preference for populating the S2(TICT) state is pertinent if we consider that the activation energy, Ea, for the formation of this state is lower than that for the reverse reaction.
,28 the rate constants for the forward (kTICT) and backward (kLE) ICT reactions can be estimated. The calculated constants (Table 4) confirm the favourability of populating the S2(TICT) state, even in aprotic solvents. The preference for populating the S2(TICT) state is pertinent if we consider that the activation energy, Ea, for the formation of this state is lower than that for the reverse reaction.
Considering the expected role of the solvent polarity on the formation of these states, the inequality Ea(TICT) < Ea(LE) is perfectly plausible. A rough estimate of the energy of the S2(TICT) state can be made by considering the energy difference between the emission maxima attributed to the S1 and S2(TICT) states. Such an estimate indicates that E{S2(TICT)} is at least 11 kJ mol−1 lower than S1, very close the value estimated by TD-DFT calculations.26 Since low temperature measurements have given a value of 260 kJ mol−1 for E(S1),33 the expected value for E{S2(TICT)} is 249 kJ mol−1.
The formation of a HICT state in equilibrium with the S2(TICT) state, as proposed by Kwok et al.,34 justifies this. This state involves hydrogen bonding between the diethylamino group and the solvent. Since the S1(LE), S2(TICT) and S2(HICT) states must coexist, the fluorescence of I in protic solvents should be triple and not dual. However, considering that the S2(HICT) state must decay preferentially by internal conversion due to the increase in the vibronic coupling between the excited molecule and the solvent, its direct observation by fluorescence spectroscopy could well be very difficult. In spite of this, an obvious reduction in the ΦF values can be observed for alcohols more susceptible to forming hydrogen bonds with the diethylamino group (methanol, ethanol and ethylene glycol), and for some other protic solvents (water, N,N-dimethylformamide and dimethyl sulfoxide) and SDS micelles, proportional to their polarities (Table 1 and Fig. 5). In recent work, Kwok et al. used time-resolved IR spectroscopy to demonstrate the existence of a hydrogen-bonded excited state, designated HICT, in equilibrium with the S2(TICT) state for 4-dimethylaminobenzonitrile in methanol.34
In water–dioxane mixtures, fluorescence suppression is observed, principally if the proton concentration in the medium increases.33 Estimating the fluorescence quenching constant due to the interaction between coumarin and water in these mixtures (Fig. 7) gives a value of 3.09 × 106 L mol−1 s−1, typical of charge-transfer processes,21,22 indicating that the formation of the S2(HICT) state from S2(TICT) is a very favourable process in protic solvents.
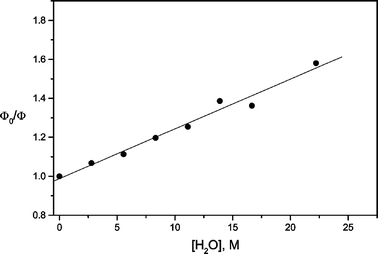 | ||
| Fig. 7 Stern–Volmer plot for the fluorescence quenching of I by water, in water–dioxane mixtures. KSV = 2.56 × 10−2 L mol−1. | ||
On the other hand, the polar interactions between the solvent and other parts of the molecule are not determining for the induction of routes capable of competing with the solvent–diethylamino group interactions for excited-state deactivation, as shown by the fluorescence data obtained for 3-(benzoxazol-2-yl)chromen-2-one.19
3.3 Excited states
The evaluation (PM3 CI) of the energy gap between S1 and T1 states (ca. 81 kJ mol−1) shows that the population of the triplet state should, in principle, be unfavourable. However, although no detectable phosphorescence could be observed in measurements at low temperature, analysis of the ΦF values at 77 and 298 K in methylcyclohexane shows that ΦST is not negligible. Since ΦF = 0.90 at 77 K, whereas it is 0.64 at 298 K, and ΦIC ≈ 0.26, then ΦST = 0.10.Since ΦST is not that small and the singlet oxygen quantum efficiency, measured in chloroform, is very small (ΦΔ = 0.06), it is possible to state that the T1 and S1 orbital symmetries must be different.22,23 Thus, as S1 is a 1(π,π*) state, T1 must be a short-lived 1(n,π*) state. This is reasonable considering that 1(π,π*) → 3(n,π*) intersystem crossing is a spin–orbit favoured process,22,23 and that this compensates for the considerable energy gap between T1 and S1. On the other hand, it is known that T1(n,π*) ketones are not usually good singlet oxygen sensitisers due to their short lifetimes.35 Therefore, considering the above-mentioned results, the occurrence of intersystem crossing from the electronically excited aminocoumarins cannot be generalised as being negligible, as proposed by Priyadarshini et al.36
4 Conclusions
The S0 → S1 transition of I is 1(π,π*) with a 1(n,π*) perturbative component, due to the electronic coupling between the nitrogen of the diethylamino group and the coumarin ring, combined with the electron-withdrawing action of the benzoxazole in the 3-position. The 1(n,π*) component must be related to an S2 state very close to S1. The small energy gap between these two states (ca. 0.8 kJ mol−1) makes vibronic interactions between them feasible, enabling configurational changes in the S2 state, which leads to an S2(TICT) state. The formation of this state causes partial electronic decoupling between the diethylamino group and the rest of the molecule, which can be increased by specific interactions with the solvent. These interactions, mainly hydrogen bonding involving the diethylamino group, result in an S2(HICT) state in equilibrium with the S2(TICT) state. In this case, internal conversion is favoured as the excited-state deactivation pathway due to the increase in vibronic coupling. The twist angle for the rotation of the diethylamino group relative to the coumarin plane was estimated as 56° for an isolated molecule in the S2(TICT) state.Compound I is fluorescent, exhibiting dual fluorescence, usually with high fluorescence quantum yields and considerable solvatochromism. The dual fluorescence is more evident in aprotic solvents. In protic solvents, only the band attributed to the emission from the S2(TICT) state, red-shifted towards the S1(LE) emission, is observed. TCSPC data show that the formation of the S2(TICT) state is favoured in both kinds of solvents. An estimate of the energy difference between these states gave a value around 11 kJ mol−1, a result which is supported by quantum chemical calculations.26 Furthermore, E{S1(LE)} ≫ E(T1). Considering that ΦST is not negligible (ΦST = 0.10), and based on the small ΦΔ value in chloroform, T1 must be a 3(n,π*) state.
The behaviour observed for compound I in different kinds of solvents makes this compound suitable for application as a fluorescent probe or laser dye; it exhibits the requisite lasing characteristics (i.e. high fluorescence yield, low intersystem crossing and low tendency to aggregate), making useful as an active medium for a tunable laser for a wide range of solvents.
A more detailed theoretical investigation of the state energies and the relationship to solvent polarity, aimed at obtaining detailed information on the solvent effects on ΔE(S1,S2) and, consequently, on the formation of the S2(TICT) and S2(HICT) states, is under way.
Acknowledgements
The authors acknowledge the FAPEMIG, FAPESP and CAPES Foundations and the CNPq for research grants and Fellowships. The authors are grateful to Professors Ira M. Brinn and David E. Nicodem (LERT/IQ/UFRJ, Brazil) for access to time-resolved measurement facilities. Thanks are also due to Dr Ana Maria F. Oliveira-Campos (Univ. Minho, Portugal), who kindly furnished the coumarin derivative studied herein.References
- J. K. Thomas, Characterization of surfaces by excited states, J. Phys. Chem., 1987, 91, 267–276 CrossRef CAS.
- G. A. Reynolds and K. H. Drexhage, New coumarin dyes with rigized structure for flashlamp-pumped dye lasers, Opt. Commun., 1975, 222–225 CrossRef CAS.
- U. Brackmann, Lambdachrome Laser Dyes, Lambda Physik, Gmbh, Göttingen, 1986 Search PubMed.
- B. M. Krasovitskii, in Organic Luminescent Materials, ed. B. M. Krasovitskii and B. M. Bolotin, VCH, Weinheim, 1988, ch. 7 Search PubMed.
- K. H. Drexhage, Topics in Applied Physics, Vol. 1, Dye Lasers, ed. F. P. Schäfer, Springer, Berlin, 1973 Search PubMed.
- F. Dall'Acqua, D. Vedaldi and S. Caffieri, in The Fundamental Bases of Phototherapy, ed. H. Hönigsmann, G. Jori and A. R. Young, OEMF, Milan, 1996, pp. 1–16 Search PubMed.
- M. Maeda, Laser Dyes, Academic Press, New York, 1984 Search PubMed.
- R. M. Christie, Fluorescent dyes, a review, Rev. Prog. Color. Relat. Top., 1993, 23, 1–18 Search PubMed.
- R. M. Christie and H. Lui, Studies of fluorescent dyes: part 1. An investigation of the electronic spectral properties of substituted coumarins, Dyes Pigm., 1999, 42, 85–93 CrossRef CAS.
- N. Chandrasekharan and L. Kelly, Fluorescent molecular thermometers based on monomer/exciplex interconversion, The Spectrum, 2002, 15, 1–7 Search PubMed.
- J. Sokolowska, W. Czajkowski and R. aw Podsiadly, The photostability of some fluorescent disperse dyes derivatives of coumarin, Dyes Pigm., 2001, 49, 187–191 CrossRef CAS.
- C. E. Wheelock, The fluorescence of some coumarins, J. Am. Chem. Soc., 1959, 81, 1348–1352 CrossRef CAS.
- G. Jones II, in Dye Laser – Principles and Applications, ed. F. D. Duarte and L. W. Hillman, Academic Press, New York, 1990, pp. 287–345 Search PubMed.
- X. H. Luan, N. M. F. S. A. Cerqueira, A. M. A. G. Oliveira, M. M. M. Raposo, L. M. Rodrigues, P. Coelho and A. M. F. Oliveira-Campos, Synthesis of fluorescent 3-benzoxazol-2-yl-coumarins, Adv. Colour Sci. Technol., 2002, 5, 18–23 Search PubMed.
- D. F. Eaton, Fluorescence materials for fluorescence measurement, Pure Appl. Chem., 1988, 60, 1107–1114 CAS.
- R. Schmidt, C. Tanielian, R. Dunsbach and C. Wolff, Phenalenone, a universal reference compound for the determination of quantum yields of singlet oxygen O2(1Δg) sensitization, J. Photochem. Photobiol., A, 1994, 79, 11–17 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, Jr., R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, N. Rega, P. Salvador, J. J. Dannenberg, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, A. G. Baboul, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, J. L. Andres, C. Gonzalez, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian 98W, Revision A11.4, Gaussian, Inc., Pittsburgh, PA, USA, 2002.
- HyperChem 5.11, Computational Chemistry Program, Hypercube Inc., Gainesville, FL, USA, 1996.
- A. E. H. Machado, J. A. Miranda, S. Guilardi, D. E. Nicodem and D. Severino, Photophysics and spectroscopic properties of 3-benzoxazol-2-yl-chromen-2-one, Spectrochim. Acta, Part A, 2003, 59, 345–355 CrossRef.
- A. E. H. Machado and J. A. Miranda, Photophysical/quantum mechanical characterization of the compound 3-benzoxazol-2-yl-7-hydroxy-chromen-2-one, J. Photochem. Photobiol., A, 2001, 141, 109–116 CrossRef.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Kluwer Academic/Plenum Publishers, New York, 2nd edn., 1999 Search PubMed.
- N. J. Turro, Modern Molecular Photochemistry, University Science Books, New York, 1991 Search PubMed.
- A. Gilbert and J. Baggott, Essentials of Molecular Photochemistry, Blackwell, Oxford, 1991 Search PubMed.
- A. B. J. Parusel, G. Kökler and S. Grimme, Density functional study of excited charge transfer state formation in 4-(N,N-dimethylamino)benzonitrile, J. Phys. Chem. A, 1998, 102, 6297–6306 CrossRef CAS.
- A. B. J. Parusel, Excited state intramolecular charge transfer in N,N-heterocyclic-4-aminobenzonitriles: a DFT study, Chem. Phys. Lett., 2001, 340, 531–537 CrossRef CAS.
- A. E. H. Machado, R. De Paula, J. Ribeiro, Formação de estado S2 (TICT) a partir da excitação eletrônica do composto 3-benzoxazol-2-il-7-(N,N-dietilamino)-cromen-2-ona, Presentation to be given at XII Simpósio Brasileiro de Química Teórica, Caxambu, MG, Brazil, November, 2003 Search PubMed.
- J. Seixas de Melo, R. S. Becker and A. L. Maçanita, Photophysical behaviour of coumarins as a function of substitution and solvent: experimental evidence for the existence of a lowest lying 1(n,π*) state, J. Phys. Chem., 1994, 98, 6054–6058 CrossRef.
- C. Reichardt, Solvatochromic dyes as solvent polarity indicators, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- P. Suppan, Solvatochromic shifts: the influence of the medium on the energy of electronic states, J. Photochem. Photobiol., A, 1990, 50, 293–330 CrossRef CAS.
- (a) Z. R. Grabowski, K. Rotkiewcz, A. Siemiarczuk, D. J. Cowley and W. Baumann, Twisted intramolecular charge transfer state (TICT) – New class of excited states with a full charge separation, Nouv. J. Chim., 1979, 3, 443–454 Search PubMed; (b) K. Rotkiewcz, K. H. Grellmann and Z. R. Grabowski, Reinterpretation of the anomalous fluorescence of p-N,N-dimethylamino-benzonitrile, Chem. Phys. Lett., 1973, 19, 315–318 CrossRef CAS.
- K. A. Zachariasse, M. Grobys, Th. von der Haar, A. Hebecker, Yu.-V. Il'ichev, O. Morawski, I. Rückert and W. Kühnle, Photoinduced intramolecular charge transfer and internal conversion in molecules with a small energy gap between S-1 and S-2. Dynamics and structure, J. Photochem. Photobiol., A, 1997, 105, 373–383 CrossRef.
- K. A. Zachariasse, M. Grobys, Th. von der Haar, A. Hebecker, Yu. V. Il'ichev, Y.-B. Jiang, O. Morawski and W. Kuhnle, Intramolecular charge transfer in the excited state. Kinetics and configurational changes, J. Photochem. Photobiol., A, 1996, 102, 59–70 CrossRef CAS.
- J. A. de Miranda, Caracterização Fotofísica de Cumarinas, MSc Dissertation, Universidade Federal de Uberlândia, MG, Brazil, 2001 Search PubMed.
- W.-M. Kwok, M. W. George, D. C. Grills, C. Ma, P. Matousek, A. W. Parker, D. Phillips, W. T. Toner and M. Towrie, Direct observation of a hydrogen-bonded charge-transfer state of 4-dimethylamino-benzonitrile in methanol by time-resolved IR spectroscopy, Angew. Chem., Int. Ed., 2003, 42, 1826–1830 CrossRef CAS.
- R. W. Redmond and S. E. Braslavsky, Time-resolved thermal lensing and phosphorescence studies on photosensitizer singlet molecular oxygen formation. Influence of the electronic configuration of the sensitizer on sensitisation efficiency, Chem. Phys. Lett., 1988, 148, 523–529 CrossRef CAS.
- K. I. Priyadarshini, B. Naik and P. N. Murthy, A study of the triplet state of 7-amino coumarin laser dye by the nanosecond pulse radiolysis technique, J. Photochem. Photobiol., A, 1990, 54, 251–261 CrossRef.
| This journal is © The Royal Society of Chemistry and Owner Societies 2004 |