Immunochemical study of DNA modifications in the nuclei of UV-damaged lymphocytes
Sergei A.
Snopov
*a,
Frank R.
de Gruijl
b,
Len
Roza
c and
Jan C.
van der Leun
d
aInstitute of Cytology of the Russian Academy of Sciences, Tikhoretsky ave 4, 194064 St Petersburg, Russia. E-mail: snopov@mail.cytspb.rssi.ru
bSylvius Laboratories, Department of Dermatology, Leiden University Medical Centre, Wassenaarseweg 72, NL-2333 AL Leiden, The Netherlands. E-mail: F.R.de_Gruijl@lumc.nl.
cDepartment of Nutritional Physiology, TNO Nutrition and Food Research, Utrechtseweg 48, NL-3704 HE Zeist, The Netherlands. E-mail: Roza@voeding.tno.nl
dEcofys, Kanaalweg 16G, NL-3526 KL Utrecht, The Netherlands. E-mail: j.vanderleun@ecofys.nl
First published on 19th August 2003
Abstract
Studies of UV-induced skin cancers show that malignisation of skin cells, as well as alterations in anti-tumor immune control, are triggered by UV-induced lesions in cellular DNA. Such lesions can probably appear in the human mononuclear leukocytes (lymphocytes) during exposure of skin to sunlight. With the aim of studying the processing of UV-induced DNA lesions in these cells, we used flow cytometry and labelling of their partially denatured nuclei with the monoclonal antibody (H3) that binds cyclobutane pyrimidine dimers in single-stranded DNA. After the first few hours of cultivation of the irradiated cells, we found an increase in H3-specific fluorescence from cellular nuclei, while there was a decrease in the number of H3-positive sites in isolated DNA from these cells. We examined cells cultured under different conditions and concluded that the effect of enhancement of H3 labelling of nuclei did not result from changes in temperature and culture medium. Furthermore, we have found that this effect, as well as the decrease in H3 labelling in isolated DNA, are both prevented by pretreatment of the cells with Novobiocin, which we used as an inhibitor for the topoisomerase II-induced relaxation of supercoiled DNA prior to repair-specific incision. The inhibition by Novobiocin of the above-mentioned changes in H3 labelling in cellular nuclei and isolated DNA of the irradiated cells clearly indicate the association of both effects with an excision repair-related DNA modification. While the partial loss of H3-binding sites from isolated DNA is obviously a result of excision of some fraction of pyrimidine dimers, the enhancement of the H3 labelling of nuclei might be due to the formation of open structures at dipyrimidine-containing DNA fragments in preparation for incision. We suggest that formation of open structures predominates quantitatively over dual incision and excision of these fragments, and leads to enhanced exposure of the pyrimidine dimers in nuclei to H3 binding. Thus, unstimulated human lymphocytes appear to be capable of performing pre-incision steps for removal of these DNA lesions.
Introduction
Ultraviolet radiation from the sun is a prominent environmental agent with genotoxic activity. Long before UV-induced primary skin cancers begin to appear in experimental animals, a systemic alteration of their immunity is detectable. The alteration results in the failure of UV-irradiated mice to reject highly antigenic, transplanted UV-induced tumors that are rejected by unirradiated syngeneic recipients.1 From studies of animal and human skin cells, it has been shown that UV-induced DNA lesions, predominantly cyclobutane pyrimidine dimers (CPDs), initiate one or more steps leading to impaired immune responsiveness: i.e. altered gene expression, release of cytokines, or decrease in antigen-presenting cell activity.2–10 Effective repair of these DNA lesions in skin cells can prevent such steps from occurring and decrease the suppression of immunity.11,12It is assumed that sunlight can directly affect not only human skin cells, but also peripheral blood mononuclear leukocytes (PBMLs).13 Ninety-nine percent of PBMLs are resting, unstimulated cells, mainly lymphocytes. The possible cellular and immune function consequences of induction, repair and ‘unrepair’ of CPDs in these cells have not been studied extensively. Whereas, the difference in processing of DNA lesions between unstimulated and stimulated PBMLs is obvious; it has been shown that unstimulated lymphocytes have a decreased capacity, compared to skin fibroblasts, keratinocytes and stimulated lymphocytes, to repair CPDs by their nucleotide excision repair system,14,15 due to a defective excision of CPDs16 and low deoxyribonucleotide pool sizes.17 It remains, however, unclear whether the pre-excision enzyme treatment of CPDs is deficient or not in unstimulated lymphocytes.
With the aim of studying the processing of CPDs in the nuclei of unstimulated PBMLs, we tried to quantify the kinetics of CPD removal using anti-CPD and FITC-conjugated antibodies and flow cytometry. This technique has been shown to be sensitive for the detection of these lesions in the nuclei of PBMLs after doses as low as 1–2 J m−2 of UVC.18 Contrary to our expectations of observing a decrease in specific CPD fluorescence from the nuclei of UV-irradiated cells due to a repair removal of CPDs, in fact, we observed an increase in this fluorescence signal during the first four hours of cultivation. In our subsequent experiments, we examined whether the cell culture conditions could affect anti-CPD antibody binding to DNA. Our results led us to conclude that the enhancement of specific CDP antibody binding to the nuclei of unstimulated PBMLs occurred due to DNA modification at the non-excised CPD-containing fragments after the pre-incision enzyme activity.
Experimental
Peripheral blood mononuclear leukocytes
Freshly drawn blood from healthy donors was stabilised with heparin sodium (Organon Teknika B.V., Oss, The Netherlands), with 25 I.E. per ml of blood. Mononuclear cells were isolated with Ficoll-Hepaque (Pharmacia, Stockholm, Sweden) by gradient centrifugation and washed twice with phosphate buffer solution (PBS). Cell concentration was measured using a microcell counter (Sysmex, Kobe, Japan).UV irradiation
Isolated PBMLs suspended in PBS with a concentration of 0.5–1.0 × 106 cells ml−1 were placed in glass Petri dishes (9 cm diameter) with accurately levelled bottoms to ensure uniform distribution of the suspension over the dish. The dishes were exposed to a 40 W TUV lamp (Philips, Eindhoven, The Netherlands) from above for different lengths of time with dose rates of 0.25 or 0.40 W m−2. The dosimetry was performed with a calibrated thermopile Kip E11 (Delft, The Netherlands) instrument. During the irradiation, the dishes containing cell suspensions were kept either at a temperature close to 0 °C, by being placed on ice, or at room temperature, depending on the particular experiment. After irradiation, PBMLs were pelleted by centrifugation at the same temperature and then either fixed immediately or transferred to plastic plates with PBS or supplemented RPMI medium for cultivation at 37 °C for different periods of time and fixed afterwards.Cultivation
In some of the experiments, PBMLs were cultivated in a CO2 incubator in 6-well polystyrene multidishes (Nunclon, Roskilde, Denmark) in RPMI medium (GIBCO, Paisley, UK) supplemented with glutamine (2 mM), penicillin (100 units ml−1), streptomycin (100 µg ml−1) and 20% autologous plasma. In other experiments, the cultivation of irradiated cells was performed in PBS without the additives. In special tests, Novobiocin (Sigma, St. Louis, MO, USA) was added to PBS in concentrations of 0.175–0.250 mM.Immuno-fluorostaining
The procedure used by Berg et al.,19 with the minor modifications reported previously,18 was applied. In brief, PBMLs were fixed with 70% ice-cold ethanol and stored overnight. After incubation with 0.02% pepsin (Sigma) in 2 M HCl for 40 min at room temperature, the cells were washed once in 0.1 M Na2B4O7 and then twice in PBS–0.5% Tween 20 (Sigma) (0.5% PBT). Incubation with monoclonal anti-CPD antibody (1 ∶ 40 dilution of culture supernatant of hybridoma H3 cells20) or with anti-single-stranded DNA antibody (1 ∶ 60 dilution of culture supernatant of hybridoma D1B cells21) was performed at room temperature for 60 min. After that, the PBMLs were washed twice in PBT and incubated with 1% FITC-conjugated rabbit-anti-mouse antibodies (Dakopats, Glostrup, Denmark) at room temperature for 60 min in the dark. After another two washes in PBT, the cells were resuspended in 400 µl PBS containing 25 µg ml−1 7-aminoactinomycin D (7-AAD; Sigma) and incubated for 15 min at 37 °C with 50 µl 1% RNAse (Sigma). Centrifugation steps after the denaturing treatment were performed for 5 s at 110g.Flow cytometry
10–20 × 103 cells stained with FITC and 7-AAD were analysed with a flow cytometric system equipped with an argon laser (488 nm) (FACSscan, Becton-Dickinson Immunocytometry Systems, Mountain View, CA, USA). FITC fluorescence was detected in the fluorescence 1 channel (530 ± 15 nm) and stored after logarithmic amplification. 7-AAD fluorescence was recorded in the fluorescence 3 channel (>651 nm) after linear amplification. The data obtained was evaluated by means of the FACSscan software (LYSYS II, Becton-Dickinson) and an analysis program produced by E. Rozemuller, M.D. (Utrecht University Hospital, The Netherlands).Immuno-slot blot
The protocol used is based on that described in ref. 22. Heat-denatured DNA samples were slot-blotted with Bio-Dot SF (Bio-Rad Laboratories B.V., Veenendaal, The Netherlands; well size 7 × 0.75 mm) apparatus onto nitrocellulose membrane of 0.1 µm pore size (ref. no. 402096, Schleicher & Schuell, Dassel/Relliehausen, Germany). The membrane and filters underneath (ref. no. 162-0161, Bio-Rad Laboratories, Hercules, CA, USA) were soaked in PBS prior to use. After blotting of DNA, the slots were rinsed with 0.250 ml PBS. The membrane was baked at 80 °C for 2 h to immobilise the DNA. Subsequently, it was incubated for 30 min at room temperature with 5% (w/v) skimmed-milk powder in PBS–0.1% Tween 20 (0.1% PBT) to prevent non-specific binding of the antibodies. Then, the membrane was washed 3 times with PBT for 10 min and incubated for 1 h at room temperature with either (i) 1 ∶ 200 diluted H3 cells culture supernatant (anti-CPD monoclonal antibody) or (ii) 1 ∶ 400 diluted D1B cells culture supernatant (anti-ssDNA monoclonal antibody). After 3 washes with 0.1% PBT, the membrane was incubated with rabbit anti-mouse antibodies conjugated with peroxidase (Dakopats) diluted 1 ∶ 1000 in 0.5% milk powder in 0.1% PBT for 1 h and again washed 3 times with PBT. Peroxidase activity was revealed using a LumiGLO™ chemiluminescent substrate kit (Kirkegaard & Perry Laboratories, Gaithersburg, MD, USA). Peroxidase-labelled antibodies catalyse the oxidation of luminol, resulting in the emission of light in the presence of H2O2. This emission was registered by exposure of photographic film to the membrane for 20–60 s.Results
Flow cytometry of specific anti-CPD fluorescence in partially denatured nuclei
Our aim was to quantify possible CPD removal from nuclei of unstimulated PBMLs after UVC irradiation with doses of 0, 2, 4, 7, 10 or 30 J m−2. The irradiated PBML suspension was transferred in several aliquots from Petri dishes to centrifuge tubes, pelleted by centrifugation, placed in plastic plates for cultivation for 0, 0.5, 1, 2, 4, 6–7 or 18–24 h, and then fixed. The fixed cells were treated with pepsin–HCl and their rests, containing partially denatured nuclei, were stained first with anti-CPD and then with FITC-conjugated antibodies.Unirradiated cells showed a background autofluorescence, recorded in the green channels of the flow cytometer, which was independent of cultivation period [Fig. 1(a), lowest line]. In the UV-irradiated cells fixed directly after irradiation, the fluorescence level was clearly higher due to a specific signal from anti-CPD antibodies bound to the nuclei and showed an apparent dependency on UV dose [Fig. 1(a), three upper lines], confirming the sensitivity achieved in our previous study.18
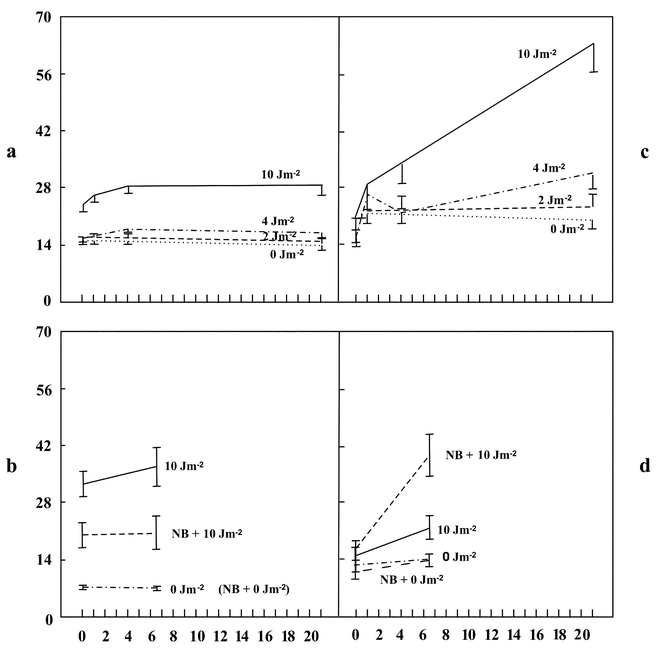 | ||
| Fig. 1 Intensities of anti-CPD (a, b) and anti-ssDNA (c, d) specific fluorescence from partially denatured nuclei of mononuclear leukocytes exposed to UVC radiation: (a, c) cells were irradiated in ice-cold PBS and then cultivated in supplemented RPMI medium at 37 °C; (b, d) cells were preincubated over a period of 1 h at room temperature in PBS with or without Novobiocin (0.175 mM), then irradiated at room temperature and cultivated in PBS at 37 °C. Doses of UVC and Novobiocin (NB) in the culture medium are shown alongside the corresponding lines. Abscissa: cultivation time/h; ordinate: scaled geometrical mean of the specific fluorescence (channel number average) from 104 nuclei measured with the flow cytometer. Bars show the standard deviation of the mean. | ||
In the first few hours of cultivation of the UV-irradiated cells, the intensity of CPD fluorescence became unexpectedly higher in 13 of 16 blood samples examined. Fig. 1(a) (upper three lines) shows the typical dynamic of the CPD signal after UV irradiation and cultivation of the cells. The CPD fluorescence increased in intensity up to 4 h of cultivation and then remained practically constant during the whole period of observation. Next, we attempted to ascertain the reason for this increase in the CPD signal via the following experiments.
Influence of cultivation factors on immuno-labelling
We speculated that cultivation of UV-irradiated cells facilitates the access of H3 antibody to nuclear DNA. Since H3 antibody binds CPD in single-stranded DNA (ssDNA),21 we checked whether the transfer of PBMLs from cold PBS to a warm humidified medium could cause enhancement of ssDNA in PBML nuclei. To detect ssDNA, we stained the nuclei with the anti-ssDNA antibody using a technique similar to that employed for anti-CPD staining.First, we found that incubation of PBMLs at different temperatures (0, 20 and 37 °C) for 1 h before fixation led to considerable changes in ssDNA-fluorescence emission from the prepared cell nuclei; these changes were completely reversible after cultivation of the cells at 37 °C for 18 h (Fig. 2). This result indicates that the strandedness of the nuclear DNA of the fixed cells is dependent on the temperature conditions of the preceding cell cultivation. To avoid any possible influence of temperature changes on the DNA strands in the following experiments, we no longer cooled the cells down to 0 °C and kept them at room temperature (about 20 °C) from the moment of drawing the blood sample, before and during UV irradiation and cultivation.
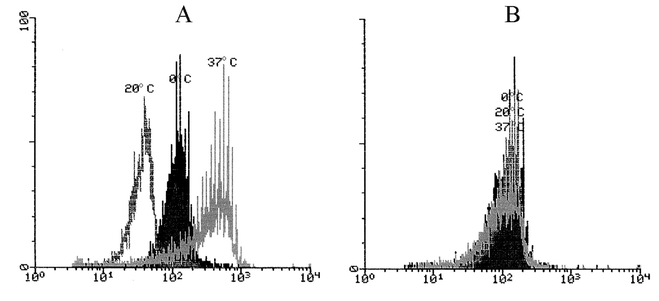 | ||
| Fig. 2 Histograms of anti-ssDNA-specific fluorescence from partially denatured nuclei of mononuclear leukocytes stored for 1 h at different temperatures (shown above the corresponding peaks) and then transferred and cultivated in supplemented RPMI medium at 37 °C for 0 (A) or 17 h (B). Abscissa: geometric mean of the specific ssDNA fluorescence; ordinate: relative cell count. | ||
Next, we found that the cultivation of PBMLs in the supplemented RPMI medium used here could also influence the DNA strands. This was manifested in changes in ssDNA fluorescence from the cellular nuclei of unirradiated cells. This fluorescence rose noticeably after 1 h cultivation and then gradually returned to the initial level [Fig. 1(c), lowest line]. In the UV-irradiated cells, the initial rapid rise in ssDNA fluorescence was followed by a further increase up to 21 h of observation. This second enhancement of the ssDNA signal appeared to be dependent on the UV dosage [Fig. 1(c), upper three lines].
To avoid any influence of culture medium exchange on the DNA strandedness in our further experiments we kept the cells in PBS during the entire cultivation period. Under these conditions, the unirradiated and UV-irradiated cells showed a somewhat lower increase in ssDNA fluorescence [Fig. 1(d), lines for 0 and 10 J m−2]. Despite this, the cultivation of UV-irradiated cells still led to an increase in CPD fluorescence [Fig. 1(b), top line]. The changes in specific ssDNA and CPD fluorescences in the nuclei of all the studied cell samples during cultivation reveal conformity to some extent, but their absolute means varied greatly between the different experiments and cell samples, which resulted in insignificant overall correlation between them.
Novobiocin effects on immuno-labelling of isolated DNA and nuclei of PBMLs
Having found an increase in CPD fluorescence from the nuclei of UV-irradiated cells, we tried to check whether a possible initial DNA repair step contributed to this effect. Consequently, 30–60 min before the irradiation of cells, we added Novobiocin in non-cytotoxic concentrations to the suspension with the aim of suppressing the initial phase of nucleotid excision repair, since Novobiocin has been found to be an inhibitor of nuclear enzymes involved in the stages preceding repair-specific DNA incision in human cells.23–27 We compared the effects of Novobiocin on CPD labelling of isolated DNA and nuclei in the same cell samples.The influence of the Novobiocin on the CPD labelling was already obvious at the 0 h cultivation time point, i.e. at the time of fixation of the UV-irradiated cells, which was about 18 min after the irradiation. (This period was required to collect the cells of all samples in centrifuge tubes and to sediment them and remove the supernatant). By this time, the CPD luminescence of DNA isolated from PBMLs pretreated with Novobiocin was discernibly higher than that for cells not treated with Novobiocin (Fig. 3, compare line 4 to line 3). This difference indicates that Novobiocin is indeed effective in inhibiting rapid excision of a considerable bulk of CPD that occurred during that short period following UV irradiation in PBMLs untreated with Novobiocin. After 6.5 h cultivation, the CPD luminiscence of DNA isolated from UV-irradiated cells not treated with Novobiocin decreased to a slight extent (Fig. 3, line 7) from the pre-cultivation level (Fig. 3, line 3), indicating that a further amount of CPDs was excised during the hours of cultivation. This latter excision seemed not to be greatly influenced by the pretreatment of cells with Novobiocin: the DNA isolated from these cells showed a similar slight decrease in CPD luminescence (Fig. 3, line 8) from its pre-cultivation level (Fig. 3, line 4).
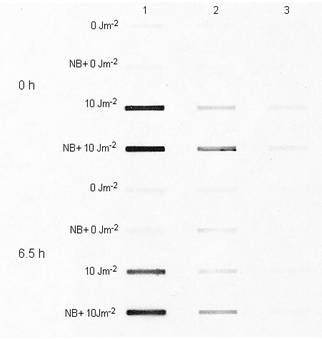 | ||
| Fig. 3 Photograph of the developed film exposed to CPD-specific luminescence from heat-denatured isolated DNA from UVC-irradiated mononuclear leukocytes. Cells were preincubated for a period of 1 h at room temperature in PBS, with or without 0.200 mM Novobiocin (NB), and then irradiated at room temperature. UVC and NB doses in the culture medium are shown next to the corresponding lines. The four upper lines correspond to DNA from irradiated cells fixed without cultivation (0 h, i.e. ∼18 min after UV irradiation), while the four lower lines were obtained for DNA from cells cultivated in PBS at 37 °C for 6.5 h. The amounts of DNA used to obtain the lines in columns 1, 2 and 3 were 900, 300 and 100 ng, respectively. | ||
In contrast to isolated DNA, the nuclei of the Novobiocin-pretreated PBMLs showed a lower CPD fluorescence at the moment of fixation after UV irradiation than the nuclei of cells that were not pretreated with Novobiocin [Fig. 1(b), 0 h]. Furthermore, after 6.5 h cultivation with Novobiocin, there was hardly any increase in CPD labelling of PBML nuclei, compared to a clear increase for PBMLs cultivated without Novobiocin [Fig. 1(b), 6.5 h]. Consequently, Novobiocin prevented the increase in CPD labelling of nuclei, despite the fact that DNA isolated from Novobiocin-pretreated cells contained significantly more anti-CPD antibody binding sites then their untreated counterparts.
Discussion
We used immuno-labelling of CPDs in nuclei of individual PBMLs after their UVC irradiation and cultivation. Unexpectedly, we have found that within the first 4 h of cultivation, the CPD-specific fluorescence signal from the cell nuclei increased [Fig. 1(a) and (b)]. As far as we are aware, this effect has not been reported previously. Considering that post-exposure formation of additional CPDs is improbable, we attempted to find a possible reason for the increase in specific CPD antibody binding to lymphocyte nuclei. We hypothesised that this increase might be a result of DNA modifications induced either by the cell culture conditions (e.g. changes in temperature and/or medium conditions) or by nucleotide excision repair enzymes. In further experiments, we examined both these possibilities.Since the H3 antibody used here binds to CPD in ssDNA,20 we decided to check first whether the ssDNA increase could be due to the conditions employed to culture the cells. Using an antibody against ssDNA, we determined that the cultivation, in fact, resulted in an increase in ssDNA-specific fluorescence intensity [Fig. 1(c)]. Because modifications of chromatin structure and stress protein synthesis have been reported to be associated with changes in temperature from 22 to 37 °C and from 37 to 20 °C28,29 we checked whether DNA strandedness might be changed in our experiments as a result of the transfer of cells from cold PBS to a warm humidified supplemented RPMI. We did indeed detect changes in ssDNA fluorescence after storage of cells at 0, 20 and 37 °C; and these changes were reversible when the cells were cultivated at a constant temperature [Fig. 2(a) and (b)]. We speculated that these temperature-related changes in DNA strandedness occurring in the first 4 h of cell cultivation might contribute to the increase in the ssDNA signal for both unirradiated and UV-irradiated cells over this period. However, when we kept the cells at constant room temperature during irradiation and cultivation they still showed changes in DNA strandedness and increased CPD labelling of UV-irradiated cell nuclei.
Furthermore, our experiments showed that UV-irradiation definitely evoked specific formation of anti-ssDNA antibody binding sites, since the ssDNA fluorescence signal did not decline after the initial increase, as for the unirradiated cells, but continued to rise in a dose-dependent manner from 4 to 21 h of cultivation [Fig. 1(c)]. However, these late changes in DNA strandedness were not accompanied by any detectable increase in CPD labelling of cellular nuclei.
From these results, we conclude that two types of ssDNA increase take place in nuclei during cultivation of UV-irradiated cells. The first increase recorded during the first 4 h of cultivation is transient, independent of UV dose and attributable to changes in culture conditions. The second increase gradually develops over 21 h, is not reversible and is most likely caused by UV irradiation, since it clearly showed dependency on the UV dosage. During the first 4 h of cultivation, both types of ssDNA increase probably occur simultaneously. In spite of the fact that we were not able to determine a significant correlation between ssDNA- and CPD-specific fluorescence, we cannot rule out the possibility of such a correlation during the first 4 h. In any case, the observed UVC-dose dependent ssDNA rise in cellular nuclei indicated the specific character of the ongoing DNA modifications, which could be associated with enzymatic repair of DNA.
Direct confirmation of the ongoing excision repair in UV-irradiated cells was provided by further experiments using CPD labelling of isolated DNA. Using isolated DNA, we clearly determined an obvious decrease in CPD-containing sites after 6.5 h of cultivation (Fig. 3). This decrease is not particularly great, and it has been reported that even after 24 h cultivation of UV-irradiated unstimulated peripheral lymphocytes, the number of CPDs detected with a dimer-specific endonuclease remains as high as 85% of their initial count.14 However, unlike this study, that investigation reported no detectable removal of CPDs within 6 h after irradiation. Another study found evidence for incision–excision in this time period after UV irradiation in unstimulated blood leukocytes.30 In our experiments, removal of CPDs not only over 6.5 h, but also at shorter cultivation times has been observed. We conclude this from the experiments with PBMLs pretreated with Novobiocin in non-cytotoxic concentrations. Novobiocin inhibits topoisomerase II, which relaxes supercoiled DNA in the stages preceding repair-specific DNA incision.23–27 We have found that Novobiocin pretreatment leads to higher CPD labelling of DNA isolated from UV-irradiated PBMCs in a period from 18 min to 6.5 h after irradiation of cells (Fig. 3). This higher CPD labelling appears to be attributable to inhibition by Novobiocin of the CPD removal by excision repair in the cells. Detectable removal of CDPs in the first 18 min after irradiation in cells not treated with Novobiocin coincides well with immediate activation of nucleotide excision repair after DNA damage and substantial incision–excision actions within the first 30–60 min. The fact that, along with rapid excision of CPDs from isolated DNA, there was a considerable enhancement in the CPD-specific labelling of partially denatured nuclei [Fig. 1(b)] supports our assumption that this latter effect is associated with DNA modification in the early phase of repair, inhibited by Novobiocin.
It is to be noted that Novobiocin did not seem to inhibit the eventual removal of some CPD sites from isolated DNA after 6.5 h of cultivation. This removal was observed to a comparable extent with both Novobiocin-treated and untreated cells (Fig. 3). Hence, the later CPD removal was not inhibited by Novobiocin and, remarkably, was not accompanied by any detectable changes in CPD labelling of nuclei, despite the considerable increase in the ssDNA signal emitted by Novobiocin-treated cells during this period [Fig. 1(c) and (d)]. This fact is in favour of the suggestion that the increase in anti-CPD antibody binding of nuclei is due to a DNA modification which takes place in the initial phases of nucleotide excision repair.
It is assumed that for efficient DNA lesion detection by the global nucleotide excision repair system, the chromatin has to be relaxed. The UV-induced global chromatin relaxation is extended over the whole nucleus and this process requires the tumour-suppressor protein p53.31 Nucleotide excision repair of lesions in human cells begins with ATP-dependent formation of an open DNA structure of approximately 25 nucletides around the DNA adduct, which is brought about by the action of topoisomerase II relaxing the supercoiled DNA structure. This opening phase is followed by dual incision of that fragment executed by two endonucleases, XPG on the 3′ side of the lesion and ERCC1-XPF on the 5′ side, with 3′ cleavage occurring first.32,33 Unstimulated human lymphocytes have been reported to show defective global repair of CPDs due to an insufficiency in excision, rather than low deoxyribonucleotide pool sizes.16 Our findings suggest that unstimulated PBMLs perform substantially more pre-incision enzymatic opening of CPD-containing DNA fragments than the subsequent dual incision and excision of these fragments. Dominance of such DNA opening over complete excision of CPD leads to enhanced anti-CPD antibody binding to DNA in nuclear structures resistant to the pepsin–HCl denaturing treatment. The PBMLs appear to be capable of pre-incision preparation phases for the enzymatic repair of damaged DNA, while showing a diminished capacity to excise the damage.
Conclusion
Immunostaining of CPDs and ssDNA in UV-irradiated non-stimulated human PBML nuclei has revealed a Novobiocin-inhibited DNA modification occurring due to the activity of enzymes in the pre-incision phase of nucleotide excision repair. This modification is manifested by a considerable enhancement in the labelling of PBML nuclei with anti-CPD antibody, which is likely to be the result of opening up stretches of CPD-containing double-stranded DNA while the excision of the damage is lacking.Acknowledgement
This work was partially supported by grant no. 07-13-064 of the Dutch Organisation for Scientific Research (NWO).References
- M. S. Fisher and M. L. Kripke, Systemic alteration induced in mice by ultraviolet light irradiation and its relationship to ultraviolet carcinogenesis, Bull. W. H. O., 2002, 80, 908–912 Search PubMed.
- L. A. Applegate, R. D. Ley, J. Alcalay and M. L. Kripke, Identification of the molecular target for the suppression of contact hypersensitivity by ultraviolet radiation, J. Exp. Med., 1989, 170, 1117–1131 CAS.
- M. L. Kripke, P. A. Cox, L. G. Alas and D. B. Yarosh, Pyrimidine dimers in DNA initiate systemic immunosuppression in UV-irradiated mice, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 7516–7520 CAS.
- D. B. Yarosh, L. Alas, J. Kibitel, A. O'Connor, F. Carrier and A. G. Fornace, Jr., Cyclobutane pyrimidine dimers in UV-DNA induce release of soluble mediators that activate the human immunodeficiency virus promoter, J. Invest. Dermatol., 1993, 100, 790–794 CAS.
- M. L. Kripke, P. A. Cox, C. Bucana, A. A. Vink, L. Alas and D. B. Yarosh, Role of DNA damage in local suppression of contact hypersensitivity in mice by UV radiation, Exp. Dermatol., 1996, 5, 173–180 CAS.
- C. Nishigori, D. B. Yarosh, S. E. Ullrich, A. A. Vink, C. D. Bucana, L. Roza and M. L. Kripke, Evidence that DNA damage triggers interleukin 10 cytokine production in UV-irradiated murine keratinicytes, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 354–10359 CrossRef CAS.
354–10359 CrossRef CAS. - A. A. Vink, A. M. Moodycliff, V. Shreedhar, S. E. Ullrich, L. Roza, D. B. Yarosh and M. L. Kripke, The inhibition of antigen-presenting activity of dendritic cells resulting from UV irradiation of murine skin is restored by in vitro photorepair of cyclobutane pyrimidine dimers, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 5255–5260 CrossRef CAS.
- H. M. Hurks, C. Out-Luiting, B. J. Vermeer, F. H. Claas and A. M. Mommaas, In situ action spectra suggest that DNA damage is involved in ultraviolet radiation-induced immunosuppression in humans, Photochem. Photobiol., 1997, 66, 76–81 CAS.
- J. Kibitel, V. Hejmadi, L. Alas, A. O'Connor, B. M. Sutherland and D. B. Yarosh, UV-DNA damage in mouse and human cells induces the expression of tumor necrosis factor alpha, Photochem. Photobiol., 1998, 67, 541–546 CAS.
- C. Petit-Frere, P. H. Clingen, M. Grewe, J. Krutmann, L. Roza, C. F. Arlett and M. H. Green, Induction of interleukin-6 production by ultraviolet radiation in normal human epidermal keratinocytes and in a human keratinocyte cell line is mediated by DNA damage, J. Invest. Dermatol., 1998, 111, 354–359 CAS.
- H. Stege, L. Roza, A. A. Vink, M. Grewe, T. Ruzicka, S. Grether-Beck and J. Krutmann, Enzyme plus light therapy to repair DNA damage in ultraviolet-B-irradiated human skin, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 1790–1795 CrossRef CAS.
- D. B. Yarosh, Enhanced DNA repair of cyclobutane pyrimidine dimers changes the biological response to UV-B radiation, Mutat. Res., 2002, 509, 221–226 CrossRef CAS.
- M. H. Green, C. Petit-Frere, P. H. Clingen, G. Bentham, J. Cole and C. F. Arlett, Possible effects of sunlight on human lymphocytes, J. Epidemiol., 1999, 9(6), S48–S57 Search PubMed.
- S. E. Freeman and S. L. Ryan, Excision repair of pyrimidine dimers in human peripheral blood lymphocytes: comparison between mitogen stimulated and unstimulated cells, Mutat. Res., 1988, 194, 143–150 CrossRef CAS.
- P. H. Clingen, C. F. Arlett, J. Cole, A. P. Waugh, J. E. Lowe, S. A. Harcourt, N. Hermanova, L. Roza, T. Mori and O. Nikaido, Correlation of UVC and UVB cytotoxicity with the induction of specific photoproducts in T-lymphocytes and fibroblasts from normal human donors, Photochem. Photobiol., 1995, 61, 163–170 CAS.
- P. Han, P. H. Clingen, J. E. Lowe, A. Katsuya, C. F. Arlett and M. H. Green, Repair of cyclobutane pyrimidine dimers in unstimulated human mononuclear cells is deficient at very low fluences of ultraviolet B and is not enhanced by addition of deoxyribonucleosides, Mutagenesis, 1998, 13, 353–356 Search PubMed.
- M. H. Green, A. P. Waugh, J. E. Lowe, S. A. Harcourt, J. Cole and C. F. Arlett, Effect of deoxyribonucleosides on the hypersensitivity of human peripheral blood lymphocytes to UV-B and UV-C irradiation, Mutat. Res., 1994, 315, 25–32 CrossRef CAS.
- S. A. Snopov, R. J. W. Berg, H. Van Weelden, K. A. Samoilova, J. C. van der Leun and F. R. de Gruijl, Molecular dosimetry by flow cytometric detection of thymine dimers in mononuclear cells from extracorporally UV-irradiated blood, J. Photochem. Photobiol., 1995, 28, 33–37 Search PubMed.
- R. J. W. Berg, F. R. de Gruijl, L. Roza and J. C. van der Leun, Flow cytometric immunofluorescence assay for quantification of cyclobutildithymine dimers in separate phases of the cell cycle, Carcinogenesis, 1993, 14, 103–106 CAS.
- L. Roza, K. J. M. van der Wulp, S. J. MacFarlane, P. H. M. Lohman and R. A. Baan, Detection of cyclobutane thymine dimers in DNA of human cells with monoclonal antibodies against a thymine dimer-containing tetranucleotide, Photochem. Photobiol., 1998, 48, 627–633.
- G. P. Van der Schans, A. A. W. M. van Loon, R. H. Groenendijk and R. A. Baan, Detection of DNA damage in cells exposed to ionizing radiation by use of anti-single-stranded DNA monoclonal antibody, Int. J. Radiat. Biol., 1989, 55, 747–760 CAS.
- A. A. Vink, J. B. A. Bergen Henegouwen, O. Nikaido, R. A. Baan and L. Roza, Removal of UV-B-induced DNA lesions in mouse epidermis soon after irradiation, J. Photochem. Photobiol., 1994, 24, 25–31 Search PubMed.
- S. L. Dresler and R. M. Robinson-Hill, Direct inhibition of u.v.-induced DNA excision repair in human cells by novobiocin, coumermycin and nalidixic acid, Carcinogenesis, 1987, 8, 813–817 CAS.
- P. J. Wood and A. G. Stansfield, Inhibition of T-cell mediated cytotoxicity by Novobiocin suggests multiple pathways for both CD4+ and CD8+ cytotoxic T cells, Immunology, 1992, 76, 460–464 CAS.
- H. W. Thielmann, O. Popanda, H. Gersbach and F. Gilberg, Various inhibitors of DNA topoisomerases diminish repair-specific DNA incision in UV-irradiated human fibroblasts, Carcinogenesis, 1993, 14, 2341–2351 CAS.
- F. Ali-Osman, M. S. Berger, S. Rajagopal, A. Spene and R. B. Livingstone, Topoisomerase II inhibition and altered kinetics of formation and repair of nitrosouria and cisplastin-induced DNA interstand cross-links and cytotoxicity in human glioblastoma cells, Cancer Res., 1993, 53, 5663–5668 CAS.
- F. Rosselli, E. Duchaud, D. Averbeck and E. Moustacchi, Comparison of the effects of DNA topoisomerase inhibitors on lymphoblasts from normal and Fanconi anemia donors, Mutat. Res., 1994, 325, 137–144 CrossRef CAS.
- J. T. Westwood, J. Clos and C. Wu, Stress-induced oligomerization and chromosomal relocalization of heat-shock factor, Nature, 1991, 353, 822–827 CrossRef CAS.
- D. B. Holland, S. G. Roberts, E. J. Wood and W. J. Cunliffe, Cold shock induces the synthesis of stress proteins in human keratinocytes, J. Invest. Dermatol, 1993, 101, 196–199 CAS.
- M. H. Lankinen, L. M. Vilpo and J. A. Vilpo, UV- and gamma-irradiation-induced DNA single-strand breaks and their repair in human blood granulocytes and lymphocytes, Mutat. Res., 1996, 352, 31–38 CrossRef CAS.
- C. P. Rubbi and J. Milner, p53 is a chromatin accessibility factor for nucleotide excision repair of DNA damage, EMBO J., 2003, 22, 975–986 CrossRef CAS.
- T. Matsunaga, D. Mu, C. H. Park, J. T. Reardon and A. Sankar, Human DNA repair excision nuclease. Analysis of the roles of the subunits involved in dual incisions by using anti-XPG and anti-ERCC1 antibodies, J. Biol. Chem., 1995, 270, 20862–20869 CAS.
- E. Evans, J. G. Moggs, J. R. Hwang, J. M. Egly and R. D. Wood, Mechanism of open complex and dual incision formation by human nucleotide excision repair factors, EMBO J., 1997, 16, 6559–6573 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and Owner Societies 2004 |