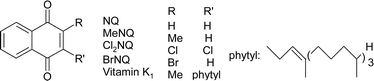
Photoprocesses of p-naphthoquinones and vitamin K1: effects of alcohols and amines on the reactivity in solution
Received
11th June 2003
, Accepted 8th September 2003
First published on 30th September 2003
Abstract
The photochemistry of 1,4-naphthoquinone (NQ), the 2-methyl, 2,3-dichloro and 2-bromo derivatives, and vitamin K1 was studied in non-aqueous solvents by time-resolved UV-vis spectroscopy after ns laser pulses at 248 and 308 nm. The triplet state of the NQs reacts with alcohols and amines, e.g. triethylamine (TEA) and DABCO, yielding semiquinone radicals (HQ˙/Q˙−). They are the major intermediates and their second-order decay kinetics depend on the properties of the additives and the medium. Transient conductivity measurements suggest the occurrence of photoinduced electron transfer from amines to the triplet state of NQs in acetonitrile. The photoconversion (λirr
= 254 nm) of NQs to the 1,4-dihydroxynaphthalenes (H2Q) was measured in the absence and presence of varying concentrations of electron and H-atom donors, and the quantum yield was found to increase with increasing electron- or proton-donor concentration. The mechanisms of photoreduction of NQs by propan-2-ol and TEA in acetonitrile exhibit a number of similarities. Oxygen quenches the triplet state, thereby forming singlet molecular oxygen. Oxygen also reacts with the semiquinone radical, thereby forming HO2˙/O2˙− radicals, and reacts with H2Q, thereby re-forming the quinone. A different pattern, involving intramolecular H-atom transfer, holds for vitamin K1, where 1,3-quinone methide (1,3-QM) diradicals were observed in acetonitrile prior to formation of two 1,2-QM tautomers, but a triplet was not. The decay of the 1,3-QM intermediates becomes faster in the presence of alcohols and amines due to proton-transfer reactions.
1 Introduction
Quinones are of great photobiological importance and serve as electron acceptors in photosynthesis.1 The photoreduction of 1,4-benzoquinone and its derivatives, such as 1,4-naphthoquinone (NQ), 2-methyl-1,4-naphthoquinone (MeNQ) and 9,10-anthraquinone (AQ), has attracted widespread attention.1–26 Generally, quinones exhibit a high quantum yield of intersystem crossing (Φisc), e.g.Φisc
= 0.74 for NQ in acetonitrile.2,10 MeNQ (vitamin K3 or menadione) was frequently used to generate radical cations, especially the radical cation of thymine.7 Cross-linking with a quantum yield of <10−3 was reported for single-stranded DNA.8 FTIR spectroscopic studies of quinones have suggested new avenues for investigation and yielded structural information.1,12 This is important for the understanding of quinone–protein interactions in photosynthetic reaction centers.
The photochemistry of vitamin K1 has been the subject of various studies.27–34 In contrast to other quinones without isoprenoid side chains, vitamin K1 undergoes intramolecular H-atom and proton-transfer processes. The products have been identified as the E and Z tautomers of 1,2-quinone methide (1,2-QM).28–31 Two 1,3-quinone methide (1,3-QM) diradical intermediates have been characterized as the singlet and triplet states of 1,3-QM.34
The photoreduction of quinones by amines has been intensively studied.11–15,21–23 The rate constant for triplet quenching (kD) of NQ and MeNQ in acetonitrile by N,N-dimethylaniline (DMA) and triethylamine (TEA), due to appropriate redox properties, is close to the diffusion-controlled limit.11,12 The photoreduction of quinones by propan-2-ol4,19 or TEA leads, via the corresponding semiquinone radicals (HQ˙/Q˙−), to hydroquinones (H2Q), 1,4-dihydroxynaphthalene in the case of NQ. The studies into the photochemistry of quinones are complemented by radiochemical studies.35–37 The semiquinone radical plays a key role in the photoreduction of NQs, even when no donor is added. Photoinduced electron transfer from furan to the triplet state has been reported for 2,3-dichloro-1,4-naphthoquinone (Cl2NQ).9
This paper aims to gain a deeper insight into the photoreduction reactions of NQ, MeNQ, 2-bromo-NQ (BrNQ) and Cl2NQ in solution at room temperature in the presence of amines and alcohols. Time-resolved UV-vis spectroscopy and conductivity measurements with 248 and 308 nm pulses were applied and the secondary intermediates were produced by triplet quenching. Conversion of the NQs into hydroquinones and the quantum yield of decomposition were measured by continuous irradiation at 254 nm. The photoreduction pathways in acetonitrile–propan-2-ol, benzene–TEA and acetonitrile–TEA mixtures show a close relationship. In addition, the photochemical reactions of vitamin K1 in acetonitrile in the absence and presence of amines and alcohols were studied. The data for vitamin K1 are consistent with the modified reaction mechanism recently presented by Wirz et al.34
2 Materials and methods
All reagents were obtained from Aldrich and the solvents from Merck, benzene was Uvasol grade. NQ was purified by recrystallization and methylcyclohexane (MCH), 2-methyltetrahydrofuran (MTHF), TEA and DEA were distilled prior to use. All other reagents were used as received. Note that commercial vitamin K1
(from Aldrich in our case) consists of Z and E isomer racemates in a ratio of 0.1–0.2.34 The molar absorption coefficients of NQ are ε254
= 1.7 × 104 M−1 cm−1 and ε308
= 1.5 × 103 M−1 cm−1. The absorption spectra were monitored on a HP 8453 UV-vis spectrophotometer. For photoconversion, the 254 nm line of a Hg lamp was used. The photoconversion was carried out with vigorous bubbling of argon through the solution prior to and during irradiation. The photodecomposition of the substrate was measured upon prolonged irradiation. Product analysis by HPLC was not successful due to the oxygen effect, as reported for AQs.22 The emission and excitation spectra, and phosphorescence lifetime at −196 °C were measured on a Cary Eclipse spectrofluorimeter.
Relative yields were obtained using optically matched solutions (A254
= 1.5–2.0). The quantum yield of decomposition (Φd) was determined using the uridine–water–oxygen actinometer.38 An excimer laser (Lambda Physik EMG 200, pulse width of 20 ns and energy <100 mJ) was used for excitation at 308 nm. For a few experiments, excitation at 248 nm was used. The absorption signals were measured with two digitizers (Tektronix 7912AD and 390AD). Typically, absorbances of 0.3–1.5 were used for λexc, corresponding to concentrations of 0.2–1 mM. The molar absorption coefficient of HQ˙ for NQ in aqueous solution is ε370
= 7 × 103 M−1 cm−1 that of Q˙− is ε390
= 13 × 103 M−1 cm−1.35,36 For NQ in acetonitrile, the molecular absorption coefficient of T–T absorption and the semiquinone radical anion are ε365
= 8.2 × 103 M−1 cm−1 and ε403
= 8.8 × 103 M−1 cm−1, respectively, and for MeNQ, the molar absorption coefficient of Q˙− is ε405
= 7.5 × 103 M−1 cm−1.10,11 The value for Q˙− of Cl2NQ in acetonitrile–water is ε400
= 7.9 × 103 M−1 cm−1.9 The quantum yields of formation of singlet molecular oxygen (ΦΔ) were measured as described previously.39 The measurements were made at 24 ±2 °C in deoxygenated solutions, unless otherwise indicated.
3 Results and discussion
3.1 Reactions of the triplet state with oxygen and alcohols
The T–T absorption spectra of NQ in argon-saturated acetonitrile [Fig. 1(a)] and carbon tetrachloride (λexc
= 308 nm) show a maximum at λTT
= 370 nm. Efficient population of the triplet state (reaction 1) is known to occur from literature values of Φisc
= 0.74, 0.86 and 0.90 for NQ, MeNQ and Cl2NQ in acetonitrile, respectively.9,10The decay kinetics in the absence of additives follow essentially a first-order law (rate constant: 1/τT). Variation of the NQ or MeNQ concentration in acetonitrile shows a linear dependence of 1/τT (reaction 2).10Therefore, the lifetimes presented are for relatively low concentrations, corresponding to A308
= 0.5, where τT
=1.9 µs. The triplet lifetime of Cl2NQ is much longer, in agreement with τT
= 7 µs in acetonitrile–water (4 ∶ 1).9 The λTT and τT values of the NQs examined are compiled in Table 1.
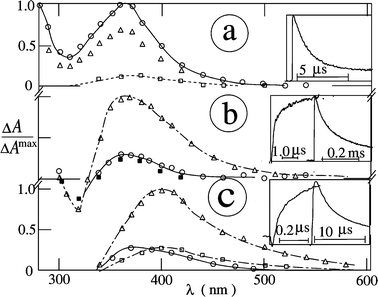 |
| | Fig. 1 Transient absorption spectra of NQ in argon-saturated acetonitrile (a) with no added donor, (b) in the presence of propan-2-ol (1 M) and (c) in the presence of DABCO (0.3 mM) at 20 ns (○), 1 µs (Δ), 10 µs (□) and 10 ms (■) after the 308 nm pulse Insets: radical formation and decay at 410 nm. | |
Table 1 Absorption maxima (λTT), triplet lifetimes (τT),a rate constants for quenching by oxygen (kox) and quantum yields of formation of singlet molecular oxygen (ΦΔ) for the NQs
| Quinone |
Solvent |
λ
TT/nm |
τ
T/µs |
k
ox/109 M−1 s−1 |
Φ
Δ
b
|
|
In argon-saturated solution, λexc
= 308 nm.
In oxygen-saturated solution.
For A308
= 0.5.
λ
exc
= 248 nm.
|
| NQ |
CCl4 |
370 |
2.5 |
0.7 |
0.6 |
| |
C6H6 |
370 |
0.2 |
>0.8 |
0.07 |
| |
CH2Cl2 |
380 |
0.9 |
0.4 |
|
| |
CH3CN |
370 |
1.9c, 3d |
0.3 |
0.4 |
| MeNQ |
CH3CN |
380 |
0.9c |
1.5 |
|
| Cl2NQ |
C6H6 |
370, 470sh |
15 |
0.6 |
0.6 |
| |
CH3CN |
370, 470sh |
8, 8d |
0.8 |
0.6 |
| BrNQ |
CCl4 |
400 |
5 |
|
0.7 |
| |
CH3CN |
400 |
3, 3d |
0.8 |
0.7 |
The rate constant for triplet quenching by oxygen (reaction 3) is kox
=
(0.3–1.5)
× 109 M−1 s−1
(Table 1).
| | | 3*Q + O2
→ Q + O2(1Δg) | (3) |
The decay of the triplet depends strongly on the kind of
solvent and/or appropriate additives (H
2D). For NQ and MeNQ in
acetonitrile, the T–T absorption is overlapped by a weak longer-lived transient with a maximum at
λrad
= 375 nm, which is assigned to the
semiquinone radical (HQ˙). In contrast, the
absorption spectra of Cl
2NQ [
Fig. 2(a)] and BrNQ show mainly the triplet state. After
reaction 4, radical termination
viareaction 5 yields Q and the corresponding
hydroquinone (H
2Q).
Further pathways to the
semiquinone radical are quenching of the triplet state by Q (self-quenching,
k2) and T–T annihilation under pulsed excitation. For NQ in
acetonitrile, the literature value is
k2
= 1 × 10
9 M
−1 s
−1.
10 Transient
absorption spectra of NQ in mixtures of
acetonitrile and
propan-2-ol are due to the triplet state and the
semiquinone radical, which are both strongly absorbing and completely overlap each other [
Fig. 1(b)]. For the NQs, the reactivity is rather high when compared to, for example,
duroquinone.
2,5 After photoconversion, triplet quenching by
hydroquinone also takes place, the rate constant for which is close to the diffusion-controlled limit.
40
 |
| | Fig. 2 Transient absorption spectra of Cl2NQ in argon-saturated acetonitrile (a) with no added donor, (b) in the presence of propan-2-ol (0.1 M) and (c) in the presence of TEA (1 mM) at 20 ns (○), 1 µs (Δ), 10 µs (□), 0.1 ms (●) and 0.1 s (♦) after the 308 nm pulse. Insets: radical formation and decay at 410 nm. | |
The rate constant for triplet decay of NQs in argon-saturated acetonitrile becomes larger on addition of propan-2-ol. The rate constants for triplet quenching (reaction 4), obtained from the linear dependences of 1/τTvs.
[propan-2-ol], are specific for each quinone and medium. The value is kD
= 3 × 107 M−1 s−1 for NQ and MeNQ, and lower for the other NQs (Table 2). In aqueous solution at pH 7, the semiquinone radical is present as a radical anion (Q˙−) with pKa
= 4.0–4.5.2,35–37
| | 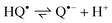 | (6) |
In contrast to AQ, where the
spectra of HQ˙ and Q˙
− are quite different, those of the NQs are only slightly shifted.
2,35 The overall photoconversion leads to H
2Q and acetone. Transient
absorption spectra of NQ in mixtures of
acetonitrile and propan-2-ol are presented in
Fig. 1(b). In contrast to NQ and MeNQ, the
spectra of
3Q* and HQ˙ are separated for Cl
2NQ [
Fig. 2(b)] and BrNQ, since
λTT is red-shifted with respect to
λrad. Decay of the
semiquinone radical under argon occurs by second-order kinetics, with a first half-life in the ms range under our conditions. In principle, the
alcohol radical could yield another
semiquinone radical
viareaction 7. A second reduction, due to
reactions 4 and
7, became unobservable and self-termination (
reaction 8) probably competes efficiently upon pulsed excitation, as already suggested for AQs.
22
Table 2 Rate constants for quenching by amines and propan-2-ol (kD), absorption maxima (λrad) and half-lives (t1/2) for the semiquinone radicals of the NQsa
| Quinone |
Solvent |
Donor |
k
D
b/109 M−1 s−1 |
λ
rad
c/nm |
t
1/2/ms |
|
In argon-saturated solution, λexc
= 308 nm.
[Amine] = 0.2–10 mM, [propan-2-ol] = 0.02–1 M.
The first and second values correspond to [amine]
= 0.3 and 20 mM, respectively.
With DEA.
λ
exc
= 248 nm.
|
| NQ |
C6H6 |
TEA |
3 |
380 |
0.002 |
| CH3CN |
DABCO |
9 (15)d |
360, 400 |
0.01 |
| TEA |
16 |
370, 405 |
2 |
| Propan-2-ol |
0.03 |
370 |
0.2 |
| MeNQ |
C6H6 |
TEA |
3 |
380 |
0.02 |
| CH3CN |
TEA |
12 |
380 |
2 |
| Propan-2-ol |
0.03 |
375 |
1 |
| Cl2NQ |
C6H6 |
TEA |
2 |
460 |
0.005 |
| CH3CN |
TEA |
10 |
460 |
>50 |
| Propan-2-ol |
0.005 |
450 |
0.09 |
| BrNQe |
CH3CN |
TEA |
3 |
390 |
2 |
| Propan-2-ol |
0.015 |
385 |
0.05 |
3.2 Reactions of the quinone triplet state with amines
The triplet decay of NQs in argon-saturated acetonitrile (λexc
= 308 nm) is accelerated on addition of TEA or DABCO (1,4-diazabicyclo[2.2.2]octane), and kD
=
(0.3–1.6)
× 1010 M−1 s−1
(Table 2). For NQ–TEA in acetonitrile, the literature value is kD
= 1.9 × 1010 M−1 s−1.10 The high rate constant for triplet quenching by amines is due to a negative free energy change, resulting from their low oxidation potentials and the reduction potentials (SCE) of −Ered
= 0.71, 0.77 and 0.45 V for NQ, MeNQ and Cl2NQ in acetonitrile, respectively.9,11 Triplet quenching by an amine occurs viareaction 4′, leading to Q˙− and the radical cation of the amine (H2D˙+).| | | 3*Q + H2D → Q˙−
+ H2D˙+ | (4′) |
The molar absorption coefficient of the DMA radical cation in acetonitrile is ε465
= 6.3 × 103 M−1 cm−1.11 With DABCO, back electron transfer (reaction 9) accounts for the second-order decay of the semiquinone radical within ca. 10 µs, as shown in Fig. 1(c) for NQ.Alternatively, a proton transfer could lead to neutral radicals (HQ˙ and HD˙); however, this does not occur.22,23 The first half-life for the NQs–TEA–acetonitrile system is relatively large (Table 2). The dependence of the yield of the semiquinone radical of NQ vs.
[TEA] can be fitted using the kD and τT values (not shown).
The transient absorption spectra of NQ and Cl2NQ in acetonitrile in the presence of TEA are shown in Fig. 3(a) and 2(c), respectively. For TEA in acetonitrile, equilibrium 10, with efficient formation of the α-aminoethyl radical is known to occur.22,23,41
| | 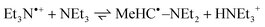 | (10) |
 |
| | Fig. 3 Transient absorption spectra (in argon-saturated solution) of (a) NQ in acetonitrile in the presence of TEA (10 mM), (b) NQ in benzene in the presence of TEA (1 mM) (both with λexc
= 308 nm) and (c) BrNQ in acetonitrile in the presence of propan-2-ol (0.1 M) (λexc
= 248 nm) at 20 ns (○), 1 µs (Δ), 10 µs (□), 0.1 ms (●) and 10 ms (■) after the pulse. Insets: (a, b) radical formation and decay at 410 nm; (c) triplet (above) and radical (below) formation and decay at 400 nm. | |
Thus, it is conceivable that on increasing the TEA concentration, deprotonation is in competition with a second reaction leading to semiquinone radicals.
| | Q + MeHC˙–NEt2
→ Q˙−
+ CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHNEt2
+ H+ CHNEt2
+ H+ | (11) |
Reaction 11 accounts for the second grow-in of the absorption within a few µs, due to formation of Q˙
− in the case of NQ [
Fig. 3(a)]. The second reduction step
via the α-aminoethyl radical was observed only at higher
TEA concentrations, corresponding to a triplet lifetime of <50 ns.
3.3 Transient conductivity
An increase in conductivity (Δκ: positive signal) was observed within 1 µs after the pulse for NQ in argon-saturated neat acetonitrile in the presence of DABCO, TEA or DEA (>1 mM), λexc
= 308 nm. The time dependences of representative cases are shown in Fig. 4. The rate constant of the fast increase depends linearly on [amine] and the signal approaches a maximum at an amine concentration of ca. 0.1 mM. The dependence of the conductivity yield of NQ vs.
[TEA] is similar to that of the semiquinone radical (not shown). These findings are in full agreement with reactions 1 and 4′of photoinduced electron transfer. For DABCO (inset b in Fig. 4) or DEA, the conductivity signal decays by second-order kinetics within 10 µs due to back electron transfer (reaction 9). The conductivity signal of NQ in argon-saturated acetonitrile in the presence of TEA (>1 mM) is much longer lived. It decays by second-order kinetics, at least for the major part. After subtraction of the (minor) remaining part, the half-lives (in the 3–30 ms range) increased by a factor of ca. 5–8 when the laser intensity was decreased ten-fold. This is consistent with a termination process (reaction 5′) and subsequent protonation.Interestingly, the radical termination of the NQ–TEA–acetonitrile system is slowed down on addition of water (Fig. 4), indicating a smaller rate constant for Q˙− than HQ˙ radicals. The conductivity increase for NQ in wet acetonitrile (5 M water, pH 7) also depends also on the propan-2-ol concentration, and the signal, which is mainly due to proton release viareaction 6, approaches a maximum at about 1 M propan-2-ol. The conductivity signal at pH 3–4 is much lower, since H-atom transfer without deprotonation does not result in a conductivity change. Moreover, a delayed increase in conductivity due to reactions 10 and 11 was not observed in any case, even at higher TEA concentrations of 3–10 mM. The signals for MeNQ, BrNQ and Cl2NQ in dry acetonitrile in the presence of DABCO or TEA (not shown) are comparable to those presented for NQ.
![Conductivity signals for NQ in argon-saturated acetonitrile in the presence of DABCO (1 mM; □), DEA (1 mM; ◇) and TEA [>1 mM; without (○) and with 5 M water (●)] after the 308 nm pulse. Insets: signals for (a) TEA (0.2, 0.8 and 5 mM, left to right) and (b) DABCO.](/image/article/2004/PP/b306670n/b306670n-f4.gif) |
| | Fig. 4 Conductivity signals for NQ in argon-saturated acetonitrile in the presence of DABCO (1 mM; □), DEA (1 mM; ◇) and TEA [>1 mM; without (○) and with 5 M water (●)] after the 308 nm pulse. Insets: signals for (a) TEA (0.2, 0.8 and 5 mM, left to right) and (b) DABCO. | |
3.4 Reactions upon 248 nm excitation
When the wavelength of excitation was changed from 308 to 248 nm, lower NQ concentrations could be used and, apart from the triplet decay of NQ and MeNQ, essentially the same results were obtained. An example of the spectra and kinetics in argon-saturated acetonitrile in the presence of propan-2-ol is shown in Fig. 3(c) for BrNQ. Owing to the above-mentioned linear dependence of 1/τTvs. NQ concentration, the longest triplet lifetime was achieved with λexc
= 248 nm. However, this reaction seems to play no discernible role for BrNQ and Cl2NQ (Table 1). Photoionization does not contribute at the applied low intensities, as no solvated electrons were detected for NQs in alcohols, where solvated electrons are not scavenged within the pulse width (as in acetonitrile). Note that the hydrated electron has been observed for MeNQ in aqueous solution.25
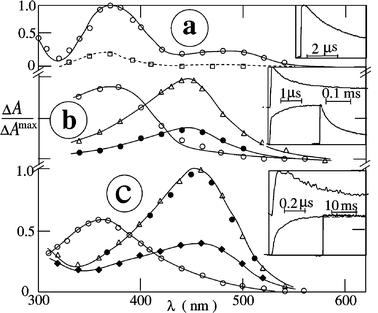
Examples of the transient absorption spectra and kinetics are shown for vitamin K1 in acetonitrile [Fig. 5(a) and (b)], and in the presence of an alcohol [Fig. 5(c)] or amines (Fig. 6). These photolysis results, which were obtained under similar conditions, seem to resemble those of the four NQs. However, the detected primary and secondary transients of vitamin K1
(1,3-QM and 1,2-QM, respectively, see section 3.9) and the NQs are not identical to those detected for NQs.
 |
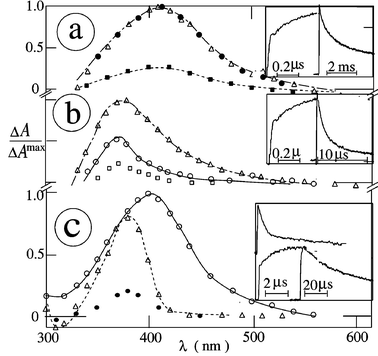
| | Fig. 5 Transient absorption spectra of vitamin K1 in acetonitrile: (a) air-saturated, no added donor; (b) argon-saturated, no added donor; (c) argon-saturated, in the presence of propan-2-ol (0.2 M). Spectra were measured at 20 ns (○), 1 µs (Δ), 10 µs (□), 0.1 ms (●) and 0.1 s (◆) after the 248 nm pulse. Insets: decay kinetics at 390 nm (above) and grow-in at 470 nm (below). | |
 |
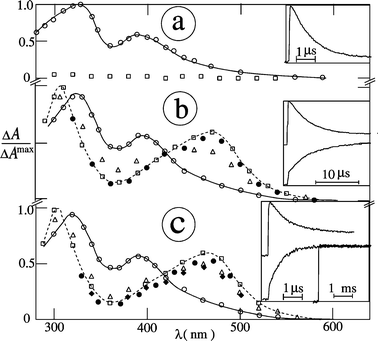
| | Fig. 6 Transient absorption spectra of vitamin K1 in argon-saturated acetonitrile in the presence of (a) DABCO (0.5 mM) and (b) TEA (2 mM) at 20 ns (○), 1 µs (Δ), 10 µs (□), 0.1 ms (●) and 0.1 s (♦) after the 248 nm pulse; insets: decay kinetics at 390 nm (above) and grow-in at 470 nm (below). | |
3.5 Continuous irradiation
UV irradiation of NQ in argon-saturated acetonitrile leads to substrate decomposition, as shown by isosbestic points at 200, 245, 272 and 342 nm, a decrease in absorption between 245 and 342 nm, and above 342 nm, and increases in the other ranges (Fig. 7, inset). The photodecomposition of NQ (Fig. 8, inset), MeNQ and Cl2NQ is more efficient in the presence of propan-2-ol (0.05–1 M). The spectral changes observed upon irradiation resemble those arising from intramolecular H-atom abstraction.42 The molar absorption coefficient of the H2Q form of Cl2NQ in acetonitrile–water (4 ∶ 1) is ε328
= 5.8 × 103 M−1 cm−1.9
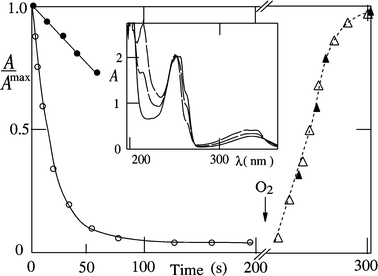 |
| | Fig. 7 Photoconversion of NQ; maximum change in A260vs. time in argon-saturated acetonitrile without (blocked symbols) and with (open symbols) added propan-2-ol (1 M) during irradiation (circles) and without irradiation after addition of oxygen (triangles). Inset: spectra in neat acetonitrile at 0 (full line), 100 and 300 s (broken line). | |
![Plots of Φdvs.
[propan-2-ol] (○) and [TEA] (□) for NQ in argon-saturated acetonitrile. Inset: spectra in the presence of propan-2-ol (1 M) at 0 (full line), 10 and 50 s (broken line).](/image/article/2004/PP/b306670n/b306670n-f8.gif) |
| | Fig. 8 Plots of Φdvs.
[propan-2-ol] (○) and [TEA] (□) for NQ in argon-saturated acetonitrile. Inset: spectra in the presence of propan-2-ol (1 M) at 0 (full line), 10 and 50 s (broken line). | |
The quantum yield of decomposition (Φd) was obtained from the decrease in absorbance at an appropriate wavelength, e.g. 260 nm. Examples are shown in Fig. 7 for NQ in acetonitrile and in presence of propan-2-ol. For NQ and MeNQ in neat methanol or propan-2-ol, Φd is significantly smaller than in mixtures of acetonitrile and alcohol (Table 3). This is due to competing steps which do not yield H2Q, e.g.reaction 8. Φd of NQ in acetonitrile increases with increasing propan-2-ol concentration; 50% of the maximum Φd value is reached at a half-concentration of ca. 0.02 M (Fig. 8). This is in line with the estimate of a half-concentration of 0.01 M obtained using a quenching rate constant of kD
= 3 × 107 M−1 s−1 and a limiting triplet lifetime of 3 µs (Tables 1 and 2). For NQ in acetonitrile, the presence of TEA (0.05–1 mM) also leads to substrate decomposition and the spectral changes for NQs are reminiscent of those observed with alcohols. The Φd values of NQ increase with increasing TEA concentration, approaching a maximum value of Φd
= 0.5 (Fig. 8). The low half-concentration of ca. 0.02 mM is consistent with the high kD value of 1.6 × 1010 M−1 s−1
(Table 2).
| Quinone |
Donor |
| None |
TEA |
Propan-2-ol |
| 0.2 mM |
2 mM |
0.1 M |
1 M |
13 M |
|
In argon-saturated acetonitrile, λirr
= 254 nm.
Values in parentheses are for oxygen-saturated solution.
Values in square brackets are for neat methanol.
|
| NQ |
0.08 (0.006)b |
0.2 |
0.45 |
0.6 |
0.7 (0.5) |
0.2 [0.2]c |
| MeNQ |
0.06 |
0.3 |
0.42 |
0.6 |
0.7 (0.5) |
0.3 [0.2] |
| Cl2NQ |
<0.002 |
0.2 |
0.3 |
0.08 |
0.12 |
0.2 |
| BrNQ |
0.005 |
|
0.3 |
0.06 |
0.1 |
|
3.6 Effects of oxygen
The above measurements were made in deoxygenated solutions, unless otherwise indicated. Triplet quenching of quinones by oxygen (reaction 3) yields singlet molecular oxygen, O2(1Δg).16,43 The quantum yield of formation (ΦΔ) of O2(1Δg) can be regarded as a minimum for Φisc. The values are substantial for NQs in several solvents (Table 1) and only low for vitamin K1, where ΦΔ in acetonitrile is below 0.02. The lower value for NQ in benzene, in contrast to Cl2NQ in benzene, is due to the short triplet lifetime (see section 3.1). The literature value for NQ in aqueous solution is ΦΔ
= 0.27.16
In the presence of TEA or alcohols, reactions 4 and 4′ could compete successfully with triplet quenching by oxygen, e.g. for amine concentrations above 0.1 M in air-saturated acetonitrile. One important reaction of oxygen is trapping of the semiquinone radical (equilibrium 12, forward reaction).
| | 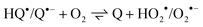 | (12) |
In fact, for duroquinone in aqueous solution, both rate constants for equilibrium 12 have been determined.37 When 9,10-dihydroxyanthracene is exposed to oxygen, complete recovery of AQ can occur,22i.e. photodecomposition of AQ is thermally reversible. This was also found for NQ in the presence of TEA (Fig. 8) and the other NQs, in contrast to 1,4-benzoquinone.23 Several possibilities may be considered; the most probable is reaction 13, based on the kinetics of oxidation of hydroquinones by molecular oxygen.44
| | | H2Q + O2
→ Q˙−
+ O2˙−
+ 2 H+ | (13) |
Owing to the thermal reversibility, the Φd values are sensitive to trace amounts of oxygen. Examples of the reversibility are shown in Fig. 7 for NQ in the absence and presence of propan-2-ol. After photoconversion (in the absence of oxygen) into 1,4-dihydroxynaphthalene, reaction 13
(after addition of oxygen) completely restores NQ. Note that the time of recovery depends on the conditions, e.g. the amount of water present.
3.7 Effects of NQ structure and medium
The rate constant for the reaction of NQ and MeNQ in acetonitrile towards H-atom abstraction from propan-2-ol is kD
= 3 × 107 M−1 s−1. The kD value is the same for AQ21,22 and lower for Cl2NQ and BrNQ (Table 2). The secondary transients are shown for NQ in acetonitrile–propan-2-ol [Fig. 1(b)]. On the other hand, the reactivity for electron transfer from TEA is close to the diffusion-controlled limit for most NQs in acetonitrile, but lower in benzene and for BrNQ in acetonitrile. No triplet with τT >20 ns was detected for NQ and MeNQ in THF, which is an example of an H-atom donating solvent of medium polarity. Intermolecular H-atom abstraction prevails also in cyclohexane, toluene and butyronitrile, whereas in carbon tetrachloride and dichloromethane, the reaction yielding the semiquinone radical is negligible at low NQ concentrations. In principle, the substantial ΦΔ
(Table 1) and Φisc values for NQ and MeNQ10 provide the possibility that up to two semiquinone radicals are formed per absorbed photon, thus giving Φd values between 0.5 and 1. Note that under otherwise the same conditions, the second reduction step, reaction 7 for alcohols or reaction 11 for TEA, which accounts for Φd >0.5, could kinetically be observed for AQs.22
In the presence of an amine in polar solvents, Q˙− and the radical cation of the amine are separate intermediates. The mechanism of photoreduction of NQs changes when the medium is less polar. In carbon tetrachloride, benzene and toluene, no free radical ions are present. An intermediate with a peak at 380 nm for the NQ–TEA–benzene system is formed by triplet quenching [Fig. 3(b)] and is attributed to the semiquinone radical. Reaction 4″ and electron back transfer, rather than formation of free radicals, accounts for the second-order decay occurring within a few µs.13,22
| | | 3Q*
+ NEt3
→
(Q˙−/Et3N˙+) | (4″) |
For NQ and MeNQ, the spectra of 3Q* and HQ˙/Q˙− are strongly overlapping, but the reactivity of a given solvent with the triplet state can be judged from the grow-in kinetics of the longer-lived semiquinone radical [Table 1, Fig. 1(b), (c) and 3(a), (b)]. In contrast, the λTT and λrad values are rather different for Cl2NQ (Fig. 2) and BrNQ [Fig. 3(c)] and triplet decay and radical grow-in are both accessible. Benzene with τT <1 µs is not an ‘inert’ medium for NQ and MeNQ as well as for AQ,22 in contrast to carbon tetrachloride. A specific triplet quenching reaction in benzene, known for NQ or MeNQ,2,3 also takes place for BrNQ, but is less effective for Cl2NQ.
3.8 Phosphorescence and transient absorption at low temperatures
The T–T absorption spectrum of Cl2NQ in MTHF at −180 °C is shown in Fig. 9(a). The transient absorption spectra of NQs in glassy media at low temperatures (Table 4) resemble those at room temperature. This is different for vitamin K1, where the species in fluid solution are not of triplet nature, see section 3.9. The relatively long lifetime in the case of vitamin K1 at −180 °C [Fig. 9(b)] indicates formation of the lowest triplet state, as supported by phosphorescence. The maximum triplet lifetime of NQs in low temperature glasses is in the 0.4–6 ms range.
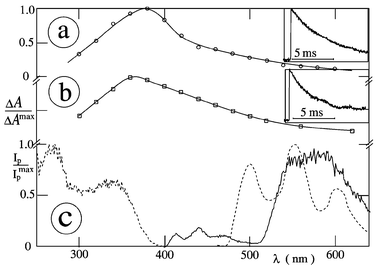 |
| | Fig. 9 Transient absorption spectra (in argon-saturated solution) of (a) Cl2NQ and (b) vitamin K1 in MTHF at −180 °C (0.1 µs, λexc
= 248 nm). (c) Phosphorescence emission (right) and excitation (left) spectra of vitamin K1 in air-saturated MCH (––) and NQ (⋯) at −196 °C (λexc
= 320 nm, λem
= 600 nm). | |
Table 4 Triplet lifetimes (τ) for the NQs and vitamin K1 using emission and absorption
| Quinone |
Solvent |
τ
p
a/ms |
λ
p
b/nm |
τ
T
c/ms |
λ
TT
d/nm |
|
Air-saturated at −196 °C.
Phosphorescence onset.
Argon-saturated at −180 °C, λexc
= 248 nm.
Absorption maximum.
|
| NQ |
MCH |
0.4 |
475 |
|
|
| |
MTHF |
0.4 |
480 |
0.4 |
370 |
| |
Ethanol |
0.4 |
480 |
|
|
| Cl2NQ |
MCH |
6 |
530 |
6 |
380 |
| |
MTHF |
6 |
540 |
6 |
380 |
| BrNQ |
MCH |
0.4 |
505 |
|
|
| |
MTHF |
0.3 |
510 |
0.4 |
400 |
| |
Ethanol |
0.4 |
520 |
|
|
| Vitamin K1 |
MCH |
3 |
520 |
3 |
360 |
| |
MTHF |
3 |
520 |
4 |
370 |
| |
Ethanol |
3 |
530 |
|
370 |
The emission and excitation spectra of vitamin K1 in MCH at −196 °C are shown in Fig. 9(c). The major band centered at 580 nm is due to phosphorescence; however, fluorescence, centered at 440 nm, also seems to play a minor role. The phosphorescence lifetime (τp) is 3 ms and the phosphorescence onset (λp), as a measure of the triplet energy, is at 520 nm. The spectra, as well as the λp and τp values, of a given quinone are similar in the different glasses (Table 4) and the phosphorescence results are in agreement with the literature.26,27
The photolysis results for vitamin K1 do not fit in with the above discussion for the four NQs examined, which deals with intermolecular transfer processes. Intramolecular H-atom transfer from the olefinic side chain to the quinone moiety has been documented for plastoquinones and vitamin K1.28–34 The photochemistry of vitamin K1 therefore differs completely from that of other quinones without side chains. Two 1,3-quinone methide (1,3-QM) diradicals have been suggested as observed intermediates, and a short-lived (non-observed) precursor (Q˙−–Phy˙+) can also be considered (reactions 14′ and 15, Scheme 1).34 The idea of a diradical existing prior to formation of the 1,2-QM tautomers (reaction 16) was first proposed by Porter and co-workers for plastoquinones.28,29 The two 1,3-QM diradicals of vitamin K1 in solution at room temperature are in equilibrium and can be quenched by oxygen (reaction 17).34
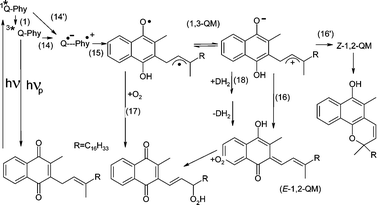 |
| | Scheme 1 | |
The first intermediate on the ns timescale of vitamin K1 in acetonitrile, in contrast to NQs without side chains, is the equilibrium of the 1,3-QM diradicals, which behave kinetically as single species.34 The major and minor maxima (λdir) of the 1,3-QM diradicals of vitamin K1 in acetonitrile are at 330 and 390 nm. The results are for λexc
= 248 nm; no marked difference was found with λexc
= 308 nm, except for faster decay of the 1,3-QM diradicals at higher laser intensities. The lifetime (τdir) under argon is up to 15 µs and the rate constant for quenching by oxygen (reaction 17, Scheme 1) is k17
= 5 × 108 M−1 s−1, in excellent agreement with the data from Wirz et al.34 The corresponding values in benzene are λdir = 330 and 390 nm, τdir = 3 µs, and k17 = 2.0 × 108 M−1 s−1.
For vitamin K1 in solution at room temperature, intersystem crossing seems to be too slow to compete with intramolecular proton transfer. It is therefore reasonable to propose reaction 14′, bypassing the triplet state, rather than reactions 1 and 14 (Scheme 1). On the other hand, phosphorescence was observed at −196 °C [Fig. 8(c)]. Radical intermediates of vitamin K1 in acetonitrile have been reported based on time-resolved resonance Raman spectroscopy.33 This identification may be doubtful and the postulated H-atom transfer from acetonitrile to the triplet state should be revised. The photochemical pathway is: excited singlet state → Q˙−–Phy˙+
→ diradical → 1,2-QM tautomers via reactions 14′, 15 and 16 (Scheme 1), respectively.
In the presence of appropriate additives, τdir of vitamin K1 in acetonitrile becomes shorter [Fig. 5(c) and 6]. Plots of 1/τdir increases linearly with the amine concentration; the rate constants for quenching are 2 × 109 M−1 s−1 for DABCO and 4 × 108 M−1 s−1 for TEA. The conversion of 1,3-QM into 1,2-QM in the presence of 10 mM acetic acid occurs with kq
= 2.5 × 108 M−1 s−1, corresponding to τdir
= 0.4 µs, which is in agreement with the literature value.34 The presence of water and alcohols (including tert-butanol) in the acetonitrile also substantially enhanced the conversion, but the plots curve upward. Extrapolation of the initial linear dependence with kq
≈ 3 × 106 M−1 s−1 to neat methanol would lead to τdir
= 12 ns, which is much longer than the directly measured value of 0.7 ns.34 The enhanced decay of the strong acid 1,3-QM in the presence of alcohols,34 amines and water is due to proton transfer through these additives, where the kq values refer to reaction 18 (Scheme 1).
4 Conclusion
The photoreduction of four NQs by propan-2-ol or TEA and related photoprocesses of vitamin K1 were analyzed. Photoinduced electron transfer from amines to the triplet state of the NQs was studied by transient UV-vis absorption spectroscopy and conductivity measurements. In acetonitrile, no equilibrium between the HQ˙ and Q˙− radicals is established. For Cl2NQ and BrNQ, the spectra of the semiquinone radicals and T–T absorptions are different, in contrast to the parent NQ. The rather long half-life of the TEA-derived semiquinone radicals excludes back electron transfer as a major reaction, although it does occur in the presence of DABCO. The photoconversion to 1,4-dihydroxynaphthalenes is efficient for NQ and MeNQ in the presence of either propan-2-ol or TEA, and still moderate for the other NQs. Addition of oxygen reverts the dihydroxynaphthalenes back to the respective NQs. The photoreactions of vitamin K1 involve two 1,3-quinone methide diradicals prior to formation of the E and Z tautomers of 1,2-QM. In the presence of alcohols and amines, proton transfer through these additives leads to faster conversion of 1,3-QM into 1,2-QM.
Acknowledgements
The author thanks Professor Wolfgang Lubitz for his support and Mrs Corinna Kesseler, Mr Leslie J. Currell and Lars Kalender for technical assistance.
References
- J.-R. Burie, A. Boussac, C. Boullais, G. Berger, T. Mattioli, C. Mioskowski, E. Nabedryk and J. Breton, FTIR spectroscopy of UV-generated quinone radicals: evidence for an intramolecular hydrogen atom transfer in ubiquinone, naphthoquinone, and plastoquinone, J. Phys. Chem., 1995, 99, 4059–4070 CrossRef CAS and references cited therein..
-
K. Maruyama and A. Osuka, Recent advances in the photochemistry of quinones, in The Chemistry of the Quinoide Compounds, ed. S. Patei and Z. Rappaport, Wiley, New York, 1988, pp. 759–878 and references cited therein Search PubMed.
-
K. Maruyama and Y. Kubo, CRC Handbook of Organic Photochemistry and Photobiology, ed. W. M. Horspool and P.-S. Song, CRC Press, Boca Raton, 1995, p. 748 Search PubMed.
- D. Schulte-Frohlinde and C. von Sonntag, Zur photoreduktion von chinonen in lösung, Z. Phys. Chem., Neue Folge, 1965, 45, 314–327.
- R. Scheerer and M. Grätzel, Laser photolysis studies of duroquinone triplet state electron transfer reactions, J. Am. Chem. Soc., 1977, 99, 865–871 CrossRef CAS.
- A. Pochon, P. P. Vaughan, D. Gan, P. Vath, N. V. Blough and D. E. Falvey, Photochemical oxidation of water by 2-methyl-1,4-benzoquinone: Evidence against the formation of free hydroxyl radical, J. Phys. Chem. A, 2002, 106, 2889–2894 CrossRef CAS.
- J. R. Wagner, J. E. van Lier and L. J. Johnston, Quinone sensitized electron transfer photooxidation of nucleic acids: chemistry of thymine and thymidine radical cations in aqueous solution, Photochem. Photobiol., 1990, 52, 333–343 CAS.
- T. Melvin, E. Bothe and D. Schulte-Frohlinde, The reaction of triplet 2-methyl-1,4- naphthoquinone (menadione) with DNA and polynucleotides, Photochem. Photobiol., 1996, 64, 769–776 CAS.
- M. Yamaji, M. Kurumi, H. Kimura and H. Shizuka, Hydration effects on the triplet exciplex between 2,3-dihalo-1,4-naphthoquinone and furan studied by steady-state and laser flash photolyses, Phys. Chem. Chem. Phys., 1999, 1, 1859–1865 RSC.
- I. Amada, M. Yamaji, M. Sase and H. Shizuka, Laser flash photolysis studies on hydrogen atom abstraction from phenol by triplet naphthoquinones in acetonitrile, J. Chem. Soc., Faraday Trans., 1995, 91, 2751–2759 RSC.
- I. Amada, M. Yamaji, S. Tsunoda and H. Shizuka, Laser photolysis studies of electron transfer between triplet naphthoquinones and amines, J. Photochem. Photobiol., A, 1996, 95, 27–32 CrossRef CAS.
- M. Mac and J. Wirz, Salt effects on the reactions of radical ion pairs formed by electron transfer quenching of triplet 2-methyl-1,4-naphthoquinone by amines. Optical flash photolysis and step-scan FTIR investigations, Photochem. Photobiol. Sci., 2002, 1, 24–29 RSC.
- X. Ci, R. S. da Silva, J. L. Goodman, D. E. Nicodem and D. G. Whitten, A reversible photoredox reaction: electron-transfer photoreduction of β-lapachone by triethylamine, J. Am. Chem. Soc., 1988, 110, 8548–8550 CrossRef CAS.
- X. Ci, R. S. da Silva, D. E. Nicodem and D. G. Whitten, Electron and hydrogen atom transfer mechanisms for the photoreduction of o-quinones. Visible light induced photoreactions of β-lapachone with amines, alcohols, and amino alcohols, J. Am. Chem. Soc., 1989, 111, 1337–1343 CrossRef CAS.
- H. Gan, X. Zhao and D. G. Whitten, Amine photoredox reactions: a photoinduced methylene shuttle initiated via two-electron oxidation of a tertiary amine by anthraquinone, J. Am. Chem. Soc., 1991, 113, 9409–9411 CrossRef CAS.
- A. E. Alegría, A. Ferrer, G. Santiago, E. Sepúlveda and W. Flores, Photochemistry of water- soluble quinones. Production of the hydroxyl radical, singlet oxygen and the superoxide ion, J. Photochem. Photobiol., A, 1999, 127, 57–65 CrossRef CAS.
- Y. Nishioku, K. Ohara, K. Mukai and S. Nagoaka, Time-resolved EPR investigation of the photo-initiated intramolecular antioxidant reaction of vitamin K – vitamin E linked molecule, J. Phys. Chem. B, 2001, 105, 5032–5038 CrossRef CAS.
- G. Balakrishnan, P. Mohandas and S. Umapathy, Time-resolved resonance Raman spectroscopic studies on the radical anions of menaquinone
and naphthoquinone, J. Phys. Chem., 1996, 100, 16
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 472–16478 CrossRef CAS.
472–16478 CrossRef CAS.
- F. Wilkinson, Transfer of triplet state energy and the chemistry of excited states, J. Phys. Chem., 1962, 66, 2569–2573 CAS.
- I. Loeff, S. Goldstein, A. Treinin and H. Linschitz, Reactions of formate ion with triplets of anthraquinone-2-sulfonate, 1,4-naphthoquinone, benzophenone-4-carboxylate, and benzophenone-4-sulfonate, J. Phys. Chem., 1991, 95, 4423–4430 CrossRef CAS.
- T. Kausche, J. Säuberlich, E. Trobitzsch, D. Beckert and K. P. Dinse, Photoreduction of 9,10-anthraquinone by triethylamine: a Fourier-transform EPR study, Chem. Phys., 1996, 208, 375–390 CrossRef CAS.
- H. Görner, Photoreduction of 9,10-anthraquinone derivatives: transient spectroscopy and effects of alcohols and amines on reactivity in solution, Photochem. Photobiol., 2003, 77, 171–179 CAS.
-
H. Görner, Photoreduction of p-benzoquinones: effects of alcohols and amines on the intermediates and reactivities in solution, Photochem. Photobiol., in press Search PubMed.
- M. Yamaji, T. Itoh and S. Tobita, Photochemical properties of the π,π* triplet state, anion and ketyl radicals of 5,12-naphthacenequinone in solution studied by laser flash photolysis: electron transfer and phenolic H-atom transfer, Photochem. Photobiol. Sci., 2002, 1, 869–876 RSC.
- J. F. Chen, X. W. Ge, G. S. Chu, Z. C. Zhang, M. W. Zhang, S. D. Yao and N. Y. Lin, Nanosecond laser photolysis studies of vitamin K3 in aqueous solution, Radiat. Phys. Chem., 1999, 55, 35–39 CrossRef CAS.
- T. Shimokage, T. Ikoma, K. Akiyama, S. Tero-Kubota, M. Yamaji and H. Shizuka, Substituent and matrix effects on the excited triplet states of 1,4-naphthoquinones, J. Phys. Chem., 1997, 101, 9253–9256 Search PubMed.
- J. J. Aaron and J. D. Winefordner, Phosphorescence study of excited triplet state properties of some K vitamins and their analytical usefulness, Anal. Chem., 1972, 44, 2122–2127 CrossRef CAS.
- G. Leary and G. Porter, The photochemistry of two phytyl quinones: α-tocopherylquinone and vitamin K1, J. Chem. Soc. A, 1970, 2273–2278 RSC.
- D. Creed, B. J. Hales and G. Porter, Photochemistry of plastoquinones, Proc. R. Soc. London, Ser. A, 1973, 334, 505–521 CAS.
- D. Creed, H. Werbin and E. T. Strom, Photochemistry of electron-transport quinones III. Characterization of in vitro, photoproducts of the photosynthetic plant quinone, plastoquinone-9, Tetrahedron, 1974, 30, 2037–2042 CrossRef.
-
R. M. Wilson, The trapping of biradicals and related photochemical intermediates, in Organic Photochemistry, ed. A. Padwa, Marcel Dekker, New York, 1985, vol. 7, pp. 339–466 Search PubMed.
- R. P. Gandhi, M. P. S. Ishar, R. Srivastava and R. Sarin, Nuclear magnetic resonance and UV spectral probe of photochemical steady states of vitamin K1, J. Photochem. Photobiol., A, 1994, 78, 153–160 CrossRef CAS.
- G. Balakrishnan and S. Umapathy, Photogenerated radical intermediates of vitamin K1: a time-resolved resonance Raman study, J. Mol. Struct., 1999, 475, 5–11 CrossRef CAS.
- M.-A. Hangarter, A. Hörmann, Y. Kamdzhilov and J. Wirz, Primary photoreactions of phylloquinone (vitamin K1) and plastoquinone-1 in solution, Photochem. Photobiol. Sci., 2003, 2, 524–535 RSC.
- K. B. Patel and R. L. Willson, Semiquinone free radicals and oxygen. Pulse radiolysis study of one electron transfer equilibria, J. Chem Soc., Faraday Trans. 1, 1973, 69, 814–825 RSC.
- P. S. Rao and E. Hayon, Ionization constants and spectral characteristics of some semiquinone radicals in aqueous solution, J. Phys. Chem., 1973, 77, 2274–2276 CrossRef CAS.
- D. Meisel and G. Czapski, One-electron transfer equilibria and redox potentials of radicals studied by pulse radiolysis, J. Phys. Chem., 1975, 79, 1503–1509 CrossRef CAS.
-
G. J. Fisher and H. E. Johns, Pyridine photohydrates, in Photochemistry and Photobiology of Nucleic Acids, ed. S. Y. Wang, Academic Press, New York, 1976, pp. 169–224 Search PubMed.
- H. Görner, Photochemical ring opening in nitrospiropyrans: triplet pathway and the role of singlet molecular oxygen, Chem. Phys. Lett., 1998, 282, 381–390 CrossRef CAS.
- E. Amouyal and R. Bensasson, Duroquinone triplet reduction, in cyclohexane, ethanol and water and by durohydroquinone, J. Chem. Soc., Faraday Trans. I, 1976, 72, 1274–1287 RSC.
- A. Demeter L. Biczók, T. Bérces, V. Wintgens, P. Valat and J. Kossanyi, Laser photolysis studies of transient processes in the photoreduction of naphthalamides by aliphatic amines, J. Phys. Chem., 1993, 97, 3217–3224 CrossRef CAS.
- Y. Chiang, A. J. Kresge, B. Hellrung, P. Schünemann and J. Wirz, Flash photolysis of 5-methyl-1,4-naphthoquinone in aqueous solution: kinetics and mechanism of photoenolization and of enol trapping, Helv. Chim. Acta, 1997, 80, 1106–1121 CrossRef CAS.
- I. Gutiérrez, S. G. Bertolotti, M. A. Biasutti, A. T. Soltermann and A. A. García, Can. J. Chem., 1997, 75, 423–428 CAS.
- V. Roginsky and T. Barsukova, Kinetics of oxidation of hydroquinones by molecular oxygen. Effect of superoxide dismutase, J. Chem. Soc., Perkin Trans. 2, 2000, 1575–1582 RSC.
|
| This journal is © The Royal Society of Chemistry and Owner Societies 2004 |
Click here to see how this site uses Cookies. View our privacy policy here.