Bimolecular processes on silica gel surfaces: energetic factors in determining electron-transfer rates
David R.
Worrall
*,
Iain
Kirkpatrick
and
Siân L.
Williams
Department of Chemistry, Loughborough University, Loughborough, Leicestershire, UK LE11 3TU. E-mail: d.r.worrall@lboro.ac.uk
First published on 18th September 2003
Abstract
Triplet state and radical cation formation is observed following laser excitation of anthracene, phenanthrene and naphthalene (and their derivatives) adsorbed on silica gel. Energy- and electron-transfer reactions of these compounds with co-adsorbed azulene have been studied using a time-resolved diffuse reflectance laser flash photolysis technique. Triplet energy transfer from the arene derivative to azulene and electron transfer from azulene to the arene radical cation have been investigated in order to distinguish between diffusional and energetic control in these systems. Energy and electron transfer can be studied independently due to differing absorption properties and energy dependencies of production of the triplet states and radical cations. Transient decay kinetics for both electron and energy transfer have been modelled using two different rate constant distributions: a log Gaussian and a symmetrical Lévy stable distribution. The latter model has also been demonstrated to be applicable to the decay of radical cations in the absence of an electron donor, which cannot be adequately described by the Gaussian model. Energy-transfer rates between the arene derivatives and azulene have been found to be close to the diffusion-controlled limit; however, in most cases, the rate of electron transfer is considerably lower. A correlation between the bimolecular rate constant and free energy of electron transfer has been found, indicating a Marcus inverted region. Compounds with bulky substituents show a further reduction in the rate of electron transfer, suggesting that an additional steric factor is involved in this process.
Introduction
The photochemistry of substrates on oxide surfaces has been the subject of a number of studies.1–26 In particular, silica gel is widely used in industry as a support for catalysts, and a greater understanding of reaction kinetics on these surfaces would be invaluable. Photoionisation of arenes on support surfaces such as silica gel1–8, γ-alumina9–14 and zeolites15–18 has previously been reported. The mechanism of radical cation decay (in the absence of electron donors) is usually via electron–radical recombination, regenerating the ground state. The radical cation decay kinetics are complex, with components on timescales ranging from microseconds to hours. We and others have shown previously that at low loadings of arenes on silica gel3,4 and γ-alumina,4,19 the initial radical cation decay can be described by a mechanism based on geminate recombination, but at later times, bulk electron diffusion becomes the dominant recombination pathway.3 The decay of excited states on silica gel is not a straightforward process, due to the heterogeneity of the surface, and rates rarely conform to a single exponential. Kinetic models often used to analyse decays on surfaces include the log Gaussian distribution of rate constants of Albery et al.,2,3,21,22,27–30 and a model involving the use of a fractal dimensional rate constant.3,9,10,23,24The rapid mobility of a number of molecules adsorbed on oxide surfaces has been reported2,3,22,31–34 and studied through either energy- or electron-transfer reactions. We have previously reported the quenching of the anthracene radical cation via electron-transfer reactions from various amines and azulene.3 We attributed the differences in the observed rates as due to either slow electron-donor diffusion or a Marcus-type inverted region.
Co-adsorption of azulene with the arenes on silica gel allows the study of both energy transfer (from arene to azulene) and electron transfer (from azulene to the radical cation) following laser excitation. We have recently shown that both energy- and electron-transfer reactions between anthracene and azulene are predominantly diffusion controlled,8 with little energetic involvement. This was interpreted as being a result of the azulene diffusing much faster than the anthracene, and the observed rate being governed by the azulene diffusion. Electron transfer within the encounter complex would then be fast relative to the diffusion rate.
In this paper, we present a study carried out using phenanthrene and several derivatives of naphthalene and anthracene as energy donors, with their radical cations as electron acceptors, and azulene as an energy acceptor or electron donor. We have found that the triplet–triplet energy-transfer reactions are essentially diffusion controlled, while for naphthalenes and some substituted anthracenes, the rate of electron transfer from azulene is considerably lower than the diffusion-controlled limit. We have applied a Marcus model35,36 to the electron-transfer data and shown an apparent Marcus inverted region. This has been demonstrated previously for back electron transfer in solution,37–42 rigid matrices,43 metal oxide systems44–46 and on silica gel.47
Experimental
Samples of anthracene adsorbed on silica gel were prepared as follows. Silica gel (Davisil grade 635, 60–100 mesh, 6 nm pore size, surface area 480 m2 g−1; Aldrich Chemical Co.) was dried at 125 °C under vacuum to 5 × 10−5 mbar for 8 h, and the vessel re-pressurised with dry nitrogen. Naphthalene (scintillation grade, 99+%; Aldrich), 9-chloroanthracene (97%; Aldrich), 2-methylanthracene (HPLC grade, ≥99%; Fluka Chemie GmbH), 2,3-dimethylnaphthalene (98%; Aldrich), 2,6-dimethylnaphthalene (99%; Aldrich), 2-methylnaphthalene (99.8%; Lancaster), 2-isopropylnaphthalene (95%; Frinton Laboratories, Inc.), or phenanthrene (zone refined, 99.5+%; Aldrich) were dissolved in n-hexane or acetonitrile (spectrophotometric grade; Aldrich) and a known weight of the resulting solution added to the dried silica gel. The mixture was allowed to equilibrate for a period of 1 h, with periodic agitation. The solvent was then removed under vacuum to a pressure of 5 × 10−5 mbar. The resulting sample was then sealed under vacuum into a cylindrical glass cuvette (22 mm diameter × 10 mm path length). Sample loadings were determined from the mass of solution added to the silica gel. Incorporation of azulene (99%; Aldrich) was achieved by its addition to the anthracene, phenanthrene or naphthalene solution prior to adsorption. Sample loadings of azulene were in the range 0.5–10 µmol g−1 (0.6–12.5 × 10−3 molecules nm−2), corresponding to homogenous 2D surface concentrations in the range 0.01 to 0.21% of a monolayer, while the loading of anthracenes, phenanthrene and naphthalenes was kept constant at 1.0 µmol g−1, corresponding to homogenous 2D surface concentrations of approximately 0.02% of a monolayer.Ground state diffuse reflectance spectra were recorded using a Perkin-Elmer Lambda Bio 40 spectrophotometer equipped with a Spectralon integrating sphere, using barium sulfate as a reference. Fluorescence spectra were recorded using a Spex FluoroMAX spectrofluorimeter using front surface geometry.
In the flash photolysis experiments, excitation of the samples was with the third (355 nm, 110 mJ pulse−1) or fourth (266 nm, 50 mJ pulse−1) harmonic of a Continuum Surelite I Nd:YAG laser. The pulse energy was attenuated using solutions of sodium nitrite in water to ensure that transient reflectance changes were kept below 10%, where the change in reflectance is directly proportional to the concentration of transient species.48–50 Diffusely reflected analysing light from a 275 W Oriel xenon arc lamp was collected and focussed onto the entrance slit of an Applied Photophysics ƒ/3.4 grating monochromator and detected with a Hamamatsu R928 side-on photomultiplier tube. Signal capture was by a LeCroy LT364 Waverunner digitising oscilloscope, interfaced to an IBM-compatible PC.
Transient decay data were analysed using a previously described model3,6,8,51 which assumes a log Gaussian distribution of rate constants, according to eqn. 1,27
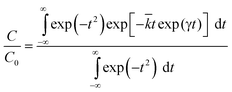 | (1) |
![[k with combining macron]](https://www.rsc.org/images/entities/i_char_006b_0304.gif) is the mean rate constant. At low sample loadings and small reflectance changes (less than 10%), C and C0 can be replaced by ΔR and ΔR0, the reflectance changes at times t and t
= 0 (relative to the laser pulse), respectively. The method for fitting transient decays using this model has been described previously in ref. 8. Transients have also been analysed assuming a Lévy stable distribution of rate constants51 by calculating the relative amplitudes of an evenly spaced set of exponentials, described by the eqn. 2,52
is the mean rate constant. At low sample loadings and small reflectance changes (less than 10%), C and C0 can be replaced by ΔR and ΔR0, the reflectance changes at times t and t
= 0 (relative to the laser pulse), respectively. The method for fitting transient decays using this model has been described previously in ref. 8. Transients have also been analysed assuming a Lévy stable distribution of rate constants51 by calculating the relative amplitudes of an evenly spaced set of exponentials, described by the eqn. 2,52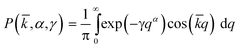 | (2) |
Measurement of oxidation potentials was carried out using an EcoChemie Autolab PGSTAT30 system in a conventional three-electrode glass cell. A 0.5 mm Pt wire served as the working electrode with Ag wire as the reference and Pt wire as the counter electrode. The electrolyte was tetrabutylammonium hexafluorophosphate (≥ 99.0%, puriss, electrochemical grade; Fluka) and was prepared as a melt under vacuum to remove impurities. The vessel was then purged with argon and anhydrous acetonitrile (99.8%; Aldrich) added to give an electrolyte concentration of 0.1 mol dm−3. To this solution, the compound to be measured was added (2 × 10−3 mol dm−3) with 1 × 10−3 mol dm−3 ferrocene (98%; Aldrich) as an internal standard. Scan rates of 1 and 10 V s−1 were employed and the signals compensated for IR drop as described in ref. 53.
Results and discussion
Transient absorption data
Transient difference spectra were recorded using either 266 or 355 nm excitation. Assignment of absorption bands due to triplet states or radical cations was based on spectral positions compared to previously characterised anthracene species,2 from solution data,54 on the basis of the observed kinetics and from energy dependence studies (vide infra). Wavelengths of maximum absorbance for triplet states and radical cations are given in Table 1. An example of a transient difference spectrum is shown in Fig. 1 for 2-isopropylnaphthalene. It can be seen that the triplet state (410 nm) shows a significantly faster rate of decay than the radical cation (670 nm). The changing shape of the 410 nm band is due to the underlying radical cation absorption at 380 nm.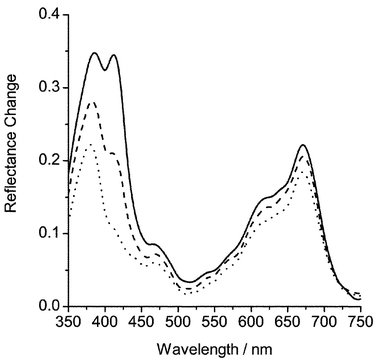 | ||
Fig. 1 Transient difference spectra at 0.4 (––), 2.0 (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ), and 8.0 ms (⋯) after the laser pulse for 2-isopropylnaphthalene (1.0 µmol g−1) adsorbed on silica gel. ), and 8.0 ms (⋯) after the laser pulse for 2-isopropylnaphthalene (1.0 µmol g−1) adsorbed on silica gel. | ||
| Compound | λ max(triplet)/nm | λ max(radical cation)/nm |
|---|---|---|
| Anthracene | 420 | 400, 710 |
| 2-Methylanthracene | 420 | 400, 710 |
| 9-Chloroanthracene | 420 | 385, 690 |
| 9-Anthracenecarboxylic acid | 420 | 380, 715 |
| Azulene | 360 | |
| Naphthalene | 410 | 380, 680 |
| 2-Methylnaphthalene | 410 | 380, 670 |
| 2-Isopropylnaphthalene | 410 | 380, 670 |
| 2,3-Dimethylnaphthalene | 420 | 395, 680 |
| 2,6-Dimethylnaphthalene | 420 | 390, 650 |
| Phenanthrene | 480 | 400, 700 |
The laser energy dependence of the naphthalene and phenanthrene triplet and radical absorption bands was determined by attenuation of the laser pulse using sodium nitrite filters. Fig. 2 shows plots of reflectance change versus laser fluence for naphthalene. The triplet-state data were corrected for the absorption due to the radical cation at the triplet wavelength. The correction factor was determined by examination of the reflectance spectra for naphthalene and phenanthrene, recorded before and after laser excitation. Fig. 3 shows the remission function spectrum of naphthalene following excitation with a single 50 mJ pulse of 266 nm laser radiation. The spectrum was corrected for absorption by silica gel. The ratio of remission function due to the radical cation absorption at 410 and 680 nm is 1 ∶ 3.2. This information has been used to subtract the radical cation absorbance from the triplet-state data at 410 nm shown in Fig. 2. Initially, it can be seen that for the triplet state, there is a linear correlation between laser energy and reflectance change with a negative deviation at higher laser fluence, typical of monophotonic absorption and saturation of the triplet state. The decrease in triplet-state absorption at higher laser intensities is due to competition between photo-ionisation and intersystem crossing of the first excited singlet state. For the radical cation, there is no linear relationship over the entire laser energy range. The upward curvature indicates that a multiphotonic process55 is responsible for the formation of the radical cation. Generation of naphthalene radical cations is known to be a multiphotonic process56 and these observations agree with previously published findings on the anthracene radical cation.2
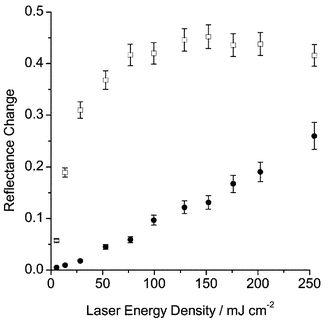 | ||
| Fig. 2 Plots of reflectance change versus laser fluence for the triplet state (□) and radical cation (●) of naphthalene (1.0 µmol g−1) adsorbed on silica gel. | ||
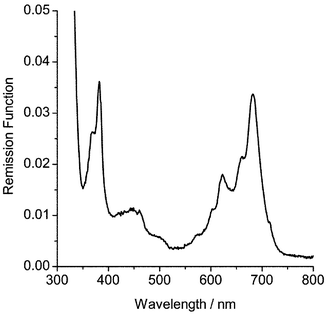 | ||
| Fig. 3 Remission function versus wavelength for naphthalene (1.0 µmol g−1) adsorbed on silica gel after a single pulse (50 mJ) of 266 nm laser radiation. | ||
Transient absorption data: kinetics of the unquenched radical cation
The kinetics of radical cation decay in the absence of molecular quenchers has been examined by application of the Lévy model. The radical cations of anthracene, naphthalene, phenanthrene and azulene were generated via direct excitation at either 355 or 266 nm. The azulene radical cation was found to have an absorption maximum at 360 nm, which compares well with the value of 368 nm reported in butyl chloride.57 In the absence of a quencher, the radical cations exhibit decays that are significantly slower than in the presence of a suitable electron donor. The decay kinetics were measured after excitation of the samples with a single pulse of 266 nm laser radiation at approximately 50 mJ by following the subsequent reflectance change using conventional reflectance spectroscopy. Fig. 4 shows the decay of the naphthalene radical cation monitored at 680 nm plotted on a logarithmic scale. Both Albery and Lévy models were applied to these data. It can be seen that the Albery model fails for the initial fast component of the trace and that the Lévy model provides a reasonable fit over the entire timescale of the decay. The Lévy model generates a symmetrical distribution which lends much greater probability to the tails of the distribution (in this case, to fast and slow rate constants) than does a conventional Gaussian distribution, as assumed in the model of Albery et al.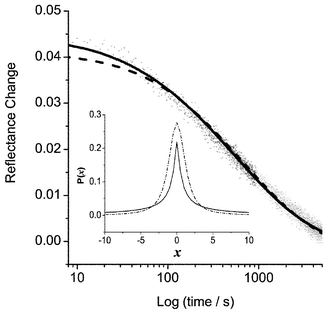 | ||
Fig. 4 Fitted kinetic decay of naphthalene radical cation at 680 nm using Lévy (––) and Albery () models; naphthalene concentration 1.0 µmol g−1. The inset shows Lévy distributions calculated from parameters in Table 3 for naphthalene on silica gel; laser fluences 130 (—) and 250 mJ cm−2
(![[dash dot, graph caption]](https://www.rsc.org/images/entities/char_e090.gif) ). ). | ||
In a previous publication, we demonstrated a technique for obtaining a fit to the decay corresponding to a global minimum on a reduced χ2 surface. This process has been adapted for the Lévy model, as shown in Fig. 5. We have found the Lévy χ2 space to be more complex than previously shown for the Albery model, but with a clearly defined global minimum.
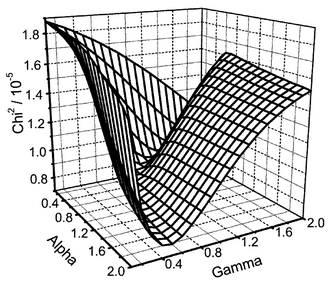 | ||
| Fig. 5 Three-dimensional plot showing reduced χ2versusα and the distribution width (γ) for anthracene on silica gel (1.0 µmol g−1). | ||
The Lévy distribution gives an increasing weighting to the tails of the distribution as the distribution width decreases, and can be described as a random walk consisting of long jumps followed by several short walks. It has been shown that this type of foraging behaviour is more efficient at seeking out randomly distributed targets than a simple random walk.58,59 Furthermore, it is suggested that this type of behaviour may be more characteristic of unquenched radical cation decay where, at longer times, bulk diffusion of electrons is the dominant factor6 and, hence, the observed kinetics are those which most efficiently seek out the randomly distributed radical cations. We have found that the decays of radical cation species in the absence of an electron donor are dependent on the excitation energy, becoming faster at high laser intensities, which supports the case for bulk diffusion of electrons as the rate-controlling factor. In the Lévy model, we suggest that the series of small motions is analogous to a trapped electron and the intermediate long jumps represent escape from potential wells with subsequent long range diffusion. There may also be a sample morphology factor here, i.e. pores versus surface diffusion. Table 2 shows extracted fitting parameters for the unquenched decay of the radical cations of the naphthalene, anthracene and phenanthrene parent compounds. In all three cases, it can be seen that is reduced as laser energy decreases. The distribution width also decreases as laser energy decreases, as shown in Table 2 and the inset in Fig. 4. In the Lévy model, as in the Gaussian model, the distribution is symmetrical around some mean rate constant. If we have components corresponding to very slow decays, then they must have counterparts on the opposing side of the distribution, and applying large weightings to fast decay components is not supported by the data collected. The origin of the change in distribution width therefore probably lies in the symmetry of the distribution, since, at high laser fluences, there will be a greater proportion of fast decay constants, which, due to the form of the distributions fitted, necessarily results in additional fast and slow components being introduced into the model. We are currently working to modify the Lévy stable distribution with a degree of asymmetry in order to overcome this deficiency in the model. The aim is to produce a model which not only provides a consistent way of evaluating the mean rate constant, but is also closer to allowing a physical interpretation of the data. In some applications of the Lévy flight model, a degree of asymmetry has been introduced into the distribution, for example, in the case of foraging animals, where there is a finite maximum time they can spend between feeding.58,60
| Compound (wavelength, energy) | Model |
/10−4 s−1 |
γ | α |
|---|---|---|---|---|
| Anthracene (355 nm, 280 mJ cm−2) | Albery | 2.0 | 1.1 | |
| Lévy | 2.1 | 0.4 | 1.4 | |
| Anthracene (355 nm, 560 mJ cm−2) | Albery | 2.2 | 1.4 | |
| Lévy | 2.3 | 0.5 | 1.3 | |
| Naphthalene (266 nm, 130 mJ cm−2) | Albery | 8.0 | 1.6 | |
| Lévy | 8.2 | 0.5 | 1.7 | |
| Naphthalene (266 nm, 250 mJ cm−2) | Albery | 11 | 2.0 | |
| Lévy | 14 | 1.2 | 1.1 | |
| Phenanthrene (266 nm, 130 mJ cm−2) | Albery | 3.5 | 1.1 | |
| Lévy | 3.6 | 0.3 | 1.6 | |
| Phenanthrene (266 nm, 250 mJ cm−2) | Albery | 4.3 | 1.4 | |
| Lévy | 4.5 | 1.3 | 0.4 |
Transient absorption data: kinetics of energy and electron transfer
For studies of the triplet state, the laser energy was attenuated such that multiphoton ionisation was essentially eliminated (Fig. 2), thereby preventing any radical cation production and allowing the exclusive examination of the triplet-state kinetics. In the study of the radical cation kinetics, the triplet state has no absorption at the radical cation absorption maximum; therefore, the radical cation may be exclusively monitored at this wavelength. Transient decay data have been analysed using the dispersive kinetic model of Albery et al. Transient decay data for both the triplet and radical cation were obtained in the presence of co-adsorbed azulene, over a range of azulene concentrations. A detailed account of our analysis methodology has been published previously.3,6,8 From this modelling, we are able to extract a mean value for the decay rate constant,, and by application of eqn. 3, we can obtain bimolecular quenching rate constants:| k = k0 + kq′[azulene] | (3) |
is the observed rate constant for the triplet state or radical cation decay in the presence of azulene and 0 is the unquenched rate constant in the absence of azulene. This may be measured for triplet-state decay, and is treated as zero within experimental error for the radical cation due to the long lifetime of this species.8 Where significant ground-state association between the arene and donor is observed, as has been shown previously in the case of the anthracene–azulene system, the free concentration of azulene was calculated from the sample loading and the radical cation quenching data, as described in a previous publication.8
Using this methodology, quenching constants have been measured for two bimolecular processes involving the arene derivatives and azulene. These are triplet–triplet energy transfer between the excited hydrocarbon derivative and azulene, and electron transfer from azulene to the radical cations of the arene derivatives, both of these processes being exoergic, as shown by the data in Table 3.
| Compound | Energy-transfer quenching constant/109 g mol−1 s−1 | Electron-transfer quenching constant/109 g mol−1 s−1 | Solution energy-transfer quenching constant/109 dm3 mol−1 s−1 | E T/kJ mol−1 | E P/V vs. Ag/AgCl |
|---|---|---|---|---|---|
| Anthracene | 9.5 ± 0.3 | 10.0 ± 0.4 | 12.0 ± 0.5 | 17861 | 1.26 |
| 2-Methylanthracene | 7.6 ± 0.3 | 5.9 ± 0.3 | 9.6 ± 0.1 | 1.19 | |
| 9-Chloroanthracene | 10.5 ± 0.1 | 7.6 ± 0.1 | 5.6 ± 0.1 | 1.38 | |
| 9-Anthracenecarboxylic acid | 9.8 ± 0.3 | 9.4 ± 0.5 | 7.8 ± 0.2 | 17762 | 1.49 |
| Azulene | 16363 | 1.04 | |||
| Naphthalene | 22.1 ± 0.1 | 4.3 ± 0.4 | 25564 | 1.64 | |
| 2-Methylnaphthalene | 14.9 ± 0.1 | 5.3 ± 0.3 | 1.61 | ||
| 2-Isopropylnaphthalene | 2.8 ± 0.3 | 0.7 ± 0.1 | 1.63 | ||
| 2,3-Dimethylnaphthalene | 7.4 ± 0.8 | 0.9 ± 0.1 | 1.54 | ||
| 2,6-Dimethylnaphthalene | 7.4 ± 0.2 | 0.7 ± 0.1 | 1.53 | ||
| Phenanthrene | 4.8 ± 0.9 | 3.0 ± 0.3 | 25765 | 1.69 |
A typical quenching plot is shown in Fig. 6 for the naphthalene–azulene system. Quenching constants are reported in Table 3, along with measured oxidation potentials. Fig. 7 shows a cyclic voltammogram for 9-chloroanthracene. A reversible signal was obtained for the methyl, chloro and carboxylic acid derivatives of anthracene; however, all other compounds employed in this study exhibited an irreversible oxidation step, consistent with short radical cation lifetimes in solution.66,67 The reported potentials in Table 3 were measured at the oxidation peak only in order to provide a suitable comparison of values and are quoted relative to the ferrocene internal standard versus Ag/AgCl.
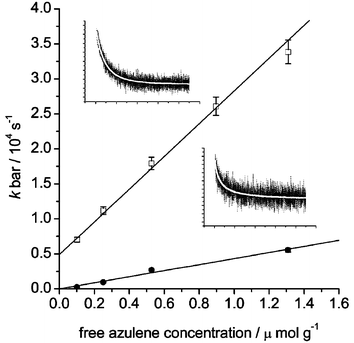 | ||
| Fig. 6 Plots of average rate constants () for triplet–triplet energy transfer (□) and electron transfer (●) for naphthalene co-adsorbed with azulene on silica gel versus free azulene concentration. The insets show transient decay of naphthalene (1.0 µmol g−1
) on silica gel co-adsorbed with azulene (0.55 µmol g−1) at 410 (top, 100 µs per division) and 680 nm (bottom, 1 ms per division). | ||
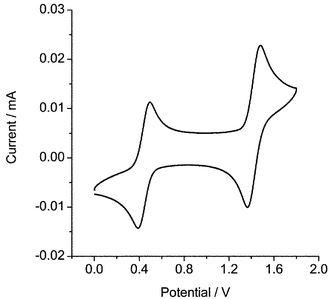 | ||
| Fig. 7 Plot of current versus potential for 9-chloroanthracene and ferrocene in anhydrous acetonitrile with tetrabutylammmonium hexafluorophosphate as electrolyte and employing a scan rate of 10 V s−1. | ||
We have previously reported a diffusion-controlled regime for the quenching of anthracene and the carboxylic acid derivative by azulene for both energy and electron transfer. It can be seen from Table 3 that this is clearly not always the case, and we have found rates of electron transfer to be lower than those of energy transfer for all of the additional compounds investigated in this study, sometimes by as much as an order of magnitude.
Firstly, considering the rates of energy transfer, these are faster for naphthalene and 2-methylnaphthalene as compared to anthracene and 2-methylanthracene. When product formation is faster than dissociation of the encounter complex, the observed rate of reaction (electron or energy transfer) is governed by the rate of diffusion. The diffusion-controlled rate constant is then expected to be proportional to the sum of the diffusion coefficients for the two reacting species. We have shown previously that the data obtained for anthracene and its derivatives are consistent with diffusion of anthracene being slow relative to that of azulene, and, hence, diffusion of the latter dominates the observed rate. Hashimoto et al.68 have developed a model for molecular diffusivity in zeolites and showed that, in their system, the diffusion coefficient of anthracene was 14 times less than that of azulene. Our observations are consistent with a similar situation prevailing in the silica gel system. Naphthalene, however, is a structural isomer of azulene, and so these may both be expected to diffuse at similar rates; therefore, the observed diffusion rate may be expected to be twice that observed for the anthracene–azulene couple. Hence, the triplet–triplet energy-transfer rate observed for naphthalene–azulene is entirely consistent with diffusion control dominating in this system.8
Substitution of anthracene in the 9-position has little effect on the rate of energy transfer. However, substitution of anthracene or naphthalene in the 2-position results in a marked decrease in the rate of energy transfer (Table 3). This cannot be attributed exclusively to a decrease in donor mobility, since this does not play a role in the anthracene–azulene system. Rather, it is suggestive of steric hindrance to the energy-transfer process, introducing an entropic factor, since the efficiency of energy transfer may not be the same for every angle of approach. This is supported by the fact that changing the substituent from methyl to isopropyl in the 2-position of naphthalene results in a decrease in both the energy- and electron-transfer rates. Multiple substitution in the naphthalene system also decreases the energy-transfer rate to below the diffusion-controlled limit. An explanation based on entropic factors may also explain the result obtained for phenanthrene. Such an effect would be expected to be more marked in a system which is less than three-dimensional, such as those studied here, than in solution.
From the measured oxidation potentials, it was possible to calculate the standard free energy of the electron-transfer reaction between the aromatic hydrocarbon radical cation and azulene using eqn. 4, where n = 1 for a one-electron oxidation and F is the Faraday constant.
| ΔGet = nF(Eoxp − Eredp) | (4) |
Here, we use the measured oxidation potentials, since the reduction potentials of the radical cations are unavailable for most of the compounds studied.
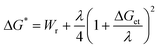 | (5) |
Fig. 8 shows a plot of ln(ket) versus free energy change, ΔGet. The free energy of activation, ΔG*, may be calculated using the Marcus equation (eqn. 5), where Wr is the electrostatic work required to bring the reactants together and λ is the reorganisation energy. This model has been used to fit the data in Fig. 8 according to the Arrhenius expression shown in eqn. 6, where Z is the collision frequency.
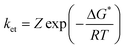 | (6) |
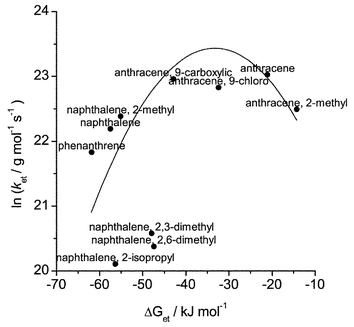 | ||
| Fig. 8 Plot of lnketversus ΔGet for anthracenes, naphthalenes and phenanthrene co-adsorbed with azulene on silica gel. The solid line shows fit according to the Marcus equation with Z = 1.65 × 1010 s−1, λ = 33.3 kJ mol−1 and Wr = 0.095 J mol−1. | ||
Phenanthrene is expected to have similar diffusion characteristics to anthracene, i.e. any diffusional character is expected to be dominated by the azulene diffusion rate. However, its redox properties are more akin to those of naphthalene (Table 3). Fig. 8 shows that the rate of the reaction between the phenanthrene radical cation and azulene is entirely consistent with that of energetic (reaction) control in this system.
Clearly the electron-transfer reactions between 2-isopropyl-, 2,3-dimethyl- and 2,6-dimethylnaphthalene derivatives and azulene do not lie on the Marcus curve. The rationale for this is the same as for the energy-transfer reactions, in that the substituents introduce additional steric factors which reduce the rate of electron transfer within the encounter complex. This is consistent with the energy-transfer data (vide supra), where energy-transfer rate constants are substantially lower than can be explained purely on the basis of diffusional arguments.
Conclusions
Energy- and electron-transfer kinetics have been measured for a range of anthracene and naphthalene derivatives and phenanthrene co-adsorbed with azulene on silica gel, and bimolecular rate constants obtained. Previously, we have demonstrated that both energy and electron transfer between anthracene or 9-anthracenecarboxylic acid and azulene proceed at the diffusion-controlled rate. For the other compounds in this study, we have found that the rate of electron transfer is consistently lower than the rate of energy transfer. The redox potentials of the compounds were measured and the free energy change for the electron transfer calculated. The data have been fitted using a Marcus model, revealing a Marcus-type inverted region. The dimethyl-substituted naphthalenes and the 2-isopropyl derivative do not lie on the Marcus curve and exhibit greatly reduced rates of energy transfer relative to the parent compounds. This reduction in both energy- and electron-transfer rates indicates that a steric factor may also be influencing them in these systems.The decay kinetics of the radical cation species, in the absence of a quencher, have been modelled successfully using a symmetrical Lévy stable distribution, which takes into account components decaying over relatively long times by applying a greater weighting to these components as compared with a Gaussian distribution. We suggest that a modification of this distribution with a degree of asymmetry may yield more information about the physical nature of diffusion on the silica surface.
Acknowledgement
The authors would like to thank Dr Frank Marken of Loughborough University for his assistance with the measurements of oxidation potentials.References
- J. K. Thomas and E. H. Ellison, Various aspects of the constraints imposed on the photochemistry of systems in porous silica, Adv. Colloid Interface Sci., 2001, 89, 195–238 CrossRef.
- F. Wilkinson, D. R. Worrall and S. L. Williams, Primary photochemical processes of anthracene adsorbed on silica-gel, J. Phys. Chem., 1995, 99(17), 6689–6696 CrossRef CAS.
- D. R. Worrall, S. L. Williams and F. Wilkinson, Electron transfer reactions of anthracene adsorbed on silica gel, J. Phys. Chem. B, 1997, 101(24), 4709–4716 CrossRef CAS.
- D. Oelkrug, S. Reich, F. Wilkinson and P. A. Leicester, Photoionization on insulator surfaces – diffuse reflectance laser flash-photolysis of distyrylbenzenes adsorbed on silica and alumina, J. Phys. Chem., 1991, 95(1), 269–274 CrossRef CAS.
- I. K. Lednev, N. Mathivanan and L. J. Johnston, Photochemistry of stilbene adsorbed on silica-gel and NaX zeolite – a diffuse-reflectance laser flash-photolysis study, J. Phys. Chem., 1994, 98(44), 11
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 444–11451 CAS.
444–11451 CAS. - D. R. Worrall, S. L. Williams and F. Wilkinson, Electron transfer on insulator surfaces: exciplex emission and the role of electron diffusion in determining radical deactivation rates, J. Phys. Chem. A, 1998, 102(28), 5484–5490 CrossRef CAS.
- D. R. Worrall, S. L. Williams, F. Wilkinson, J. E. Crossley, H. Bouas-Laurent and J. P. Desvergne, Spectroscopy and ion-electron recombination kinetics of radical ions of anthracenes and substituted anilines on silica gel, J. Phys. Chem. B, 1999, 103(43), 9255–9261 CrossRef CAS.
- D. R. Worrall, I. Kirkpatrick and S. L. Williams, Controlling factors in electron and energy transfer reactions on silica gel surfaces, Photochem. Photobiol. Sci., 2002, 1(11), 896–901 RSC.
- S. Pankasem and J. K. Thomas, Pyrene, pyrene derivatives, and 1,1′-binaphthyl as luminescent probes for photophysical studies of alumina surfaces, J. Phys. Chem., 1991, 95(19), 7385–7393 CrossRef CAS.
- S. Pankasem and J. K. Thomas, Reflectance spectroscopic studies of the cation radical and the triplet of pyrene on alumina, J. Phys. Chem., 1991, 95(18), 6990–6996 CrossRef CAS.
- G. Beck and J. K. Thomas, Photochemical processes on aluminum-oxide – a pulsed laser study of photoinduced reactions, Chem. Phys. Lett., 1983, 94(6), 553–557 CrossRef CAS.
- D. Oelkrug, G. Krabichler, W. Honnen, F. Wilkinson and C. J. Willsher, Photophysical behavior of diphenylpolyenes adsorbed on alumina by diffuse reflectance laser flash-photolysis, J. Phys. Chem., 1988, 92(12), 3589–3594 CrossRef CAS.
- Y. Mao and J. K. Thomas, Photochemical-reactions of biphenyl and its chlorinated derivatives on the surfaces of gamma-alumina and silica-alumina, J. Chem. Soc., Faraday Trans., 1992, 88(20), 3079–3086 RSC.
- Y. Mao and J. K. Thomas, Photochemical-reactions of pyrene on surfaces of gamma-alumina and silica-alumina, Langmuir, 1992, 8(10), 2501–2508 CrossRef CAS.
- K. K. Iu and J. K. Thomas, Photophysical properties of pyrene in zeolites .2. Effects of coadsorbed water, Langmuir, 1990, 6(2), 471–478 CrossRef CAS.
- L. J. Johnston, J. C. Scaiano, J. L. Shi, W. Siebrand and F. Zerbetto, Observation and modeling of the recombination kinetics of diphenylmethyl radicals in the cavities of Na-X zeolite, J. Phys. Chem., 1991, 95(24), 10018–10024 CAS.
- S. Hashimoto, N. Fukazawa, H. Fukumura and H. Masuhara, Observation and Characterization of Excimer Emission from Anthracene Included in Nax Zeolite, Chem. Phys. Lett., 1994, 219(5–6), 445–451 CrossRef CAS.
- X. S. Liu, K. K. Iu, J. K. Thomas, H. Y. He and J. Klinowski, Spectroscopic studies of protonated aromatic species and radical cations in H+-zeolites, J. Am. Chem. Soc., 1994, 116(26), 11811–11818 CAS.
- F. Wilkinson and R. Beer, Diffuse reflectance laser photolysis of adsorbed molecules, in Photochemical Processes in Organised Molecular Systems, ed. K. Honda, Elsevier Science, Amsterdam, 1991, pp. 377–398 Search PubMed.
- G. H. Zhang, J. K. Thomas, A. Eremenko, T. Kikteva and F. Wilkinson, Photoinduced charge-transfer reaction between pyrene and N,N′-dimethylaniline on silica gel surfaces, J. Phys. Chem. B, 1997, 101(42), 8569–8577 CrossRef CAS.
- D. Oelkrug, M. Gregor and S. Reich, Translational mobility in the adsorbed state – bimolecular deactivation kinetics of laser-excited azaaromatics on microporous silica, Photochem. Photobiol., 1991, 54(4), 539–546 CAS.
- D. Oelkrug, S. Uhl, F. Wilkinson and C. J. Willsher, Bonding and mobility of acridine on some metal-oxide surfaces as studied by delayed fluorescence and transient absorption-spectroscopy, J. Phys. Chem., 1989, 93(11), 4551–4556 CrossRef CAS.
- R. Krasnansky, K. Koike and J. K. Thomas, Gaussian approximation to the unique heterogeneous Langmuir-Hinshelwood type fluorescence quenching at the silica-gel gas-solid interface – pyrene and 9,10-diphenylanthracene singlet quenching by oxygen, J. Phys. Chem., 1990, 94(11), 4521–4528 CrossRef CAS.
- S. A. Ruetten and J. K. Thomas, Fluorescence and triplet quantum yields of arenes on surfaces, J. Phys. Chem. B, 1998, 102(3), 598–606 CrossRef CAS.
- R. Dabestani, J. Higgin, D. Stephenson, I. N. Ivanov and M. E. Sigman, Photophysical and photochemical processes of 2-methyl, 2-ethyl, and 2-tert-butylanthracenes on silica gel. A substituent effect study, J. Phys. Chem. B, 2000, 104(44), 10235–10241 CrossRef CAS.
- H. Garcia and H. D. Roth, Generation and reactions of organic radical cations in zeolites, Chem. Rev., 2002, 102(11), 3947–4007 CrossRef CAS.
- W. J. Albery, P. N. Bartlett, C. P. Wilde and J. R. Darwent, A general-model for dispersed kinetics in heterogeneous systems, J. Am. Chem. Soc., 1985, 107(7), 1854–1858 CrossRef CAS.
- D. Avnir, The geometry factor in photoprocesses on irregular (fractal) surfaces .1. Static considerations, J. Am. Chem. Soc., 1987, 109(10), 2931–2938 CrossRef CAS.
- T. Eliaskohav, M. Sheintuch and D. Avnir, Steady-state diffusion and reactions in catalytic fractal porous-media, Chem. Eng. Sci., 1991, 46(11), 2787–2798 CrossRef CAS.
- J. Klafter, A. Blumen and G. Zumofen, Energy-transfer in fractal structures, J. Lumin., 1984, 31–32, 627–633 CrossRef CAS.
- M. A. T. Marro and J. K. Thomas, Movement of small molecules on a silica surface – a photophysical and photochemical study, J. Photochem. Photobiol., A, 1993, 72(3), 251–259 CrossRef CAS.
- D. W. Bjarneson and N. O. Petersen, Direct diffusion measurements of naphthacene on silica as a function of silanol density, J. Am. Chem. Soc., 1990, 112(3), 988–992 CrossRef CAS.
- R. K. Bauer, R. Borenstein, P. Demayo, K. Okada, M. Rafalska, W. R. Ware and K. C. Wu, Surface photochemistry – translational motion of organic-molecules adsorbed on silica-gel and its consequences, J. Am. Chem. Soc., 1982, 104(17), 4635–4644 CrossRef CAS.
- N. J. Turro, M. B. Zimmt, I. R. Gould and W. Mahler, Triplet energy-transfer as a probe of surface-diffusion rates – a time-resolved diffuse reflectance transient absorption-spectroscopy study, J. Am. Chem. Soc., 1985, 107(20), 5826–5827 CrossRef CAS.
- R. A. Marcus, The theory of oxidation-reduction reactions involving electron transfer, J. Chem. Phys., 1956, 24, 966–978 CrossRef CAS.
- R. A. Marcus, Chemical and electrochemical electron-transfer theory, Annu. Rev. Phys. Chem., 1964, 15, 155–196 CrossRef CAS.
- H. Imahori, K. Tamaki, D. M. Guldi, C. P. Luo, M. Fujitsuka, O. Ito, Y. Sakata and S. Fukuzumi, Modulating charge separation and charge recombination dynamics in porphyrin fullerene linked dyads and triads: Marcus-normal versus inverted region, J. Am. Chem. Soc., 2001, 123(11), 2607–2617 CrossRef CAS.
- D. M. Guldi and K. D. Asmus, Electron transfer from C-76 (C-2v′) and C-78 (D-2) to radical cations of various arenes: evidence for the Marcus inverted region, J. Am. Chem. Soc., 1997, 119(24), 5744–5745 CrossRef CAS.
- I. R. Gould, J. E. Moser, D. Ege and S. Farid, Effect of molecular dimension on the rate of return electron-transfer within photoproduced geminate radical ion-pairs, J. Am. Chem. Soc., 1988, 110(6), 1991–1993 CrossRef CAS.
- I. R. Gould, R. Moody and S. Farid, Electron-transfer reactions in the Marcus inverted region – differences in solvation and electronic coupling between excited charge-transfer complexes and geminate radical ion-pairs, J. Am. Chem. Soc., 1988, 110(21), 7242–7244 CrossRef CAS.
- I. R. Gould, D. Ege, S. L. Mattes and S. Farid, Return electron-transfer within geminate radical ion-pairs – observation of the Marcus inverted region, J. Am. Chem. Soc., 1987, 109(12), 3794–3796 CrossRef CAS.
- A. Polese, S. Mondini, A. Bianco, C. Toniolo, G. Scorrano, D. M. Guldi and M. Maggini, Solvent-dependent intramolecular electron transfer in a peptide-linked Ru(bpy)32+-C-60 dyad, J. Am. Chem. Soc., 1999, 121(14), 3446–3452 CrossRef CAS.
- J. R. Miller, J. V. Beitz and R. K. Huddleston, Effect of free-energy on rates of electron-transfer between molecules, J. Am. Chem. Soc., 1984, 106(18), 5057–5068 CrossRef CAS.
- G. Ramakrishna and H. N. Ghosh, Emission from the charge transfer state of xanthene dye-sensitized TiO2 nanoparticles: a new approach to determining back electron transfer rate and verifying the Marcus inverted regime, J. Phys. Chem. B, 2001, 105(29), 7000–7008 CrossRef CAS.
- X. J. Dang and J. T. Hupp, Interfacial charge-transfer pathways: evidence for Marcus-type inverted electron transfer in metal oxide semiconductor/inorganic dye systems, J. Am. Chem. Soc., 1999, 121(36), 8399–8400 CrossRef CAS.
- H. Lu, J. N. Prieskorn and J. T. Hupp, Fast interfacial electron-transfer – evidence for inverted region kinetic-behavior, J. Am. Chem. Soc., 1993, 115(11), 4927–4928 CrossRef CAS.
- P. P. Levin, S. M. B. Costa and L. F. V. Ferreira, Kinetics of intersystem electron-transfer within triplet radical-ion pairs on silica studied by diffuse-reflectance laser flash-photolysis – bell-shaped energy-gap dependence on the surface, J. Phys. Chem., 1995, 99(4), 1267–1275 CrossRef CAS.
- R. W. Kessler, G. Krabichler, S. Uhl, D. Oelkrug, W. P. Hagan, J. Hyslop and F. Wilkinson, Transient decay following pulse excitation of diffuse-scattering samples, Opt. Acta, 1983, 30(8), 1099–1111 Search PubMed.
- H. K. A. Kan and T. P. Lin, Calculation of reflectance of a light diffuser with non-uniform absorption, J. Opt. Soc. Am., 1970, 60, 1252–1256 Search PubMed.
- D. Oelkrug, W. Honnen, F. Wilkinson and C. J. Willsher, Modeling of transient production and decay following laser excitation of opaque materials, J. Chem. Soc., Faraday Trans. 2, 1987, 83, 2081–2095 RSC.
- P. Levy, Theorie de l'Addition des Variables Aleatoires, Gauthier-Villars, Paris, 1937 Search PubMed.
- R. N. Mantegna, Levy walks and enhanced diffusion in Milan stock exchange, Physica A, 1979, 179, 232–242 CrossRef.
- F. Marken, A. Neudeck and A. M. Bond, Effects due to capacity and resistance, in Electroanalytical Methods, ed. F. Scholz, Springer-Verlag, Berlin/Heidelberg, 2002, pp. 66–68 Search PubMed.
- R. Bensasson and E. J. Land, Triplet-triplet extinction coefficients via energy transfer, Trans. Faraday Soc., 1971, 67, 1904–1915 RSC.
- U. Lachish, A. Schafferman and G. Stein, Intensity dependence in laser flash photolysis experiments: hydrated electron formation from ferrocyanide, tyrosine and tryptophan, J. Chem. Phys., 1976, 64(10), 4205–4211 CrossRef CAS.
- S. Steenken, C. J. Warren and B. C. Gilbert, Generation of radical-cations from naphthalene and some derivatives, both by photoionization and reaction with SO4− – formation and reactions studied by laser flash-photolysis, J. Chem. Soc., Perkin Trans. 2, 1990, 335–342 RSC.
- T. Shida, Electronic Absorption Spectra of Radical Ions, Physical Sciences Data Vol. 34, Elsevier Science, Amsterdam, 1988 Search PubMed.
- G. M. Viswanathan, V. Afanasyev, S. V. Buldyrev, E. J. Murphy, P. A. Prince and H. E. Stanley, Levy flight search patterns of wandering albatrosses, Nature, 1996, 381(6581), 413–415 CrossRef CAS.
- M. G. E. d. Luz, S. V. Buldyrev, S. Havlin, E. P. Raposo, H. E. Stanley and G. M. Viswanathan, Improvements in the statistical approach to random Levy flight searches, Physica A, 2001, 295, 89–92 CrossRef.
- G. M. Viswanathan, V. Afanasyev, S. V. Buldyrev, S. Havlin, M. G. E. d. Luz, E. P. Raposo and H. E. Stanley, Levy flight search patterns of biological organisms, Physica A, 2001, 295, 85–88 CrossRef.
- D. N. Dempster, T. Morrow and M. F. Quinn, Exctinction coefficients for triplet-triplet absorption in ethanol solutions of anthracene, naphthalene, 2,5-diphenyloxazole, 7-diethylamino-4-methyl coumarin and 4-methyl-7-amino-carbostyril, J. Photochem., 1974, 2, 329–341 CrossRef CAS.
- S. Hirayama, Effect of substituent on the behaviour of the excited singlet and triplet states in carbonyl derivatives of anthracene of the type 9-X.CO.A, J. Chem. Soc., Faraday Trans. 1, 1982, 78, 2411–2421 RSC.
- W. G. Herkstroeter, Triplet energies of azulene, beta-carotene and ferrocene, J. Am. Chem. Soc., 1975, 97, 4161–4167 CrossRef CAS.
- G. N. Lewis and M. Kasha, Phosphorescence and the triplet state, J. Am. Chem. Soc., 1944, 66, 439–440.
- H. J. Goerner, Phosphorescence of trans-stilbene, stilbene derivatives and stilbene-like molecules at 77K, J. Phys. Chem., 1989, 93, 1826–1832 CrossRef.
- E. Vauthey, E. Haselbach and P. Suppan, Photoionization of naphthalene and anthracene in acetonitrile, Helv. Chim. Acta, 1987, 70(2), 347–353 CAS.
- A. D. Liu, M. C. Sauer, D. M. Loffredo and A. D. Trifunac, Transient absorption-spectra of aromatic radical cations in hydrocarbon solutions, J. Photochem. Photobiol., A, 1992, 67(2), 197–208 CrossRef CAS.
- S. Hashimoto, M. Hagiri and A. V. Barzykin, Triplet-triplet energy transfer as a tool for probing molecular diffusivity within zeolites, J. Phys. Chem. B, 2002, 106, 844–852 CrossRef CAS.
- N. A. Vandantzig, H. S. Shou, J. C. Alfano, N. C. C. Yang and D. H. Levy, Photoinduced charge-transfer in bichromophoric molecules in the gas-phase, J. Chem. Phys., 1994, 100(10), 7068–7078 CrossRef CAS.
- M. Castella, J. Prochorow and A. Tramer, Vanderwaals and charge-transfer states of molecular-complexes formed in supersonic jets, J. Chem. Phys., 1984, 81(5), 2511–2512 CrossRef CAS.
- J. Jortner, M. Bixon, B. Wegewijs, J. W. Verhoeven and R. P. H. Rettschnick, Long-range, photoinduced charge separation in solvent-free donor bridge acceptor molecules, Chem. Phys. Lett., 1993, 205(4–5), 451–455 CrossRef CAS.
- J. A. Syage, P. M. Felker and A. H. Zewail, Picosecond excitation and selective intramolecular rates in supersonic molecular-beams. 3. Photochemistry and rates of a charge-transfer reaction, J. Chem. Phys., 1984, 81(5), 2233–2256 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and Owner Societies 2004 |