Reactive species formed on proteins exposed to singlet oxygen
Michael J.
Davies
*
The Heart Research Institute, 145 Missenden Road, Camperdown, Sydney, NSW 2050, Australia. E-mail: m.davies@hri.org.au
First published on 3rd September 2003
Abstract
Singlet oxygen (1O2) is believed to be generated in biological systems by a range of endogenous processes (e.g. enzymatic and chemical reactions) and exogenous stimuli (e.g. UV or visible light in the presence of a sensitiser). Kinetic data is consistent with proteins being a major target for 1O2, with damage occurring preferentially at Trp, His, Tyr, Met, and Cys side-chains. Reaction with each of these residues gives rise to further reactive species. In the case of Trp and Tyr, initial poorly characterised endoperoxides are believed to undergo ring-opening reactions to give hydroperoxides, which can be reduced to the corresponding alcohols; other products arising from radical intermediates can also be generated, particularly in the presence of UV light and metal ions. With His side-chains, poorly characterised peroxides are also formed. Reaction with Met and Cys has been proposed to occur via zwitterionic peroxy intermediates. Peroxides are also generated on isolated proteins, and protein within intact cells, via1O2-mediated reactions. The peroxides formed on Trp, Tyr, and His peptides, as well as on proteins, have been shown to induce damage to other targets, with molecular oxidation of thiol residues an important reaction. This can result in the inactivation of cellular enzymes and the oxidation of other biological targets. Protein cross-linking and aggregation can also be induced by reactive species formed on photo-oxidised proteins, though the nature of the species that participate in such reactions is poorly understood. These secondary reactions, and particularly those involving hydroperoxides, may play a key role in the induction of secondary damage (bystander effects) in systems subject to photo-oxidation.
 Michael J. Davies Michael J. Davies | Michael Davies was awarded his BSc (1981) and DPhil (1984) degrees from the University of York, UK. After post-doctoral studies at Brunel University in London, he was appointed as a lecturer at the University of York in 1987. He moved to Australia in late 1995 to take up a position as a Group Leader at the Heart Research Institute, Sydney, on an Australian Research Council QE2 Fellowship. He was promoted to a Senior/Professorial Fellowship in 2000, and the Deputy Directorship of the Institute in 2001. In 2003, he was awarded the Silver Medal of the International EPR Society for his work in biology/medicine. His primary research interests lie in mechanisms of protein oxidation, the biological consequences of such reactions, and the development of methods to quantify protein damage in vivo. |
Formation of singlet oxygen in biological systems
Singlet oxygen (molecular oxygen in its 1Δg state; 1O2) has been postulated to be an important intermediate in a range of biological systems. Evidence has been presented for the formation of 1O2 by a range of peroxidase enzymes (e.g. myeloperoxidase,1,2 lactoperoxidase,3 horseradish peroxidase,4 and chloroperoxidase5) and during lipoxygenase-catalysed reactions.6 A number of stimulated cell types (e.g. eosinophils7 and macrophages8) have been proposed to generate this oxidant, possibly as a result of the presence of the above enzymes, though other cells, such as neutrophils, do not appear to do so.1,91O2 is also formed in high yield in reactions involving ozone10 and during the reaction of H2O2 with HOCl,11 sodium molybdate,12 or peroxynitrite.13 A number of endoperoxide compounds have been synthesised which undergo thermal decomposition at room or elevated temperatures, thereby allowing the specific generation of defined yields of 1O2.141O2 has also been shown to be formed during the bimolecular termination reactions of some peroxyl radicals;15 this species may therefore be an important intermediate in lipid (and other) peroxidation reactions.Despite the wide range of non-photochemical reactions outlined above that generate 1O2, the most common source of 1O2 in biological systems is probably exposure to UV light, or visible light in the presence of an appropriate sensitiser. Cells contain a wide range of chromophores that can act as sensitising agents for 1O2 formation, and the quantum yields for the formation of this species by a wide range of agents have been collated.16 Whilst in some cases it is well established what the major 1O2-generating pathway is (e.g. within cells that have been pre-loaded with an exogenous sensitiser before light exposure), in many cases, there is considerable doubt as to what the principal sensitising agent is, with a large number of endogenous materials able to act in this capacity.
Direct generation of 1O2via the absorption of light by proteins that do not contain bound or associated co-factors is rather limited. Most proteins only absorb UV light relatively weakly in the region of the spectrum (λ > 290 nm) that is incident at the Earth's surface; shorter solar wavelengths are efficiently removed by molecules in the atmosphere. The major chromophores that absorb at wavelengths longer than 280 nm are tryptophan (Trp), tyrosine (Tyr), and cystine. Histidine (His), cysteine (Cys), and phenylalanine (Phe) only absorb UV significantly at shorter wavelengths (Phe ≤ ca. 270 nm, His ≤ ca. 240 nm, Cys ≤ ca. 250 nm). None of these side-chains has a high quantum yield for 1O2 generation.16 All other major amino acid side-chains and backbone peptide bonds do not absorb significantly at wavelengths above ca. 230 nm. A number of oxidation products of protein side-chains (particularly of Trp) are, however, reasonably efficient 1O2 sensitisers.16 Direct 1O2 formation by proteins is therefore inefficient and probably only contributes in a minor manner to the overall flux of 1O2 that might be generated within a biological system, despite the widespread abundance of proteins. The majority of 1O2 to which proteins may be exposed is therefore likely to arise via the photochemistry of other chromophores. As energy transfer to yield 1O2 usually competes with electron transfer, most photosensitisers generate both 1O2 and radicals.17 However, as energy transfer to O2 is often more rapid (k ≈ 1–3 × 109 dm3 mol−1 s−1) than electron transfer (e.g. to give O2−˙, typically k ≤ 1 × 107 dm3 mol−1 s−1), the former process often predominates, though there is obviously an enormous variation depending on the sensitiser, the excitation wavelength, and the reaction conditions.17
Proteins as a target for 1O2
Rate constants have been determined for the reaction of 1O2 with a wide range of biological targets, including DNA, RNA, proteins, lipids, and sterols.18 However, due to the abundance of proteins within most biological systems (they comprise ca. 70% of the dry mass of most cells19) and the high rate constants for reaction of 1O2 with some protein side-chains,18,20 it would be expected that these will be the major targets for 1O2. This conclusion should, however, be treated with caution, as this will obviously be affected by the potential localisation and accumulation of sensitisers within particular cellular compartments (e.g. within membranes) and the limited diffusion radius of 1O2.211O2 can interact with protein side-chains by both physical quenching and chemical reaction. The former results in energy transfer and de-excitation of the singlet state, but no chemical change in the energy acceptor. For proteins, only Trp appears to give rise to significant physical quenching,22 with k ≈ 2–7 × 107 dm3 mol−1 s−1; chemical reaction occurs with k ≈ 3 × 107 dm3 mol−1 s−1.22 The relative contributions of these processes with Trp are affected by a number of factors, including the environment of the residue and the presence and position of substituents on the indole ring.23,24The rate constants for chemical reaction of 1O2 with the side-chains of other free amino acids vary dramatically (Table 1). Of the common amino acids, only Trp, His, Tyr, Met and Cys, undergo rapid chemical reaction at physiological pH values. All other side-chains react with k < 0.7 × 107 dm3 mol−1 s−1 at neutral pH.18 At higher pH values, Arg and Lys react rapidly via their unprotonated forms.26
Formation and characterisation of peroxides on amino acid side-chains
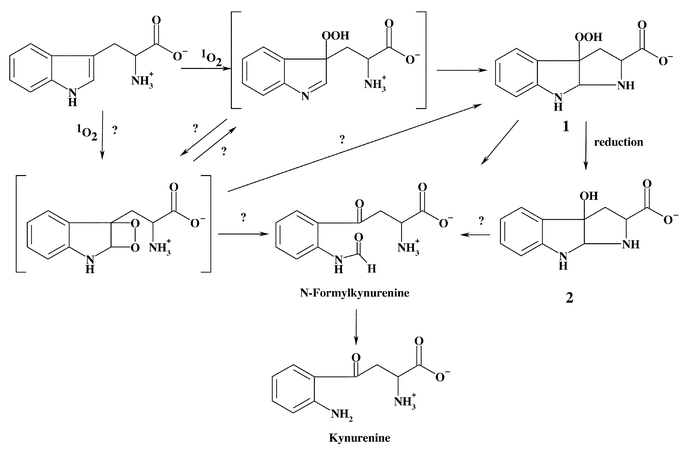
In the case of Trp, Tyr, and His, there is good evidence for the formation of multiple endo- and hydroperoxides as a result of the initial addition of 1O2 to each of these residues. In the case of Trp and Tyr, the nature of these peroxides is reasonably well established. The intermediates formed with His, Met, and Cys are less well defined.In the case of free Trp, reaction with 1O2 yields 3α-hydroperoxypyrroloindole (1) as an isolable product. The precursor species to this compound are not well defined. It has been proposed that the initial species may be a dioxetane across the C2–C3 double bond, though there is an absence of direct evidence for this, and there are other potential intermediates, including a hydroperoxide at C3 (Scheme 1).27–32 The chemistry of peptide-bound Trp appears to be similar to that of free Trp, which is somewhat surprising given that the nitrogen lone pair involved in the ring-closing reaction will be much less available in peptides, due to the presence of the amide bond. It would therefore be expected that the formation of species analogous to 1 would occur at a much slower rate (cf. data for Tyr below). Under such conditions other reaction pathways may become important, and the precursor peroxides may be isolable or detectable; this has yet to be investigated in detail.
 | ||
| Scheme 1 | ||
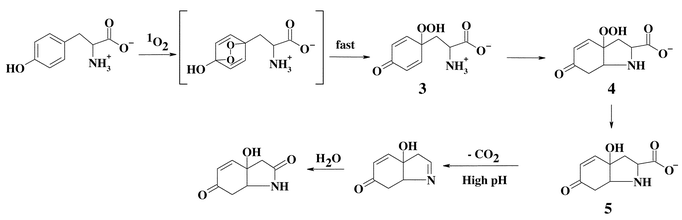
Detailed studies have been carried out with free Tyr and peptide-bound Tyr. In the case of the free amino acid, evidence has been presented for the generation of a number of peroxidic species. These are outlined in Scheme 2. The proposed initial intermediate—the endoperoxide—has not been detected. Ring opening of this species is proposed to give a hydroperoxide at C1 (3) and direct evidence has been obtained for the presence of this compound.33 Evidence has also been presented for the formation of related species with other phenols.34,35 With free Tyr, rapid ring closure involving the unprotonated amine function then ensues, resulting in the generation of the cyclised product 3a-hydroperoxy-6-oxo-2,3,3a,6,7,7a-hexahydro-1H-indol-2-carboxylic acid (4) which has been extensively characterised.33,36–39
 | ||
| Scheme 2 | ||
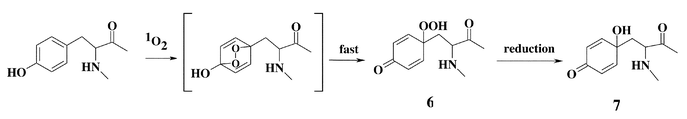
With Tyr residues in peptides, peroxide formation is also observed (Fig. 1), with the initial species believed to be an endoperoxide analogous to that outlined in Scheme 2. Subsequent ring opening of this species has been postulated to give rise to a hydroperoxide group at C1; this species has been fully characterised by NMR.33 The yield of this material, when generated using Rose Bengal and light of λ > 345 nm, is enhanced when the reaction is carried out in D2O rather than H2O, and decreased when the reaction is carried out in the presence of the 1O2 scavenger azide; these data are consistent with the peroxides being generated via1O2-mediated reactions. The ring-closure reaction with the free amino group that occurs with the free amino acid is markedly slowed in peptides where the Tyr residue is not present at the N-terminus, due to the delocalisation of the nitrogen lone pair into the amide bond, making this atom a much less efficient nucleophile in the ring-closing Michael reaction. With compounds such as 4-hydroxyphenylpropionic acid (HPPA), where such reactions cannot occur, the C1 peroxide is also the major species detected. The major peroxidic product detected with peptide-bound Tyr residues at room temperature is therefore the C1 hydroperoxide (6) formed via the reactions in Scheme 3.33
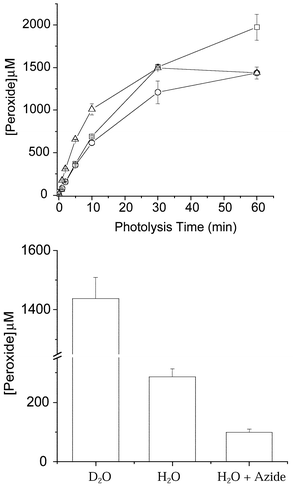 | ||
| Fig. 1 (a) Formation of peroxides on 4-hydroxyphenylpropionic acid (□), N-acetyl Tyr (○), and Gly–Tyr–Gly (△) (all 2.5 mM) in D2O, pH 7, as a result of illumination with light (from a 250 W tungsten-filament bulb passing through a 345 nm cut-off filter) in the presence of O2 and the photosensitiser Rose Bengal (10 µM) at 4 °C with increasing illumination time. (b) Comparison of Gly–Tyr–Gly peroxide yields in D2O, H2O, and H2O with added azide (5 mM) after 60 min illumination in the presence of Rose Bengal (10 µM) and O2 at 4 °C. Peroxide concentrations were measured using the FOX assay with H2O2 standards.40 Data are the means (±SD) of triplicate determinations from a single experiment representative of several, as the absolute peroxide yields varied between experiments due to slight differences in oxygenation rates. | ||
 | ||
| Scheme 3 | ||
Oxidation of free His by 1O2 is believed to occur via the initial formation of one or more endoperoxides, such as 8, and the consumption of a single molecule of O2 per mole of His (Scheme 4).41 Endoperoxides analogous to 8 have been detected directly by NMR with highly substituted derivatives in organic solvents at low temperatures.42 As the temperature was raised, these species were observed to decompose to yield further intermediates, which could not be characterised, and, subsequently, a range of stable products. Recent studies from the author's laboratory have shown that a number of peroxides are present in reaction mixtures where the illumination is carried out at 4 °C (Fig. 2).43 These peroxides have lifetimes of minutes to hours at room temperature, which is inconsistent with them being endoperoxides. It is possible that they are hydroperoxides formed by ring opening of an initial endoperoxide, by analogy with the behaviour observed for Trp and Tyr. A number of these species are sufficiently stable to allow separation from the parent compound by HPLC, and the characterisation of these species is currently being attempted. NMR analyses of these reaction systems are consistent with this hypothesis.44
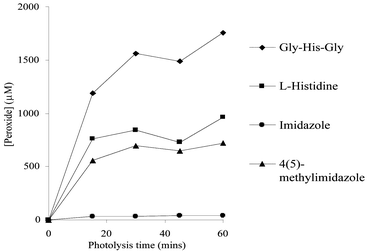 | ||
| Fig. 2 Formation of peroxides (in µM) on imidazole, histidine, and imidazole and histidine derivatives in H2O, pH 7, as a result of illumination with light (from a 250 W tungsten-filament bulb passing through a 345 nm cut-off filter) in the presence of O2 and the photosensitiser Rose Bengal (10 µM) at 4 °C with increasing illumination time. Peroxide concentrations were measured using the FOX assay with H2O2 standards.40 Data are the means (±SD) of triplicate determinations from a single experiment representative of several, as the absolute peroxide yields varied between experiments due to slight differences in oxygenation rates. | ||
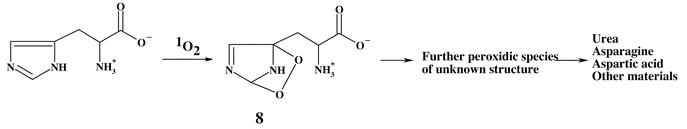 | ||
| Scheme 4 | ||
The formation of reactive species during the oxidation of free Met has been examined and evidence has been presented for the formation of a zwitterionic peroxy species (Scheme 5).45 This species undergoes subsequent reaction with a second molecule of Met to give two moles of the sulfoxide45 (reviewed in ref. 17 and 46). The stoichiometry of O2 to Met consumption is complex and varies with pH, with this being 1 ∶ 1 above pH 7 and 1 ∶ 2 below pH 5, consistent with other reactions also playing a role. A number of other intermediates, including a nitrogen–sulfur cyclic intermediate, have been reported (reviewed in ref. 17 and 46); subsequent hydrolysis of this species gives the sulfoxide.45 This cyclic intermediate is only likely to be of significance with free Met, due to the involvement of the free amino group. The situation with Met residues in peptides and proteins remain to be fully elucidated, though it has been shown that the sulfoxide is a major product.47,48
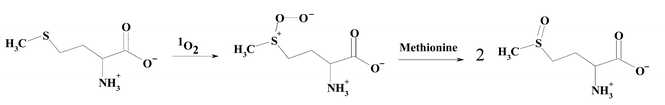 | ||
| Scheme 5 | ||
As with Met, the products arising from reaction of free Cys are well established—the disulfide is formed, though not in a quantitative manner—but the mechanism that gives rise to this product is less well established.17,25,46 In addition to the disulfide, oxyacids such as cysteic acid (RSO3H) are formed under some conditions.17,46 The quantitative yield of the products arising from Cys residues in proteins is not well established and it would be expected that the ratio of disulfide to oxyacids will vary with the protein structure, due to steric or electronic barriers to dimer formation. This remains to be explored in detail. Qualitative studies have shown that oxidation of free cystine occurs via a zwitterionic peroxy intermediate, which reacts further with another molecule of cystine to give two molecules of RSS(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)R.46,49 The effect of incorporation of cystine residues into a protein on such reactions, where steric and electronic effects are likely to inhibit subsequent processes, remains to be determined.
O)R.46,49 The effect of incorporation of cystine residues into a protein on such reactions, where steric and electronic effects are likely to inhibit subsequent processes, remains to be determined.
Peroxide formation on isolated proteins
Peroxidic species have been shown to be formed on exposure of a wide range of proteins to 1O2, irrespective of their composition.39,50 Examination of a range of proteins with different amino acid compositions has shown that peroxides can be generated on Trp, Tyr, or His residues within proteins. Thus, peroxides have been detected on melittin, which contains only Trp of the major reactive residues (i.e. no Tyr, His, Met, or Cys), on trypsin inhibitor, which contains only Tyr of the major reactive side-chains, as well as other proteins that lack one or more of these reactive residues.51 These peroxides are formed with a wide range of different sensitisers, though the overall yield varies markedly.43,52,53 The peroxide concentration can be enhanced by use of D2O as the solvent, consistent with the mediation of 1O2 (Fig. 3).50 Protein peroxide formation has also been observed with other, non-photolytic, sources of 1O2 (e.g. with a molybdate–H2O2 system; Fig. 4).50 The exact location of these peroxides within protein structures, and the nature of these species, have not been established as yet.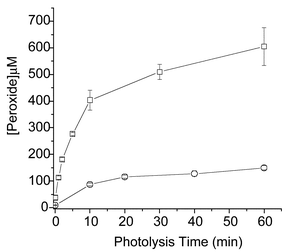 | ||
| Fig. 3 Formation of peroxides on bovine serum albumin (75 µM) as a result of illumination with light (from a 250 W tungsten-filament bulb passing through a 345 nm cut-off filter) in the presence of O2 and the photosensitiser Rose Bengal (10 µM) at 4 °C in H2O (○) and D2O (□). Peroxide concentrations were measured using the FOX assay with H2O2 standards.40 Data are the means (±SD) of triplicate determinations from a single experiment representative of several, as the absolute peroxide yields varied between experiments due to slight differences in oxygenation rates. | ||
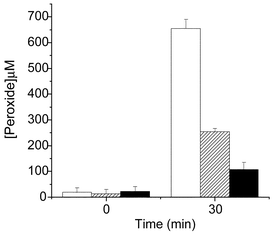 | ||
| Fig. 4 Formation of peroxides on bovine serum albumin (75 µM) treated with a MoO42−–H2O2 system. Reactions were performed at 22 °C with 12.5 mM MoO42− in D2O (open bars), H2O (stippled bars), or H2O in the presence of azide (5 mM) (black bars) at pH 7.4. The reactions were initiated by the addition of H2O2, with further aliquots of H2O2 added after 1 and 10 min; 1.62 mM H2O2 was added in total. The reactions were allowed to proceed for 30 min, then unreacted H2O2 was removed by gel filtration using a PD-10 column, and protein peroxide concentrations were measured using the FOX assay with H2O2 standards.40 Data are expressed as means (n = 6, from 2 separate experiments) ± SD. | ||
Peroxide formation on proteins within cells
Peroxides can be generated, in high yield, on bulk cellular proteins extracted from cells and subsequently exposed to visible light in the presence of a sensitiser (e.g. Rose Bengal54). Recent studies have shown that peroxides are also generated on proteins within intact viable cells on exposure to visible light in the presence of a range of sensitisers.43,54 The yield of these peroxides depends markedly on the sensitiser employed, even when identical initial sensitiser concentrations and illumination times are employed.43 This is believed to be due not only to variation in the quantum efficiency of 1O2 generation, but also differences in sub-cellular localisation and concentration. The enhanced yield of these materials observed when the cells are cultured in media made up with D2O, and decreased concentration in the presence of azide, is consistent with a role for 1O2 in the formation of these peroxides, though these data do not exclude a role for other photochemical processes in the formation of these species.54 In the case of THP-1 cells (a human monocyte-derived cell line), a quantification has been attempted with regard to protein peroxide formation compared to total O2 consumed. The data obtained are consistent with a reasonably efficient conversion of O2 into protein-bound peroxides, with ca. 15% being detected as protein peroxides.54 This figure is likely to be an underestimate of the total flux of O2 going via this reaction pathway, as no correction was made for potential decay or further O2-consuming reactions of the peroxides once formed.54 The absolute levels of protein-bound peroxide formation in these cells have been calculated as representing an incidence of peroxide groups per protein molecule of ca. 0.5.54 This figure is of a similar order of magnitude to the level of carbonyl groups detected on proteins in aged human cells or diseased brain tissue.55–58 Whether peroxide formation on proteins occurs in intact biological systems (e.g. skin exposed to UV light) has not yet been definitively established. The data presented above suggest that this is likely.Decomposition of side-chain peroxides
Compound 1, formed on oxidation of Trp by 1O2, spontaneously decomposes to the corresponding alcohol, 3α-hydroxypyrroloindole (2), at room or elevated temperatures.39 The rate of decomposition is enhanced by the presence of reducing agents such as dimethylsulfide. In the absence of reducing agents, decomposition appears to be complex and may involve both heterolytic and homolytic cleavage of the peroxide bond.27–32 Decomposition of 1 and 2 may also yield N-formylkynurenine32 and, hence, kynurenine via hydrolysis (Scheme 1). Metal ions and UV light have been shown to catalyse the decomposition of Trp-derived peroxides to radicals.31,51 Two of the major overall products of oxidation, N-formylkynurenine and kynurenine, are formed with both the free amino acid and Trp residues in proteins,59 suggesting that similar pathways occur in both cases. N-Formylkynurenine and kynurenine are better photosensitisers than Trp and, hence, can give rise to further 1O2 formation during continued light exposure.16,60–62 Thus, the formation of N-formylkynurenine and kynurenine on proteins can result in an escalating cycle of damage. Extended UV exposure may also exacerbate damage via the sensitised decomposition of the initial peroxides. The poor sensitising activity, and high physical quenching activity (compared to chemical reaction), of Trp residues is believed to play a key role in the initial protection of the human lens against UV damage,63 but after high levels of exposure, the gradual accumulation of more potent sensitisers, such as N-formylkynurenine and kynurenine and species formed by covalent addition of free Trp oxidation products to lens proteins,64–66 may result in an enhancement of damage. The role of Trp and related materials as protective or potentiating agents in the human lens has been reviewed elsewhere.63,67The hydroperoxide species formed on Tyr residues are unstable and decompose above 4 °C.33 Thermal decomposition at ambient or lower temperatures, and in the absence of added reagents, occurs in a relatively clean manner to give 5 in the case of the free amino acid (Scheme 2), and 7 with peptide-bound Tyr (Scheme 3), though decomposition at higher temperatures or in the presence of redox-active metal ions and UV light is less clean and a number of other products are formed.33,39 Radicals have been detected during some of these peroxide decomposition processes;33,39 the products of these radical reactions remain to be established, though previous studies have reported that 3,4-dihydroxyphenylalanine and di-tyrosine are not products.59 It has been reported that 5 formed via the reactions shown in Scheme 2 can be oxidised further in basic conditions, giving rise to the decarboxylated keto compound.36,38 The ultimate fate of the dienone alcohol (7) formed from the peptide-bound Tyr peroxide (Scheme 3) has yet to be established, though it has been shown that this species can undergo Michael reactions with nucleophiles such as thiols and amines.50 These reactions may contribute to protein cross-linking reactions.
With histidine and related imidazole compounds, the initial endoperoxides detected by NMR at low temperatures undergo rapid decomposition to give a number of further poorly characterised peroxide intermediates and, ultimately, in the case of histidine derivatives, products including derivatives of aspartic acid and asparagine, and urea.41,42,68 These reactions, which involve ring opening, consume further molecules of O2.69 Some of this O2 consumption may arise via the decomposition of peroxides to give carbon-centred radicals that subsequently react to form peroxyl radicals.39 The nature of these reactions are poorly understood and remain to be elucidated, though it is clear that multiple oxygenations and ring-opening reactions occur.
As might be expected on the basis of the above data, protein-bound peroxides also decay at room temperature or above to give non-peroxide products (Fig. 5).50 These decay kinetics are likely to reflect composite curves for each of the individual species present and are, therefore, dependent on the protein under study. A wide range of factors are likely to affect the stability of these protein-bound materials, including the accessibility of these species to external reductants (see also below), the presence of metal ions and other reductants, continued exposure to UV, and the location of these materials on the protein structure (e.g. within hydrophobic pockets or on the surface of the protein). Little is known as yet about the factors that control the exact lifetime of such species.
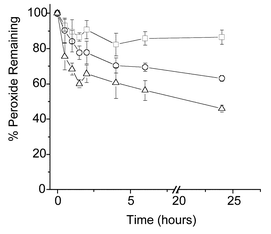 | ||
| Fig. 5 Stability of bovine serum albumin-derived peroxides at different temperatures. Peroxides were generated as outlined in Fig. 3. After the cessation of illumination, the samples were incubated in the absence of light for up to 24 h at 4 (□), 22 (○), and 37 °C (△), with the residual peroxide levels assayed at the indicated time points, as outlined in Fig. 3. Data are expressed as means of percentage initial peroxide concentration (n = 6, from 2 separate experiments) ± SD. | ||
Data has been presented on the lifetime of protein peroxides formed within viable cells after generation using the sensitiser Rose Bengal and visible light. The species detected had half-lives of ca. 4 h at 37 °C when the cells were incubated in the absence of light, but in complete growth medium.54 The kinetics of decay of these species was not affected by the presence of glucose or pre-loading of the cells with ascorbate.54 It is likely that the lifetime of intracellular protein peroxides will depend significantly on the extent of depletion of cellular reducing equivalents (which may remove these species either directly or as a result of their utilisation as enzymatic co-factors), the species on which the peroxides are formed, their intracellular location (e.g. whether these are present on membrane proteins in a hydrophobic environment or on the surface of cytosolic proteins), and the activity of protective enzymes. The influence of some of these factors on the stability of protein peroxides is discussed further below.
Transfer of damage to other biological targets
Reaction with antioxidants
Peroxides generated on peptides and proteins by 1O2 undergo relatively ready reaction with a range of low molecular mass thiols, including the physiologically relevant tripeptide GSH.70 Reaction does not appear to be stoichiometric, with large excesses of thiol (up to 10-fold) required to ensure rapid and complete removal of the peroxides.70 The products of these reactions have not been characterised in detail, though it is likely that these will include the disulfide and oxyacids (RSOH, RSO2H, and RSO3H).71 Equal excesses (i.e. 10-fold) of a range of different thiols have been shown to give rise to different kinetics and extents of peroxide removal, suggesting that there are multiple factors that influence these reactions.70 Other sulfur-containing species, such as the thioethers methionine and 3,3′-thiodipropionic acid, react much less rapidly.70 In contrast, the organoseleno compound ebselen is very efficient, and ascorbic acid and some derivatives (though not ascorbic acid-2-phosphate) are moderately effective.70 Phenolic compounds (e.g. Trolox C, BHT, and probucol) have, as expected, little effect.70Reaction with other proteins
As might be expected from the above discussion, protein thiols also react with peptide and protein peroxides. It has been shown that Trp-, Tyr- and His-derived peroxides present on small peptides can deplete thiol groups present on the enzyme glyceraldehyde-3-phosphate dehydrogenase (GAPDH), with this process paralleling the loss of the peroxides.70,72 The rate of oxidation of thiol groups on proteins is structure dependent, with both the nature and size of the peroxide and the accessibility/location of the thiol group on the target protein playing a major role.70,72,73 Thiols present on the same protein are oxidised at varying rates by different peroxides, and the same peroxide can react at different rates with a range of protein thiols. Smaller peroxides appear to induce thiol oxidation at a more rapid rate than larger peroxides (e.g. peptide-bound versus protein-bound species73), though evidence has also been obtained for electronic interactions and more subtle structural effects also playing a role (cf. the more efficient inactivation of caspase enzymes by Trp-derived peroxides compared to Tyr- and His-derived species73). As with the low molecular mass thiols, the stoichiometry of thiol oxidation to peroxides removed is variable, with values ranging between 2 and 1.70 The products generated from such thiol oxidation have not been determined, but the variation in stoichiometry is consistent with the formation of both disulfides and oxyacids, with these being generated via molecular reactions rather than radical-mediated processes. The yield of disulfide versus other products is likely to be dramatically affected by the protein structure, with both electronic and steric effects determining the accessibility of the thiol to dimerisation reactions. These thiol oxidation reactions can result in enzyme inactivation, with loss of activity of a number of enzymes detected on incubation with peptide and protein peroxides (e.g. glyceraldehyde-3-phosphate dehydrogenase, glutathione reductase, caspases, and Ca2+-ATPases72–74).There is considerable evidence for the facile formation of high molecular weight aggregates (dimers and higher species) via cross-linking reactions.75 Some of these aggregates may arise from radical–radical termination reactions of Tyr-derived phenoxyl radicals to give dityrosine,76–78 though it has been reported that this is not a major process with most photo-oxidised proteins.59 Intermolecular disulfide cross-linking has been discussed above, and appears to be a common process.17,79 UVA light has been reported to generate disulfide cross-linked proteins in the lens and in isolated proteins; this may occur via1O2-mediated80 or radical reactions,81,82 or both processes concurrently.82,83
Carbonyl groups formed as a result of side-chain oxidation52 can react with amine (Lys) and guanidine (Arg) functions on other proteins via Schiff base formation. Though such reactions are reversible, subsequent Amadori rearrangments results in irreversible cross-linking. It has also been proposed that His oxidation products can react with Lys, Cys, or other His residues to give cross-links.47,59,84–88 These reactions may account for the consumption of Lys and Arg residues in photo-oxidised proteins (reviewed in ref. 17), and may be responsible for the ‘dark’ reaction (post-cessation of illumination) inactivation observed with some enzymes (e.g. ref. 89–91). His residues may be of particular importance in aggregate formation, as proteins that lack His residues have been reported not to form cross-links.59,85
In contrast to aggregation, there are few reports on protein fragmentation induced by photo-oxidation intermediates.17,24 Photo-oxidation of lysozyme in the presence of lumiflavin has been reported to give peptide fragments, with this being proposed to occur either via oxidation of Trp residues and/or the formation of radicals.92 Radicals generated from peroxides formed on Trp, Tyr, and His residues39 have been shown to carry out intermolecular hydrogen atom abstraction reactions from α-carbon sites on model peptides to yield backbone radicals;50 these are likely progenitors of backbone fragmentation in the presence of O2.71,93,94 The significance of such reactions with proteins has yet to be determined.
Reaction with prosthetic groups
Evidence has been presented for the modification of prosthetic groups within proteins structures. Exposure of catalase to 1O2 has been shown to generate multiple conformers with more acidic isoelectric points as a result of the modification of the heme group.95,96 This damage is distinct from that induced by UV light per se,95 and has been proposed as a marker of 1O2 formation in vivo.97 Similar reactions are likely with other prosthetic groups with which 1O2 reacts rapidly (for rate constants, see ref. 18).Reaction with other targets
There is relatively little information as yet on the potential role of 1O2-mediated peptide or protein peroxides in the induction of damage to biological targets such as lipids and DNA, though preliminary evidence for this type of activity has started to appear.98,99 This type of activity, if it involves peroxides, is likely to occur via the formation of radicals from the peroxide, catalysed by continued exposure to short wavelength UV light (which has been shown to decompose Tyr-derived peroxides33) or metal ions.33,39,72 Analogous experiments with peptide- and protein-derived peroxides generated by high energy radiation (γ radiation or X-rays, reviewed in ref. 100), have shown that these species can induce DNA base oxidation, strand breaks, DNA–protein cross-links,101–104 and lipid oxidation,105 so such damage might also be expected to occur with 1O2-mediated peroxides.Consequences of 1O2-mediated damage to proteins
The formation of modified proteins as a result of 1O2-mediated photo-oxidation can result in wide-ranging biophysical and biochemical changes in both the properties and function of proteins. These include an increased or decreased susceptibility of the oxidised protein to proteolytic enzymes, alterations in mechanical properties (e.g. of silk and collagen), an increased extent of, or susceptibility to, unfolding, changes in conformation, an increase in hydrophobicity, altered light scattering properties and optical rotation, and changes in binding of co-factors and metal ions (e.g. ref. 17, 47, 59, 106, and 107). The role of reactive species formed on the oxidised proteins in these reactions is not fully understood, though it is clear that many of the products of such oxidation are poorly repaired, and the major fate of these species within cells is catabolism (reviewed in ref. 93 and 108–110). Though many oxidised proteins are known to be removed at an enhanced rate compared to normal proteins, populations exist which are poorly removed, resulting in intracellular accumulation of modified materials, particularly in the lysosomal compartments of cells.111,112 The build-up of such materials may have major effects on cellular function,93,108–110 and thereby contribute in a significant manner to the initiation and development of damage induced by sources of 1O2, for example, that seen in skin and the lens of the eye on prolonged exposure to UV light. Furthermore, the evidence that is starting to appear on the downstream effects of peptide and protein peroxide formation, such as the inhibition of key cellular enzymes and the potential induction of DNA damage, suggests that these species may be key intermediates in cellular dysfunction and disease progression.Acknowledgements
Due to space limitations, a large number of papers which are pertinent to this topic could not be cited; the author apologies for these omissions. The author gratefully acknowledges the important contributions of his co-workers cited in the reference list. Work carried out in the author's laboratory is supported by the Australian Research Council and National Health and Medical Research Council.References
- J. R. Kanofsky, J. Wright, G. E. Miles-Richardson and A. I. Tauber, Biochemical requirements for singlet oxygen production by purified human myeloperoxidase, J. Clin. Invest., 1984, 74, 1489–1495 Search PubMed.
- J. R. Kanofsky, J. Wright and A. I. Tauber, Effect of ascorbic acid on the production of singlet oxygen by purified human myeloperoxidase, FEBS Lett., 1985, 187, 299–301 CrossRef CAS.
- J. R. Kanofsky, Singlet oxygen production by lactoperoxidase, J. Biol. Chem., 1983, 258, 5991–5993 CAS.
- J. R. Kanofsky, Singlet oxygen production from the peroxidase-catalyzed oxidation of indole-3-acetic acid, J. Biol. Chem., 1988, 263, 14
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 171–14175 CAS.
171–14175 CAS. - J. R. Kanofsky, Singlet oxygen production by chloroperoxidase-hydrogen peroxide-halide systems, J. Biol. Chem., 1984, 259, 5596–5600 CAS.
- J. R. Kanofsky and B. Axelrod, Singlet oxygen production by soybean lipoxygenase isozymes, J. Biol. Chem., 1986, 261, 1099–1104 CAS.
- J. R. Kanofsky, H. Hoogland, R. Wever and S. J. Weiss, Singlet oxygen production by human eosinophils, J. Biol. Chem., 1988, 20, 9692–9696.
- M. J. Steinbeck, A. U. Khan and M. J. Karnovsky, Extracellular production of singlet oxygen by stimulated macrophages quantified using 9,10-diphenylanthracene and perylene in a polystyrene film, J. Biol. Chem., 1993, 268, 15649–15854 CAS.
- J. R. Kanofsky, Bromine derivatives of amino acids as intermediates in the peroxidase-catalyzed formation of singlet oxygen, Arch. Biochem. Biophys., 1989, 274, 229–234 CrossRef CAS.
- J. R. Kanofsky and P. Sima, Singlet oxygen production from the reactions of ozone with biological molecules, J. Biol. Chem., 1991, 266, 9039–9042 CAS.
- C. S. Foote and S. Wexler, Olefin oxidations with excited singlet molecular oxygen, J. Am. Chem. Soc., 1964, 86, 3879–3880 CrossRef CAS.
- J. M. Aubry, B. Cazin and F. Duprat, Chemical sources of singlet oxygen. 3. Peroxidation of water-soluble singlet oxygen carriers with the hydrogen peroxide-molybdate system, J. Org. Chem., 1989, 726–728 CrossRef CAS.
- P. Di Mascio, E. J. Bechara, M. H. Medeiros, K. Briviba and H. Sies, Singlet molecular oxygen production in the reaction of peroxynitrite with hydrogen peroxide, FEBS Lett., 1994, 355, 287–289 CrossRef CAS.
- M. Nakano, Y. Kambayashi, H. Tatsuzawa, T. Komiyama and K. Fujimori, Useful 1O2 (1Δg) generator, 3-(4′-methyl-1′-naphthyl)-propionic acid, 1′,4′-endoperoxide (NEPO), for dioxygenation of squalence (a skin surface lipid) in an organic solvent and bacterial killing in aqueous medium, FEBS Lett., 1998, 432, 9–12 CrossRef CAS.
- M. Nakano, K. Takayama, Y. Shimizu, Y. Tsuji, H. Inaba and T. Migita, Spectroscopic evidence for the generation of singlet oxygen in self-reaction of sec-peroxy radicals, J. Am. Chem. Soc., 1976, 98, 1974–1975 CrossRef CAS.
- R. W. Redmond and J. N. Gamlin, A compilation of singlet oxygen yields from biologically relevant molecules, Photochem. Photobiol., 1999, 70, 391–475 CAS.
- R. C. Straight and J. D. Spikes, in Singlet O2, ed. A. A. Frimer, CRC Press, Boca Raton, 1985, vol. 4, p. 91 Search PubMed.
- F. Wilkinson, W. P. Helman and A. B. Ross, Rate constants for the decay and reactions of the lowest electronically excited state of molecular oxygen in solution. An expanded and revised compilation, J. Phys. Chem. Ref. Data, 1995, 24, 663–1021 CAS.
- Geigy Scientific Tables: Physical Chemistry, Composition of Blood, Hematology, Somatometric Data, ed. C. Lentner, Ciba-Geigy Ltd, Basle, 1984, vol. 3 Search PubMed.
- B. Monroe, in Singlet O2, ed. A. A. Frimer, CRC Press, Boca Raton, 1985, vol. 1, p. 177 Search PubMed.
- J. R. Kanofsky, Quenching of singlet oxygen by human red cell ghosts, Photochem. Photobiol., 1991, 53, 93–99 CAS.
- I. B. C. Matheson, R. D. Etheridge, N. R. Kratowich and J. Lee, The quenching of singlet oxygen by amino acids and proteins, Photochem. Photobiol., 1975, 21, 165–171 CAS.
- M. C. Palumbo, N. A. Garcia and G. A. Arguello, The interaction of singlet molecular oxygen O2 (1Δg) with indolic derivatives. Distinction between physical and reactive quenching., J. Photochem. Photobiol., B, 1990, 7, 33–42 CrossRef CAS.
- A. Michaeli and J. Feitelson, Reactivity of singlet oxygen toward amino acids and peptides, Photochem. Photobiol., 1994, 59, 284–289 CAS.
- M. Rougee, R. V. Bensasson, E. J. Land and R. Pariente, Deactivation of singlet molecular oxygen by thiols and related compounds, possible protectors against skin photosensitivity, Photochem. Photobiol., 1988, 47, 485–489 CAS.
- G. Papeschi, M. Monici and S. Pinzauti, pH effect on dye sensitized photooxidation of aminoacids and albumins, Med. Biol. Environ., 1982, 10, 245–250 Search PubMed.
- M. Nakagawa, K. Yoshikawa and T. Hino, The photosensitized oxygenation of Nb-methyltryptamine, J. Am. Chem. Soc., 1975, 97, 6496–6501 CrossRef CAS.
- M. Nakagawa, H. Okajima and T. Hino, Photosensitized oxygenation of Nb-methoxycarbonyltryptophan methyl ester and Nb-methoxycarbonyl tryptamine. Isolation and novel transformation of a 3a-hydroperoxypyrroloindole, J. Am. Chem. Soc., 1976, 98, 635–637 CrossRef CAS.
- M. Nakagawa, H. Watanabe, S. Kodato, H. Okajima, T. Hino, J. L. Flippen and B. Witkop, A valid model for the mechanism of oxidation of tryptophan to formylkynurenine - 25 years later, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 4730–4733 CAS.
- M. Nakagawa, H. Okajima and T. Hino, Photosensitized oxygenation of Nb-methoxycarbonyltryptamines. A new pathway to kynurenine derivatives, J. Am. Chem. Soc., 1977, 99, 4424–4429 CrossRef CAS.
- I. Saito, T. Matsuura, M. Nakagawa and T. Hino, Peroxidic intermediates in photosensitized oxygenation of tryptophan derivatives, Acc. Chem. Res., 1977, 10, 346–352 CrossRef CAS.
- R. Langlois, H. Ali, N. Brasseur, J. R. Wagner and J. E. van Lier, Biological activities of phythalocyanines – IV. Type II sensitized photooxidation of L-tryptophan and cholesterol by sulfonated metallo phthalocyanines, Photochem. Photobiol., 1986, 44, 117–123 CAS.
- A. Wright, W. A. Bubb, C. L. Hawkins and M. J. Davies, Singlet oxygen-mediated protein oxidation: evidence for the formation of reactive side-chain peroxides on tyrosine residues, Photochem. Photobiol., 2002, 76, 35–46 CrossRef CAS.
- I. Saito, S. Kato and T. Matsuura, Photoinduced reactions. XL. Addition of singlet oxygen to monocyclic aromatic rings, Tetrahedron Lett., 1970, 3, 239–242 CrossRef.
- F. Jensen and C. S. Foote, Chemistry of singlet oxygen – 48. Isolation and structure of the primary product of photooxygenation of 3,5-di-t-butyl catechol, Photochem. Photobiol., 1987, 46, 325–330 CAS.
- E. Katsuya, K. Seya and H. Hikino, Photo-oxidation of L-tyrosine, an efficient, 1,4-chirality transfer reaction, J. Chem. Soc., Chem. Commun., 1988, 934–935 RSC.
- F. M. Jin, J. Leitich and C. von Sonntag, The photolysis (λ = 254 nm) of tyrosine in aqueous solutions in the absence and presence of oxygen – the reaction of tyrosine with singlet oxygen, J. Photochem. Photobiol., A, 1995, 92, 147–153 CrossRef CAS.
- S. Criado, A. T. Soltermann, J. M. Marioli and N. A. Garcia, Sensitized photooxidation of di- and tripeptides of tyrosine, Photochem. Photobiol., 1998, 68, 453–458 CAS.
- A. Wright, C. L. Hawkins and M. J. Davies, Singlet oxygen-mediated protein oxidation: evidence for the formation of reactive peroxides, Redox Rep., 2000, 5, 159–161 Search PubMed.
- C. Gay, J. Collins and J. M. Gebicki, Hydroperoxide assay with the ferric-xylenol orange complex, Anal. Biochem., 1999, 273, 149–155 CrossRef CAS.
- M. Tomita, M. Irie and T. Ukita, Sensitized photooxidation of histidine and its derivatives. Products and mechanism of the reaction, Biochemistry, 1969, 8, 5149–5160 CrossRef CAS.
- P. Kang and C. S. Foote, Synthesis of a C-13, N-15 labeled imidazole and characterization of the 2,5-endoperoxide and its decomposition, Tetrahedron Lett., 2000, 41, 9623–9626 CrossRef CAS.
- V. Policarpio, C. L. Hawkins and M. J. Davies, unpublished data.
- V. Policarpio, W. A. Bubb, C. L. Hawkins and M. J. Davies, unpublished data.
- P. K. Sysak, C. S. Foote and T.-Y. Ching, Chemistry of singlet oxygen – XXV. Photooxygenation of methionine, Photochem. Photobiol., 1977, 26, 19–27 CAS.
- W. Ando and T. Takata, in Singlet O2, ed. A. A. Frimer, CRC Press, Boca Raton, 1985, vol. 3, p. 1 Search PubMed.
- J. Dillon, R. Chiesa, R. H. Wang and M. McDermott, Molecular changes during the photooxidation of alpha-crystallin in the presence of uroporphyrin, Photochem. Photobiol., 1993, 57, 526–530 CAS.
- E. L. Finley, M. Busman, J. Dillon, R. K. Crouch and K. L. Schey, Identification of photooxidation sites in bovine alpha-crystallin, Photochem. Photobiol., 1997, 66, 635–641 CAS.
- C. S. Foote and J. W. Peters, Chemistry of singlet oxygen. XIV. A reactive intermediate in sulfide photooxidation, J. Am. Chem. Soc., 1971, 93, 3975–3796.
- A. Wright, C. L. Hawkins and M. J. Davies, unpublished data.
- C. L. Hawkins and M. J. Davies, unpublished data.
- J. A. Silvester, G. S. Timmins and M. J. Davies, Protein hydroperoxides and carbonyl groups generated by porphyrin-induced photo-oxidation of bovine serum albumin, Arch. Biochem. Biophys., 1998, 350, 249–258 CrossRef CAS.
- J. A. Silvester, G. S. Timmins and M. J. Davies, Photodynamically-generated bovine serum albumin radicals: evidence for damage transfer and oxidation at cysteine and tryptophan residues, Free Radical Biol. Med., 1998, 24, 754–766 CrossRef CAS.
- A. Wright, C. L. Hawkins and M. J. Davies, Photo-oxidation of cells generates long-lived intracellular protein peroxides, Free Radical Biol. Med., 2003, 34, 637–647 CrossRef CAS.
- C. N. Oliver, B. W. Ahn, E. J. Moerman, S. Goldstein and E. R. Stadtman, Age-related changes in oxidized proteins, J. Biol. Chem., 1987, 262, 5488–5491 CAS.
- L. Lyras, P. J. Evans, P. J. Shaw, P. G. Ince and B. Halliwell, Oxidative damage and motor neurone disease. Difficulties in the measurement of protein carbonyls in human brain tissue, Free Radical Res., 1996, 24, 397–406 Search PubMed.
- M. J. Davies, S. Fu, H. Wang and R. T. Dean, Stable markers of oxidant damage to proteins and their application in the study of human disease, Free Radical Biol. Med., 1999, 27, 1151–1163 CrossRef CAS.
- S. Linton, M. J. Davies and R. T. Dean, Protein oxidation in ageing, Exp. Gerentol., 2001, 36, 1503–1518 Search PubMed.
- D. Balasubramanian, X. Du and J. S. J. Zigler, The reaction of singlet oxygen with proteins, with special reference to crystallins, Photochem. Photobiol., 1990, 52, 761–768 CAS.
- P. Walrant and R. Santus, N-formyl-kynurenine, a tryptophan photooxidation product, as a photodynamic sensitizer, Photochem. Photobiol., 1974, 19, 411–417 CAS.
- M.-P. Pileni, R. Santus and E. J. Land, On the photosensitizing properties of N-formylkynurenine and related compounds, Photochem. Photobiol., 1978, 28, 525–529 CAS.
- M.-P. Pileni, M. Giraud and R. Santus, Kynurenic acid. II. Photosensitizing properties, Photochem. Photobiol., 1979, 30, 257–261 CAS.
- M. J. Davies and R. J. W. Truscott, Photo-oxidation of proteins and its role in cataractogenesis, J. Photochem. Photobiol., B, 2001, 63, 114–125 CrossRef CAS.
- J. A. Aquilina and R. J. Truscott, Cysteine is the initial site of modification of α-crystallin by kynurenine, Biochem. Biophys. Res. Commun., 2000, 276, 216–223 CrossRef CAS.
- J. A. Aquilina, J. A. Carver and R. J. Truscott, Polypeptide modification and cross-linking by oxidized 3- hydroxykynurenine, Biochemistry, 2000, 39, 16176–16184 CrossRef CAS.
- J. A. Aquilina and R. J. W. Truscott, Identifying sites of attachment of UV filters to proteins in older human lenses, Biochim. Biophys. Acta, 2002, 1596, 6–15 CrossRef CAS.
- J. E. Roberts, in Sun Protection in Man, ed. P. U. Giacomoni, Elsevier, Amsterdam, 2001, p. 155 Search PubMed.
- S. Kai and M. Suzuki, Dye-sensitized photooxidation of 2,4-disubstituted imidazoles – the formation of isomeric imidazolinones, Heterocycles, 1996, 43, 1185–1188 CAS.
- S. H. Chang, G. M. Teshima, T. Milby, B. Gillece-Castro and E. Canova-Davis, Metal-catalyzed photooxidation of histidine in human growth hormone, Anal. Biochem., 1997, 244, 221–227 CrossRef CAS.
- P. E. Morgan, R. T. Dean and M. J. Davies, unpublished data.
- W. M. Garrison, Reaction mechanisms in the radiolysis of peptides, polypeptides, and proteins, Chem. Rev., 1987, 87, 381–398 CrossRef CAS.
- P. E. Morgan, R. T. Dean and M. J. Davies, Inhibition of glyceraldehyde-3-phosphate dehydrogenase by peptide and protein peroxides generated by singlet oxygen attack, Eur. J. Biochem., 2002, 269, 1916–1925 CrossRef CAS.
- M. B. Hampton, P. E. Morgan and M. J. Davies, Inactivation of cellular caspases by peptide-derived tryptophan and tyrosine peroxides, FEBS Lett., 2002, 527, 289–292 CrossRef CAS.
- C. Schoneich, V. Sharov and M. J. Davies, unpublished data.
- J. D. Goosey, J. S. Zigler Jr. and J. H. Kinoshita, Cross-linking of lens crystallins in a photodynamic system: a process mediated by singlet oxygen, Science, 1980, 208, 1278–1280 CAS.
- P. Guptasarma and D. Balasubramanian, Dityrosine formation in the proteins of the eye lens, Curr. Eye Res., 1992, 11, 1121–1125 Search PubMed.
- J. D. Spikes, H. R. Shen, P. Kopeckova and J. Kopecek, Photodynamic crosslinking of proteins. III. Kinetics of the FMN- and rose bengal-sensitized photooxidation and intermolecular crosslinking of model tyrosine-containing N-(2-hydroxypropyl)methacrylamide copolymers, Photochem. Photobiol., 1999, 70, 130–137 CAS.
- H. Shen, J. D. Spikes, C. J. Smith and J. Kopecek, Photodynamic cross-linking of proteins V. Nature of the tyrosine-tyrosine bonds formed in the FMN-sensitized intermolecular cross-linking of N-acetyl-L-tyrosine, J. Photochem. Photobiol., A, 2000, 133, 115–122 CrossRef CAS.
- R. J. Truscott and R. C. Augusteyn, Oxidative changes in human lens proteins during senile nuclear cataract formation, Biochim. Biophys. Acta, 1977, 492, 43–52 CrossRef CAS.
- M. Linetsky and B. J. Ortwerth, Quantitation of the singlet oxygen produced by UVA irradiation of human lens proteins, Photochem. Photobiol., 1997, 65, 522–529 CAS.
- M. Linetsky, H. L. James and B. J. Ortwerth, The generation of superoxide anion by the UVA irradiation of human lens proteins, Exp. Eye Res., 1996, 63, 67–74 CrossRef CAS.
- J. Dillon, B. J. Ortwerth, C. F. Chignell and K. J. Reszka, Electron paramagnetic resonance and spin trapping investigations of the photoreactivity of human lens proteins, Photochem. Photobiol., 1999, 69, 259–264 CAS.
- F. J. Giblin, V. R. Leverenz, V. A. Padgaonkar, N. J. Unakar, L. Dang, L. R. Lin, M. F. Lou, V. N. Reddy, D. Borchman and J. P. Dillon, UVA light in vivo reaches the nucleus of the guinea pig lens and produces deleterious, oxidative effects, Exp. Eye Res., 2002, 75, 445–458 CrossRef CAS.
- H. Verweij and J. van Steveninck, Model studies on photodynamic crosslinking, Photochem. Photobiol., 1982, 35, 265–267 CAS.
- P. Guptasarma, D. Balasubramanian, S. Matsugo and I. Saito, Hydroxyl radical mediated damage to proteins, with special reference to the crystallins, Biochemistry, 1992, 31, 4296–4303 CrossRef CAS.
- H. R. Shen, J. D. Spikes, P. Kopecekova and J. Kopecek, Photodynamic crosslinking of proteins. I. Model studies using histidine- and lysine-containing N-(2-hydroxypropyl)methacrylamide copolymers, J. Photochem. Photobiol., B, 1996, 34, 203–210 CrossRef CAS.
- H. R. Shen, J. D. Spikes, P. Kopeckova and J. Kopecek, Photodynamic crosslinking of proteins. II. Photocrosslinking of a model protein-ribonuclease A, J. Photochem. Photobiol., B, 1996, 35, 213–219 CrossRef CAS.
- H.-R. Shen, J. D. Spikes, C. J. Smith and J. Kopecek, Photodynamic cross-linking of proteins IV. Nature of the His-His bond(s) formed in the rose bengal-photosensitized cross-linking of N-benzoyl-L-histidine, J. Photochem. Photobiol., A, 2000, 130, 1–6 CrossRef CAS.
- T. M. Dubbelman, C. Haasnoot and J. van Steveninck, Temperature dependence of photodynamic red cell membrane damage, Biochim. Biophys. Acta, 1980, 601, 220–227 CrossRef CAS.
- A. Spector, G. M. Wang and R. R. Wang, The prevention of cataract caused by oxidative stress in cultured rat lenses. II. Early effects of photochemical stress and recovery, Exp. Eye Res., 1993, 57, 659–667 CrossRef CAS.
- A. Spector, G. M. Wang, R. R. Wang, W. C. Li and N. J. Kleiman, A brief photochemically induced oxidative insult causes irreversible lens damage and cataract. II. Mechanism of action, Exp. Eye Res., 1995, 60, 483–493 CAS.
- T. Gomyo and M. Fujimaki, Studies on changes in protein by dye sensitized photooxidation. Part 3. On the photodecomposition products of lysozyme, Agric. Biol. Chem., 1970, 34, 302–309 CAS.
- M. J. Davies and R. T. Dean, Radical-Mediated Protein Oxidation: From Chemistry to Medicine, Oxford University Press, Oxford, 1997, pp. 1–443 Search PubMed.
- C. L. Hawkins and M. J. Davies, Generation and propagation of radical reactions on proteins, Biochim. Biophys. Acta, 2001, 1504, 196–219 CrossRef CAS.
- F. Lledias and W. Hansberg, Oxidation of human catalase by singlet oxygen in myeloid leukemia cells, Photochem. Photobiol., 1999, 70, 887–892 CAS.
- F. Lledias, P. Rangel and W. Hansberg, Oxidation of catalase by singlet oxygen, J. Biol. Chem., 1998, 273, 10630–10637 CrossRef CAS.
- F. Lledias and W. Hansberg, Catalase modification as a marker for singlet oxygen, Methods Enzymol., 2000, 319, 110–119 CAS.
- G. D. Ouedraogo and R. W. Redmond, Photosensitized membrane oxidation leads to remote DNA damage by secondary reactive oxygen species, Free Radical Biol. Med., 2002, 33(suppl. 2), S421.
- L. Booth and R. W. Redmond, Can lipid peroxidation of plasma membranes induce DNA strand breaks, Free Radical Biol. Med., 2002, 33(suppl. 2), S390.
- J. M. Gebicki, Protein hydroperoxides as new reactive oxygen species, Redox Rep., 1997, 3, 99–110 Search PubMed.
- S. Gebicki and J. M. Gebicki, Crosslinking of DNA and proteins induced by protein hydroperoxides, Biochem. J., 1999, 338, 629–636 CrossRef CAS.
- C. Luxford, B. Morin, R. T. Dean and M. J. Davies, Histone H1- and other protein- and amino acid-hydroperoxides can give rise to free radicals which oxidize DNA, Biochem. J., 1999, 344, 125–134 CrossRef CAS.
- C. Luxford, R. T. Dean and M. J. Davies, Radicals derived from histone hydroperoxides damage nucleobases in RNA and DNA, Chem. Res. Toxicol., 2000, 13, 665–672 CrossRef CAS.
- C. Luxford, R. T. Dean and M. J. Davies, Induction of DNA damage by oxidised amino acids and proteins, Biogerentology, 2002, 3, 95–102 Search PubMed.
- M. J. Davies, unpublished data.
- C. Prinsze, T. M. Dubbelman and J. Van Steveninck, Protein damage, induced by small amounts of photodynamically generated singlet oxygen or hydroxyl radicals, Biochim. Biophys. Acta, 1990, 1038, 152–157 CrossRef CAS.
- E. Silva, C. De Landea, A. M. Edwards and E. Lissi, Lysozyme photo-oxidation by singlet oxygen: properties of the partially inactivated enzyme, J. Photochem. Photobiol., B, 2000, 55, 196–200 CrossRef CAS.
- K. J. Davies, S. W. Lin and R. E. Pacifici, Protein damage and degradation by oxygen radicals. IV. Degradation of denatured protein, J. Biol. Chem., 1987, 262, 9914–9920 CAS.
- R. T. Dean, S. Fu, R. Stocker and M. J. Davies, Biochemistry and pathology of radical-mediated protein oxidation, Biochem. J., 1997, 324, 1–18 CAS.
- E. R. Stadtman and R. L. Levine, Protein oxidation, Ann. N. Y. Acad. Sci., 2000, 899, 191–208 CAS.
- A. J. Grant, W. Jessup and R. T. Dean, Inefficient degradation of oxidized regions of protein molecules, Free Radical Res. Commun., 1993, 18, 259–267 Search PubMed.
- A. J. Grant, W. Jessup and R. T. Dean, Accelerated endocytosis and incomplete catabolism of radical-damaged protein, Biochim. Biophys. Acta, 1992, 1134, 203–209 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and Owner Societies 2004 |