Bi-directional photocurrent generation dependent on the wavelength of irradiation of a mixed monolayer assembly
Tsuyoshi
Akiyama
*a,
Satoshi
Nitahara
b,
Shinobu
Inoue
c and
Sunao
Yamada
*a
aDepartment of Applied Chemistry, Kyushu University, 6-10-1, Hakozaki, Higashi-ku, Fukuoka 812-8581, Japan. E-mail: t-akitcm@mbox.nc.kyushu-u.ac.jp
bDepartment of Materials Physics and Chemistry, Kyushu University, 6-10-1, Hakozaki, Higashi-ku, Fukuoka 812-8581, Japan
cMaterials Science Laboratory, Mitsui Chemicals, Inc., Nagaura, Sodegaura, Chiba 299-0265, Japan
First published on 10th December 2003
Abstract
A mixed self-assembled monolayer consisting of a ruthenium tris-bipyridine complex linked to viologen and a palladium phthalocyanine derivative was fabricated on a gold electrode by means of pendant thiol groups. The direction of the photocurrent which is induced in the electrode when it is irradiated depends on the wavelength of the light used.
The development of novel organic thin film devices based on the assembly of molecular components has been an exceptionally active field in recent times. Self-assembled monolayers (SAMs) of donor (D)–acceptor (A) pairs on electrodes have attracted particular attention for application as photoelectric conversion devices.1–6 Photoinduced electron transfer between the donor and the acceptor is responsible for photocurrent generation. Implantation of light-harvesting molecules7 and fabrication of three-dimensional nanostructured assemblies by combining gold nanoparticles and thiol dyes8–10 have proven to be successful avenues for improving photocurrent generation. Until now, however, the development of devices based on SAMs has been directed towards the improvement of photoelectric conversion efficiency. One of the noteworthy advantages of self-assembly is that different molecules can be implanted on the electrode at the same time.1,7 This may allow SAMs to be exploited for new applications in photonic fields.
We have developed a novel photoelectric conversion device which uses a mixed SAM composed of a linked ruthenium bipyridine–viologen disulfide10 and a palladium phthalocyanine thiol derivative (Fig. 1).11 The direction (cathodic or anodic) of the photocurrent induced in the monolayer can be reversed by altering the irradiation wavelength.
 | ||
| Fig. 1 Schematic illustration of the structure of the Ru(bipy)3–viologen thiol and Pd phthalocyanine thiol mixed SAM, (RuVS + PcS)/Au. | ||
The syntheses of the linked ruthenium bipyridine complex–viologen disulfide [RuVS]210 and the phthalocyanine disulfide [PcS]211 have been described previously. The gold electrode was prepared by depositing a small amount of titanium and then gold on a glass plate by vacuum deposition. This gold electrode was immersed in a solution of [RuVS]2 (5 × 10−4 mol dm−3) in MeCN–CH2Cl2 (1 ∶ 1 v/v) for 4 days. Then, the electrode was removed from the solution, rinsed with acetonitrile and dichloromethane, and then dried in air, to give an electrode modified with RuVS (denoted RuVS/Au). An electrode modified with [PcS]2 (PcS/Au) was also prepared by the same procedure. The electrode modified with both compounds, (RuVS + PcS)/Au (Fig. 1), was prepared from a solution containing identical concentrations (5 × 10−4 mol dm−3) of [RuVS]2 and [PcS]2 by the same procedure as used for RuVS/Au. The surface coverage of RuVS in RuVS/Au (5.5 × 10−11 mol cm−2) was estimated by monitoring the reductive wave of the viologen moiety of RuVS, and the surface coverage of PcS/Au obtained from the absorption spectrum of PcS (1.6 × 10−11 mol cm−2). As to (RuVS + PcS)/Au, the surface coverage of each dye was evaluated from the reductive wave of the viologen moiety and from the adsorption ratio of RuVS and PcS on gold powder:1,12 6.0 × 10−12 mol cm−2 for RuVS and 1.9 × 10−12 mol cm−2 for PcS. The reason for the lower coverage in the case of (RuVS + PcS)/Au is uncertain at present, but the photocurrent intensity correlates well with the surface coverage (vide infra).
Photocurrent measurements on the modified electrodes were carried out using a three-electrode photoelectrochemical cell consisting of the modified electrode (working), an Ag/AgCl (sat. KCl) electrode (reference), and a platinum wire (counter) in an aqueous solution containing 0.1 mol dm−3 NaClO4 under a nitrogen atmosphere in the presence of methylviologen (MV2+; 5 × 10−3 mol dm−3) and triethanolamine (TEOA; 5 × 10−2 mol dm−3). The light from a 150 W tungsten lamp was passed through a monochromator and irradiated a 0.28 cm2 area of the modified electrode. The photocurrent action spectra were measured by changing the excitation wavelength (Δλ = ±16 nm).
When the photocurrent action spectrum of RuVS/Au was measured at E = 0 mV, anodic photocurrents were observed in the 380–750 nm region. The action spectrum showed a broad band around 450 nm, almost overlapping with the absorption band of RuVS in acetonitrile. This suggests that the ruthenium complex is the photoactive species and the anodic photocurrent generation is mainly due to photoinduced intramolecular electron transfer from the photoexcited ruthenium complex to the viologen moiety.1,4 The oxidized ruthenium complex is reduced by TEOA.
On the other hand, cathodic photocurrents were observed in the case of PcS/Au at E = 0 mV. The action spectrum also showed broad bands around 700 nm, corresponding to the absorption bands of phthalocyanine in toluene. Thus, the phthalocyanine moiety is the photoactive site and intermolecular photoinduced electron-transfer from the photoexcited phthalocyanine to MV2+ in the bulk mainly induces the cathodic photocurrent generation in PcS/Au. A small amount of residual oxygen in the electrolyte solution may also contribute to some extent to the cathodic photocurrent generation. These photocurrent mechanisms are shown in Figure 2.
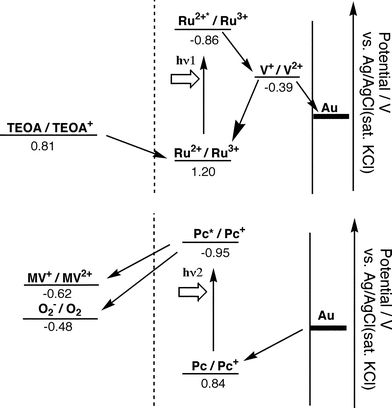 | ||
| Fig. 2 Photocurrent mechanism and electron flow. | ||
At E = −200 mV, the photocurrent from (RuVS + PcS)/Au was observed in the cathodic direction when exciting in the 400–750 nm region and the photocurrent action spectrum showed broad peaks around 630 and 700 nm; thus, the contribution from RuVS is negligible. At E = +400 mV on the other hand, the photocurrent was observed only in the anodic direction when irradiating in the 400–750 nm region, with a relatively large broad peak around 450 nm, meaning that the contribution from PcS is negligible. These results show that RuVS functions individually for anodic photocurrent generation at E = −200 mV, while PcS acts similarly for cathodic photocurrent generation at E = +400 mV. At E = 0 mV, however, photocurrents were observed in both directions, depending on the irradiation wavelength, as shown in Fig. 3. The photocurrent action spectrum for (RuVS + PcS)/Au at E = 0 mV shows a broad anodic peak around 450 nm, corresponding with the absorption peak of RuVS, and two cathodic peaks around 630 and 700 nm, corresponding with the absorption peaks of PcS, with a cross-point (zero current) around 550 nm.
 | ||
| Fig. 3 Absorption spectra of RuVS (––) and PcS (⋯), and the photocurrent action spectrum of (RuVS + PcS)/Au at E = 0 mV (●). | ||
The photocurrent measurements summarized above indicate that two kinds of photoinduced electron-transfer pathways operate almost independently in the (RuVS + PcS)/Au monolayer. Intramolecular photoinduced electron transfer occurs from the photoexcited ruthenium complex to viologen in RuVS and intermolecular photoinduced electron transfer from the photoexcited phthalocyanine to MV2+ (and residual oxygen) takes place in the bulk. The photocurrent generated by PcS/Au was compared under three different experimental conditions in terms of the sacrificial reagents: (1) [MV2+] = 5 × 10−3 mol dm−3 under aerobic conditions, (2) [MV2+] = 5 × 10−3 mol dm−3 under a nitrogen atmosphere, and (3) without MV2+ under aerobic conditions. The ratio of the photocurrent at 700 nm for the three conditions (1), (2), and (3) is 20 ∶ 4 ∶ 7. These results indicate that oxygen is a good electron acceptor for the photoexcited phthalocyanine and the presence of oxygen promotes electron transfer from the photoexcited phthalocyanine to MV2+. It is likely that oxygen accepts an electron from MV+, regenerating MV2+. In any case, one of these two pathways can be preferentially driven by irradiation at the appropriate wavelength, because the absorption bands of the ruthenium complex (around 450 nm; hν1) and phthalocyanine (600–700 nm; hν2) are widely separated from each other. At negative applied potentials, electron transfer from the electrode to the cation radical of phthalocyanine occurs easily in PcS (Eox = +0.84 V vs. Ag/AgCl), while that from the viologen moiety to the electrode becomes difficult in RuVS (Ered = −0.39 V vs. Ag/AgCl). At positive potentials, electron-transfer from the one-electron reduced viologen moiety to the electrode becomes favorable in RuVS. Accordingly, the direction of the photocurrent in (RuVS + PcS)/Au at E = 0 mV reverses at ∼550 nm.
The bi-directional photocurrent response reported here arises as a result of the implantation of two separate and individually functioning photoinduced electron-transfer systems on the electrode. This novel photocurrent behavior is promising for applications in molecular photonic devices and photoelectric logic devices.
Notes and references
- S. Yamada, H. Kohrogi and T. Matsuo, Photoelectrochemical responses of gold electrodes co-modified with sulfur-containing ruthenium tris(2,2′-bipyridine) and viologen derivatives, Chem. Lett., 1995, 639–640 CAS.
- T. Kondo, T. Ito, S. Nomura and K. Uosaki, Photoelectrochemical characteristics of a self-assembled monolayer of porphyrin-mercaptoquinone coupling molecules, Thin Solid Films, 1996, 284/285, 652–655 CrossRef.
- T. Akiyama, H. Imahori, A. Ajawakom and Y. Sakata, Synthesis and self-assembly of porphyrin-linked fullerene on gold surface using S-Au linkage, Chem. Lett., 1996, 907–908 CAS.
- N. Terasaki, T. Akiyama and S. Yamada, Structural characterization and photoelectrochemical properties of the self-assembled monolayers of tris(2,2′-bipyridine)ruthenium(II)-viologen linked compounds formed on the gold surface, Langmuir, 2002, 18, 8666–8671 CrossRef CAS.
- T. Akiyama and S. Yamada, Photocurrent generation from self-assembled monolayers of donor-acceptor pairs formed on conductive surfaces, Trends Photochem. Photobiol., 2001, 8, 67–85 Search PubMed.
- K. Uosaki, T. Kondo, X-Q. Zhang and M. Yanagida, Very efficient visible-light-induced uphill electron transfer at a self-assembled monolayer with a porphyrin-ferrocene-thiol linked molecule, J. Am. Chem. Soc., 1997, 119, 8367–8368 CrossRef CAS.
- H. Imahori, H. Norieda, H. Yamada, Y. Nishimura, I. Yamazaki, Y. Sakata and S. Fukuzumi, Light-harvesting and photocurrent generation by gold electrodes modified with mixed self-assembled monolayers of boron-dipyrrin and ferrocene-porphyrin-fullerene triad, J. Am. Chem. Soc., 2001, 123, 100–110 CrossRef CAS.
- M. Lahav, V. H-Shabtai, J. Wasserman, E. Katz, I. Willner, H. Dürr, Y-Z. Hu and S. Bossman, Photoelectrochemistry with integrated photosensitizer-electron acceptor and Au-nanoparticle arrays, J. Am. Chem. Soc., 2000, 122, 11
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 480–11487 CrossRef CAS.
480–11487 CrossRef CAS. - Y. Kuwahara, T. Akiyama and S. Yamada, Construction of gold nanoparticle-ruthenium(II) tris(2,2′-bipyridine) self-assembled multistructures and their photocurrent responses, Thin Solid Films, 2001, 393, 273–277 CrossRef CAS.
- Y. Kuwahara, T. Akiyama and S. Yamada, Facile fabrication of photoelectrochemical assemblies consisting of gold nanoparticles and a tris(2,2′-bipyridine) ruthenium(II)-viologen linked thiol, Langmuir, 2001, 17, 5714–5716 CrossRef CAS.
- S. Yamada, T. Akiyama, S. Inoue and T. Misawa, Jpn. Pat., 2003-123863, 2003.
- The surface coverage of RuVS in (RuVS + PcS)/Au was evaluated from the reductive wave of the viologen moiety. The absorption spectrum of (RuVS + PcS)/Au is very complicated, so identification of the PcS moiety proved difficult. In order to evaluate the relative ratio of surface coverage in the mixed monolayer assembly, gold powder (less than 20 mesh, 0.1 g), and 4 ml of a MeCN–CH2Cl2 solution containing identical concentrations (2 × 10−6 mol dm−3) of [RuVS]2 and [PcS]2 were stirred in the glass cell. The coverage ratio of RuVS and PcS on gold powder was evaluated from the decrease in the absorption intensities of RuVS and PcS (RuVS ∶ PcS = 3.1 ∶ 1). From this ratio and the reductive wave due to the viologen moiety in (RuVS + PcS)/Au, the surface coverage of both molecules in (RuVS + PcS)/Au was evaluated.
| This journal is © The Royal Society of Chemistry and Owner Societies 2004 |