Multiphoton-excited luminescence of a lanthanide ion in a protein complex: Tb3+ bound to transferrin
Gaye F.
White
,
Konstantin L.
Litvinenko
,
Stephen R.
Meech
,
David L.
Andrews
and
Andrew J.
Thomson
*
School of Chemical Sciences and Pharmacy, University of East Anglia, Norwich, UK NR4 7TJ
First published on 9th September 2003
Abstract
The Tb(III) complex of the iron-transport protein transferrin (Tb2–Tfr) exhibits strongly sensitised, sharp line luminescence from f–f states following multiphoton excitation via two tyrosinate residues directly co-ordinated to the lanthanide ion. Using an ultrafast Ti:sapphire laser system, a quadratic dependence of the Tb(III) luminescence intensity was observed on excitation with photons at 503 and 566 nm, and a cubic dependence with photons at 800 nm. The two-photon cross-sections at 503 and 566 nm are 7.4 × 10−50 and 0.37 × 10−50 cm4 s photon−1 mol−1, respectively, which compare favourably with values reported for the green fluorescent protein. Three-photon excitation at 800 nm gives rise to a Tb(III) emission spectrum with excellent signal to noise ratios. These results lead to a proposal that if a Tb(III)–protein complex with similar luminescent properties could be formed in vivo, an intra-cellular imaging system that uses multiphoton-excited, long-lived lanthanide ion luminescence could be developed. This offers the prospect of multiphoton imaging in tight focal planes using sharp line emission with long lifetimes for wavelength and time discrimination against background fluorescence.
Introduction
In recent years, the excitation of fluorescence by multiphoton processes has attracted much attention through the increasing availability of suitable ultrafast lasers. In particular, with sources such as the Ti:sapphire laser delivering ultrashort pulses of high instantaneous intensity, but low average power, two-photon fluorescence has become a tool of strategic importance for biomolecular spectroscopy. This has led to applications in the imaging of biological samples using optical microscopy.1 Two-photon excitation (2PE) entails the simultaneous absorption of two photons, each having half the energy of the gap between the ground and excited state. The general relationship between the incident light intensity, I, and the probability of absorption depends on Im, where m is the number of photons simultaneously absorbed.2,3 As such, multiphoton processes have a number of advantages for the imaging of biological specimens. Absorption takes place only in the region of highest intensity in the incident beam, thus giving rise to highly localised emission, permitting measurements of three-dimensional images without recourse to confocal optics.4 The use of long wavelengths for excitation allows greater depth penetration of scattering and absorbing samples for better depth profiling.5 Photolytic damage is also greatly reduced, as photo-excitation only occurs significantly at the focal point, not throughout the sample. However, these advantages can only be successfully realised for bio-imaging with dyes that have high multiphoton absorption cross-sections, are resistant to photo-degradation and can be introduced into biological cells. The green fluorescent protein (GFP), and its mutant forms, have been widely applied because the chromophore can be expressed within cells from gene fusions.6 GFP has also proved to be advantageous for two-photon fluorescence microscopy, because of its relatively large two-photon absorption cross-section.6,7At high levels of irradiance, the captured energy of two photons can migrate to an acceptor, which is thereby excited above the input photon energy, as evidenced by short-wavelength fluorescence. In biological materials, this process combines the advantages of multiphoton excitation with fluorescence resonance energy transfer (FRET), a technique that has emerged as a significant method in its own right for elucidation of tertiary structure and protein-folding mechanisms.8 The potential of two-photon resonance energy transfer was identified several years ago and the theory subsequently developed specifically for protein–chromophore complexes.9,10 Experiments on two-photon FRET fall into two main categories, mostly conducted on biomolecular systems. Innovative work by Fleming et al. on purple bacteria first employed the technique as a means of delivering excitation energy to bacteriochlorophyll directly from the first excited singlet state of ancillary carotenoid pigments.11,12 Subsequent work by Lakowicz et al. exhibited two- and three-photon FRET in a variety of nucleic acids and proteins with lanthanide fluorophores, firmly establishing the groundwork for applications in biochemistry and cellular imaging.13 Two-photon FRET studies with the red fluorescent protein as acceptor have been conducted by Miyawaki et al.,14 and the first imaging applications, on live human cells, were recently reported by Xu et al.15
Lanthanide reporter ions which are luminescent in the aqueous phase prove especially useful in FRET, and in protein arrays in high throughput screens.16–18 Their utility in this respect stems from their property of having extremely sharp emission spectra, with features readily discernible from the much broader bands associated with intrinsic protein chromophores. The ion luminescence arises from formally forbidden transitions between partially filled f orbitals that are shielded from the environment by filled shells, 6s2 and 5p6. This results in narrow spectral bandwidths that lack long series of vibrational side-bands and display small crystal field effects. These ions also have long luminescence lifetimes. These properties enable wavelength and time discrimination against the high background fluorescence often present in biological cells, and they have been exploited in a variety of biological assays.19–21 Tb3+ and Eu3+ are the most useful lanthanides for such applications, since they emit in the visible region with millisecond decay times. However, because of the forbidden nature of the f–f transitions, the single photon absorption coefficients are extremely low. This problem can, however, be overcome by sensitisation of the luminescence through co-ordination with ligands exhibiting suitably intense absorption bands at energies that permit efficient energy transfer to the f–f manifold of an acceptor lanthanide ion.22,23
Tb3+ is most often selected as a luminescent label for proteins because its luminescence can be sensitised by energy transfer from the intrinsic protein fluorophores of the aromatic amino acids, tyrosine, tryptophan, and phenylalanine.20 The efficiency of this transfer in Tb–protein complexes depends on the distance between the sensitising amino acid and the co-ordinated metal ion.16 However, when Tb3+ is directly co-ordinated to a tyrosinate residue, highly efficient sensitisation of the lanthanide ion emission takes place. Human serum transferrin (Tfr) provides an exemplar of this effect. This protein, involved in the transport of Fe3+ in the bloodstream, consists of two globular lobes, the C- and N-lobes, both of which bind Fe3+ with very high affinity, log K ≈ 20.24 For Fe3+ these co-ordination sites are octahedral, with four ligands supplied by the protein, i.e. two from tyrosinate residues, one from an aspartate residue and one from a histidine residue, plus two ligands supplied by an exogenous, bidentate carbonate ion.24Fig. 1 shows one Fe3+ binding site in the N-lobe domain of recombinant transferrin. (Tf/2N).25
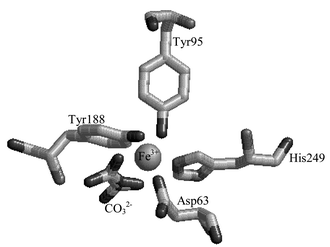 | ||
| Fig. 1 Representation of Fe3+ site in recombinant N-lobe transferrin Tf/2N, (Brookhaven ID 1A8E).25 | ||
Tfr and Tf/2N bind much more weakly to lanthanide ions than to Fe3+ by a factor of ca. log K = 13.24 Typical equilibrium constants reported are: for Gd2–Tfr, log K = 6.8; for Nd2–Tfr, log K1 = 7.3 and log K2 = 5.3; for Sm2–Tfr, log K1 = 8.4 and log K2 = 6.6.26,27 Harris et al. followed the binding of Tb3+ to C-terminal monoferric Tfr and N-terminal monoferric Tfr, and found log K = 9.0 for the C-lobe binding site and log K = 6.4 for the N-lobe site.28 These are conditional binding affinities, at pH 7.5, in the presence of ambient HCO3−. The determination of metal–Tfr binding constants is complicated by the presence of two similar binding sites and the pH and [HCO3−] dependence of the values. Raising the pH from 7 to 9 increases the affinity of transferrin for lanthanide ions.26–29 In the present study, experiments were carried out at pH 9. Under these conditions, it is possible to observe ligand-sensitised Tb3+ luminescence, excited via the protein absorption, that is 105 more intense than that observed for aqueous Tb3+.30,31
Recent evidence from EXAFS studies shows that the co-ordination number of Tb3+ is 8 in Tb2–Tfr, compared with 6 in Fe2–Tfr.31,32 Photophysical studies, comparing the effects of H2O and D2O on the Tb2–Tfr luminescence spectrum, give evidence that H2O is directly co-ordinated to Tb3+ in the protein complex.31,32 These results suggest that, in Tb2–Tfr, each metal ion has four ligands supplied by the protein plus two from a bidentate carbonate ion, as in Fe2–Tfr, with two additional H2O ligands giving the co-ordination number of 8.31Fig. 2 shows the structure proposed for the metal ion binding sites in Tb2–Tfr.
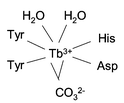 | ||
| Fig. 2 Schematic representation of the proposed structure of the metal ion binding sites in Tb2–Tfr. | ||
The present work aims to investigate the multiphoton excitation properties of the luminescence of the Tb2–Tfr complex in order to assess its potential as a chromophore and fluorophore suitable for bio-imaging.
Materials and methods
Sample preparation
In order to ensure the Tb2–Tfr binding affinity was assessed in the absence of competing metal ions, human serum apo-transferrin (apo-Tfr) was purchased from Sigma and subjected to dialysis with EDTA followed by the pH 9 buffer. The buffer, 10 mM HEPES and 10 mM TAPS at pH 9, was prepared in MilliQ H2O and passed through a column containing Chelex 100. Solutions of 10 mM Tb3+ were freshly prepared using Tb(NO3)3·6H2O (99.999%), obtained from Aldrich Inorganics and Organometallics. The amount of Tb3+ required was added to the protein solution dropwise, using a Hamilton syringe, with stirring, to aid uptake without precipitation.One-photon excitation experiment
One-photon-excited (1PE) luminescence studies were carried out on an ISA FluoroMax-2 fluorimeter. The light source was a 150 W continuous, ozone-free xenon lamp. Excitation spectra were recorded on a Perkin Elmer LS50 luminescence spectrometer. Excitation and emission band-passes were set at 5 nm in both cases.Multiphoton excitation experiment
The laser source for two-photon-excited luminescence was a non-collinear optical parametric amplifier (NOPA; Clark-MXR, Inc., Dexter, MI, USA) pumped by a regeneratively amplified Ti:sapphire laser (CPA-1000, Clark-MXR, Inc.). After attenuation, 1 kHz, 200 µJ, 100 fs pulses at 800 nm were used to seed the NOPA. A combination of continuum generation and parametric amplification allows the NOPA output to be continuously tuned over wavelengths from 500 to 650 nm with no change in optics. This covers the wavelengths required for two-photon excitation of the lowest energy electronic transition of proteins (250–325 nm). After compressing the NOPA output in a prism compressor, 60–70 fs pulses of 1–4 µJ in energy (depending on wavelength) were used for 2PE. A 5 cm lens focused the laser radiation into a 1 cm square quartz cuvette containing the sample. For intensity-dependent measurement, the intensity was controlled by neutral density filters The luminescence was collected in a direction perpendicular to the excitation radiation by focusing the emitted light onto the entrance slit of a computer-controlled ISA 270M imaging monochromator. The luminescence spectra were recorded by a multichannel detector with a spectral resolution of 6 nm. For three-photon excitation (3PE), the 2PE geometry was used, but the direct 800 nm output of the CPA-1000 was used as the excitation source. In this case, pulse energies were in the range 0.1 to 20 µJ before focusing; the lower limit being determined by the accuracy of the power meter and the upper limit by the observation of competing nonlinear effects.Results
One-photon excitation
Fig. 3 shows one-photon-excited emission spectra of 100 µM apo-Tfr, with and without Tb3+ added.![Emission spectra, one-photon excited at 283 nm, of apo-Tfr (100 µM at pH 9) with Tb3+ solution added to give 600 µM Tb3+
(solid line), 200 µM Tb3+
(dash-dot line), 60 µM Tb3+
(dashed line), 2 µM Tb3+
(short dash line), and 0 µM Tb3+
(dotted line). The arrow indicates the direction of increasing [Tb3+].](/image/article/2004/PP/b306760b/b306760b-f3.gif) | ||
| Fig. 3 Emission spectra, one-photon excited at 283 nm, of apo-Tfr (100 µM at pH 9) with Tb3+ solution added to give 600 µM Tb3+ (solid line), 200 µM Tb3+ (dash-dot line), 60 µM Tb3+ (dashed line), 2 µM Tb3+ (short dash line), and 0 µM Tb3+ (dotted line). The arrow indicates the direction of increasing [Tb3+]. | ||
The excitation wavelength of 283 nm matches an absorption maximum of the protein. Apo-Tfr shows one broad emission band centred at 330 nm, attributed to the fluorescence of the aromatic amino acids.3 Tb3+ luminescence is characterised by seven narrow emission bands at ca. 490, 545, 585, 623, 651, 670, and 681 nm corresponding to transitions from the 5D4 excited state to the 7F6, 7F5, 7F4, 7F3, 7F2,7F1, and 7F0 components of the ground states, respectively.19,33,34 The first three transitions lie within the wavelength region of the fluorimeter's detector and are clearly observed in the fluorescence spectrum of Tfr with 2 to 600 µM Tb3+ added. In each case, the 5D4 → 7F5 transition is at 548 nm, and the 5D4 → 7F6 and 5D4 → 7F4 transitions are split into two major peaks. This emission profile matches that previously reported for complexes formed between Tb3+ and Tfr, and Tb3+ and Tf/2N.28,30,31 Under the same concentration conditions, no luminescence was observed from aqueous Tb3+ alone.
The protein fluorescence intensity at 330 nm decreased as Tb3+ was added to Tfr. When the ratio of Tb3+ to Tfr was greater than 2, the emission intensity at 330 nm was 14% lower than that recorded from the same concentration of apo-Tfr. The emission intensities at 490 and 548 nm increased with the concentration of Tb3+ until the ratio of Tb3+ to Tfr approached 2, and then increased more slowly as excess Tb3+ was added. Fig. 4 shows the results of a conditional binding study where Tb3+ was added incrementally to 0.15 µM apo-Tfr in 10 mM HEPES and 10 mM TAPS at pH 9. The protein-sensitised Tb3+ emission intensity at 548 nm, excited at 283 nm, was used to monitor the formation of the Tb–protein complex. Concentrations were chosen to maintain Tb3+ additions below the solubility limits of Tb3+ with OH− and HCO3−, reported to be ca. 10−7 M at pH 7.4 in the presence of ambient HCO3−. However, it is possible that the formation of insoluble Tb3+ salts was competing with the formation of Tb2–Tfr towards the end of the titration experiment. This may explain why only approximately 90% of the possible binding capacity was achieved. The simple binding algorithm (eqn. 1) was used to estimate the overall binding constant (log K = 7.14 ± 0.27) and the binding capacity (1.75 ± 0.06 Tb3+ per protein molecule) of transferrin for Tb3+ under these conditions. This analysis does not take account of differences between the metal ion binding sites in transferrin.
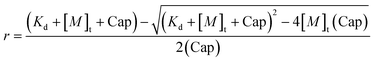 | (1) |
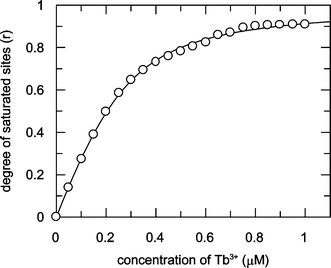 | ||
| Fig. 4 Simple binding algorithm fit (log K = 7.14 ± 0.27 for a binding capacity of 1.75 ± 0.06 Tb3+ per protein molecule) to the formation of 0.15 µM Tb2–Tfr at pH 9. The degree of saturated sites (r) was monitored as the increase in emission intensity at 548 nm, excited at 283 nm, as Tb3+ was added to 0.15 µM apo-Tfr in 10 mM HEPES and 10 mM TAPS at pH 9. | ||
Fig. 5 compares the excitation spectrum of Tb2–Tfr (monitoring Tb3+ emission at 548 nm) with the excitation spectra of tyrosine at pH 10 (tyrosinate), tyrosine at pH 7 and apo-Tfr (monitoring amino acid emission at 345, 305, and 335 nm, respectively). The profile of the Tb2–Tfr excitation most closely matches that of tyrosinate excitation. This is consistent with the postulate that energy transfer from tyrosinate residues co-ordinated to the metal ion in Tb2–Tfr is involved in the excitation of Tb3+ luminescence.
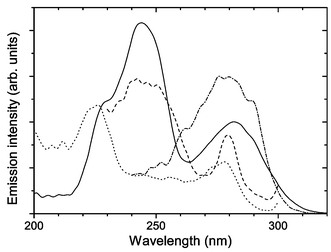 | ||
| Fig. 5 One-photon excitation spectra of Tb2–Tfr (1 µM at pH 9) monitoring Tb3+ emission at 548 nm (solid line), tyrosine (5 µM at pH 10) monitoring fluorescence emitted at 345 nm (dashed line), tyrosine (2 µM at pH 7) monitoring fluorescence emitted at 305 nm (dotted line), and apo-Tfr (3 µM at pH 9) monitoring fluorescence emitted at 335 nm (dash-dot line). | ||
Two-photon excitation
Three different wavelengths of excitation radiation were chosen to study 2PE luminescence from apo-Tfr (100 µM) and Tb2–Tfr (100 µM), prepared by the addition of 200 µM Tb3+ to 100 µM apo-Tfr. Excitation radiation at 503 and 566 nm corresponds to doubled wavelengths of 252 and 283 nm, close to the maxima observed in the Tb2–Tfr 1PE spectrum (Fig. 5). The third wavelength, 620 nm, was chosen to be off-resonant from the excitation spectra of apo-Tfr and Tb2–Tfr. The resulting 2PE luminescence spectra are presented in Fig. 6, where the intensities are reported relative to those recorded on excitation at 503 nm.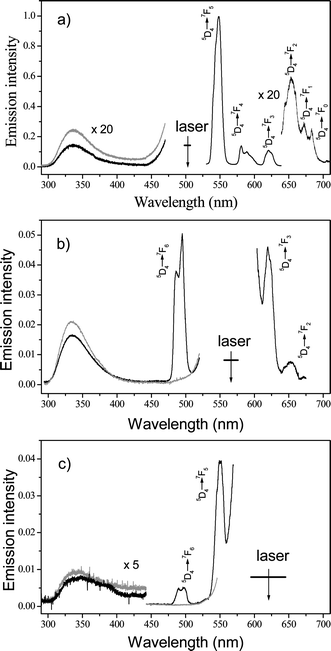 | ||
| Fig. 6 Two-photon excitation spectra of Tb2–Tfr (100 µM at pH 9; black line) and apo-Tfr (100 µM at pH 9; gray line): (a) excitation wavelength = 503 nm; (b) excitation wavelength = 566 nm; (c) excitation wavelength = 620 nm. Arrows indicate excitation wavelengths and half-width range of laser. | ||
The apo-Tfr spectra excited at wavelengths of 503 and 566 nm show the broad emission band centred at 330 nm, characteristic of apo-Tfr fluorescence. In the Tb2–Tfr spectra, this peak is reduced in intensity compared to that observed for apo-Tfr and, apart from spectral positions obscured by scattered light in the region of the excitation wavelength, the characteristically narrow bandwidth emission transitions of Tb3+ are clearly observed. In spectra excited at 620 nm, emission peaks at 330, 490, and 548 nm are evident, with intensities 10 to 20 times smaller than those excited at 503 and 566 nm. No trace of Tb3+ emission from 10 mM aqueous solutions could be observed under these experimental conditions. The fact that the intensity of the Tb3+ emission lines is comparable to the emission of the protein, which is attributed mainly to emission from the six tryptophan residues in Tfr, already suggests the potential of Tb2–Tfr for two-photon bio-imaging.
The operation of a 2PE mechanism is proved by measurement of the dependence of the luminescence intensity on the power of the excitation radiation:
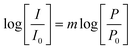 | (2) |
For 2PE, the expected slope in the double logarithmic plot of emission intensity, I, versus the incident laser power, P, where I0 and P0 are normalisation coefficients, is m = 2.35 This is also true for the 2PE followed by an intermolecular energy transfer step.9,10 The results of intensity-dependent experiments for the 2PE luminescence of Tb2–Tfr are shown in Fig. 7. It is apparent that the exponents m exhibit values very close to 2. Minor deviations leading to m < 2 are expected if the power of the incident radiation is sufficient to cause saturation of the luminescence intensity.36 Values of m > 2 are unexpected. It is difficult to comment in more detail on the data for 620 nm (m = 2.4) as the signals are weak. The available intensity range is limited to a maximum of 1 µJ, which adds further uncertainty. One possible cause of the observed positive deviation from m = 2 is that an additional channel for three-photon absorption (plus energy transfer) becomes significant relative to the expected two-photon excitation of the chelated tyrosine, which is only weakly resonant at the longer wavelength.
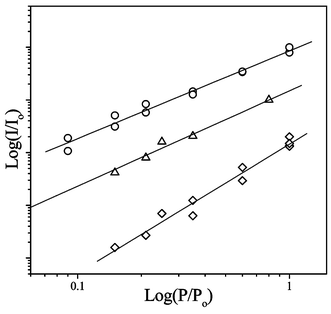 | ||
| Fig. 7 Double logarithmic plots of the two-photon-excited luminescence intensity (I) of Tb2–Tfr (100 µM at pH 9) versus the incident laser power (P), where I0 and P0 are normalisation coefficients. For excitation at 503 nm, monitoring emission at 548 nm, the slope m = 1.66 ± 0.08 (○). For excitation at 566 nm, monitoring emission at 490 nm, m = 1.81 ± 0.06 (△). For excitation at 620 nm, monitoring emission at 548 nm, m = 2.44 ± 0.13 (◇). | ||
The two-photon cross-section of Tb2–Tfr was estimated by comparing the results of this experiment to the emission intensity and quantum yield of a standard of known two-photon cross-section, according to eqn. 3.37
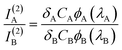 | (3) |
| σ = δϕ | (4) |
The two-photon action cross-section of the sample, σA, can be estimated using eqn. 5.
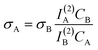 | (5) |
Aqueous tryptophan solution (100 µM) was used as the two-photon cross-section standard. Table 1 gives the quantum yields, two-photon cross-sections and two-photon action cross-sections reported for the aromatic amino acids by Meshalkin.38
As these values were determined under different experimental conditions to those used in this study, using 532 nm as the excitation wavelength, accurate comparisons are not possible and the results determined using this method can only be considered approximate. However, since the wavelength of 532 nm used for the standard falls within the range employed here, and since the same absorption band of tryptophan is involved in both systems, the correction factor should be small and constant (for a particular wavelength). With 566 nm as the excitation wavelength, the two-photon action cross-sections determined for the 330 nm emission band were σapo-Tfr{Tfr(330)} = 1.6 × 10−51 cm4 s photon−1 mol−1 for apo-Tfr and σTb2–Tfr{Tfr(330)} = 1.2 × 10−51 cm4 s photon−1 mol−1 for Tb2–Tfr. For Tb2–Tfr, a value of σTb2–Tfr{Tb(490)} = 3.7 × 10−51 cm4 s photon−1 mol−1 was determined for the 490 nm (5D4 → 7F6) Tb3+ emission line.
Table 2 compares the intensities of the seven Tb3+ emission lines observed after 2PE of Tb2–Tfr using 503, 566, and 620 nm excitation wavelengths. The most intense emission, observed at 548 nm after 2PE at 503 nm, corresponds to the 5D4 → 7F5 Tb3+ transition excited through protein absorbance. This is 20 times more intense than the 5D4 → 7F6 Tb3+ emission band excited at 566 nm; i.e. after 2PE at 503 nm, σTb2–Tfr{Tb(548)} = 7.4 × 10−50 cm4 s photon−1 mol−1.
| Tb3+ transition | λ em/nm | Intensitya | ||
|---|---|---|---|---|
| λ ex = 500 nm | λ ex = 566 nm | λ ex = 620 nm | ||
| a Values normalised to the intensity determined for the 5D4 → 7F6 Tb3+ emission band excited at 566 nm. | ||||
| 5D4 → 7F6 | 490 | — | 1 | 0.1 |
| 5D4 → 7F5 | 548 | 20.8 | — | 0.63 |
| 5D4 → 7F4 | 585 | 2.77 | — | — |
| 5D4 → 7F3 | 620 | 2.23 | 0.5 | — |
| 5D4 → 7F2 | 650 | 0.48 | 0.96 | — |
| 5D4 → 7F1 | 670 | 0.08 | — | — |
| 5D4 → 7F0 | 685 | 0.08 | — | — |
The 2PE emission from a 10 mM aqueous Tb3+ solution was investigated. When the fundamental wavelength was 566 nm, resonant with the 7F6 → 5I8 Tb3+ transition, the emission spectrum (not shown) was barely resolvable from the baseline. This suggests a two-photon action cross-section of ca. 1 × 10−55 cm4 s photon−1 mol−1.
Three-photon excitation
The result of the three-photon excitation experiment on Tb2–Tfr, prepared by the addition of 200 µM Tb3+ to 100 µM apo-Tfr, is shown in Fig. 8.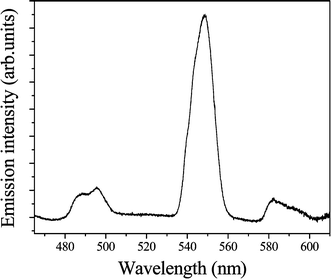 | ||
| Fig. 8 Emission spectrum, three-photon excited at 800 nm, of Tb2–Tfr (100 µM at pH 9). | ||
Emission bands at wavelengths of 490 and 548 nm, characteristic of Tb3+ and excited at 800 nm, are detected with excellent signal to noise ratios. As observed in the 1PE and 2PE experiments, the 5D4 → 7F5 transition of Tb3+ appears at 548 nm and the 5D4 → 7F6 transition as a split band at 490 nm. The double logarithmic plot of emission intensity, I, versus the incident laser power, P, shown in Fig. 9, demonstrates that the luminescence intensity exhibits a cubic dependence on the power of the excitation radiation, m = 3.4 ± 0.3. This is close to the value expected, m = 3. Possible sources of error, and additional nonlinear absorption channels, may lead to values of m greater than predicted, as discussed above.
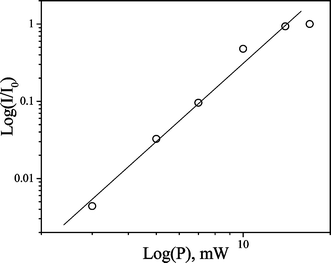 | ||
| Fig. 9 Double logarithmic plot of the three-photon-excited luminescence intensity (I) of Tb2–Tfr (100 µM at pH 9) versus the incident laser power (P), where I0 is a normalisation coefficient. For excitation at 800 nm, monitoring emission at 548 nm, the slope m = 3.4 ± 0.3. | ||
Discussion
One-photon excitation of Tb2–Tfr
The luminescence of Tb3+ in Tb2–Tfr excited by 1PE at 283 nm is 105 times more intense than that of the same concentration of free Tb3+ ions excited at 263 nm.30,31 The observation of intense Tb3+ luminescence together with protein fluorescence in the 1PE emission spectra (Fig. 3) shows that Tb3+ ions are bound to the protein. The reduction in protein fluorescence that accompanies the uptake of Tb3+ indicates that fluorescence energy is transferred from the protein to the metal ion, and the fact that the profile of the Tb2–Tfr excitation most closely matches that of tyrosinate excitation indicates that energy transfer from co-ordinated tyrosinate residues is involved in this process. After comparison of the Tb2–Tfr spectrum with that of free Tb3+, it is noted that the 5D4 → 7F6 and 5D4 → 7F4 transitions are split into two major peaks and the 5D4 → 7F6 transition is shifted from 545 to 548 nm. Protein-sensitised Tb3+ luminescence in Tf/2N has the same spectral profile as that observed in Tb2–Tfr.31 Therefore, the split transitions reflect the asymmetric nature of the Tfr binding sites, rather than an intrinsic difference between the C- and N-lobe binding sites.31The increase in Tb3+ luminescence intensity sensitised by protein binding was used to quantify the formation of the complex by plotting the increase in emission intensity at 548 nm, excited at 283 nm, against the concentration of Tb3+ added to 0.15 µM apo-Tfr (Fig. 4). This experiment estimated that, at pH 9 in the presence of ambient bicarbonate, the binding capacity of Tfr for Tb3+ is approximately 2 per mole of protein, with an average binding affinity of ca. log K = 7. Other workers have reported separate binding affinities, determined at pH 7.5, for the C- and N-lobe binding sites of log K = 9.0 and 6.4, respectively.28 Our studies included binding affinity determinations carried out at different pH values between pH 7 and pH 9, see Table 3.
These experiments show that the binding affinity of Tfr for Tb3+ is much reduced below ca. pH 8, which presents an obstacle to the study of such a Tb–protein system in vivo. Also, as Tfr interacts with a wide range of metal ions—particularly Fe3+, with a binding affinity of log K = 20—there is likely to be interference from other metal ions in vivo. However, other workers have investigated the possibility of producing genetically engineered proteins and peptides that have higher affinities and are more selective for lanthanide ions in biological systems.39–44 Tb2–Tfr possesses exemplary luminescent properties that make it suitable for multiphoton imaging. Future studies will investigate the preparation of a system to achieve tight, selective Tb3+ binding at neutral pH with luminescent properties comparable to those of Tb2–Tfr.
Multiphoton excitation of Tb2–Tfr
Tb3+ luminescence is clearly evident in the 2PE spectra of Tb2–Tfr (Fig. 6), whereas no significant emissions were observed for aqueous Tb3+ excited at 503, 566, and 620 nm, even at a concentration of 10 mM. As with 1PE, the 2PE spectra of apo-Tfr and Tb2–Tfr show that the Tfr fluorescence is quenched as Tb3+ is bound. These observations indicate that energy transfer from Tfr to co-ordinated Tb3+ also dominates the 2PE emission. It is more difficult to study the Tb2–Tfr emission profiles after 2PE because of interference due to scattering of excitation radiation. However, using two excitation wavelengths, 503 and 566 nm, it is possible to observe the emission peaks expected for ligand-sensitised Tb3+ luminescence together with the protein fluorescence, as for 1PE. The results of the three-photon excitation experiment (Fig. 8) also give evidence for energy transfer from Tfr to co-ordinated Tb3+. Using 800 nm as the excitation wavelength, ligand-sensitised emissions at 490, 548, and 585 nm are clearly visible.Examination of the excitation profiles of the Tb2–Tfr luminescence provides further evidence that ligand-sensitised Tb3+ luminescence occurs after 2PE in the same manner as observed after 1PE. From Table 2, it can be estimated that the ratio of the 2PE luminescence intensities of Tb2–Tfr excited at 503, 566, and 620 nm is 100 ∶ 25 ∶ 3. This ratio was calculated by taking into account the power of the excitation radiation and the integrating time of the multichannel detector. The result compares well with the 1PE spectrum of Tb2–Tfr, shown in Fig. 5, where the 1PE fluorescence intensity excited at 250 nm is twice that of the luminescence excited at 283 nm. More quantitative agreement between the two measurements is not expected, as 1PE and 2PE probabilities depend on different molecular parameters. Even where only the ground and one excited state are considered, the 1PE rate depends on the square of the transition moment, whereas 2PE depends quadratically on the product of that transition moment and the difference between the ground and excited state static moments.45 Indeed, for systems whose optical response is dominated by one excited state, as here, a large shift in dipole moment between the ground and excited state would lead to a more efficient 2PE cross-section and such effects could account for the efficient 2PE cross-sections observed in the Tb2–Tfr system.
Comparisons with multiphoton-excited luminescence in other systems
2PE luminescence microscopy of biological samples requires luminescent dyes that have high two-photon action cross-sections and exhibit distinctive luminescence that can be distinguished from the background fluorescence of biological samples. Table 4 compares the experimental σ values determined for 2PE Tb3+ luminescence in the Tb2–Tfr complex with typical σ values reported in the literature for 2PE fluorophores used in biochemical studies.7 Using the optimum conditions for Tb2–Tfr detection of emission at 548 nm after 2PE at 500 nm gave σTb2–Tfr{Tb(548)} = 7.4 × 10−50 cm4 s photon−1 mol−1. This two-photon action cross-section is of the same order of magnitude as those reported by Xu et al. for the commonly used fluorescent dyes DAPI and Coumarin 307, and the green fluorescent protein analogues wild-type GFP and S65T, used as fluorescent labels in genetic experiments.7 Such systems have already been successfully applied in 2PE luminescence microscopy of biological samples.6| Fluorophore | λ ex/nm | σ at emission maximum/1050 cm4 s photon−1 mol−1 | Reference |
|---|---|---|---|
| DAPI | 700 | 0.4 | 7 |
| Coumarin 307 | 800 | 10.0 | 7 |
| GFP (wild-type) | 800 | 6.5 | 7 |
| GFP (S65T) | 920 | 8.0 | 7 |
| Apo-Tfr | 566 | 0.16 | This study |
| Tb2–Tfr | 566 | 0.37 | This study |
| Tb2–Tfr | 500 | 7.4 | This study |
| Aqueous Tb3+ | 566 | 1 × 10−5 | This study |
| Gd3+ in LaF3 | 490 | 2 × 10−5 | 48 |
The σ value determined for aqueous Tb3+ solution, included in Table 4, is an approximate value estimated from the very low emission intensity observed at 490 nm by 2PE at 566 nm. In terms of the energetics, 2PE at 566 nm corresponds to 1PE at 283 nm, which closely matches the energy required for the 7F6 → 5I8 and 7F6 → 5F5 absorption transitions involving Tb3+ 4f electrons.19 Previous studies of lanthanide ions, namely, solid-state forms of Eu3+ and Gd3+, reported that 2PE emission intensities were very low due to the forbidden nature of second-order f–f transitions.46–48 For comparative purposes, an estimate of the TP cross-section reported for Gd3+ in LaF3 is given in Table 4.48 The 2PE σ value determined for Tb2–Tfr is approximately 105 times greater than that of aqueous Tb3+. This shows that, as with 1PE, excitation via energy transfer from the protein ligand greatly sensitises the observed Tb3+ luminescence. Fig. 6 shows that the intensity of Tb3+ luminescence excited through the protein is sensitised to such a degree that it is of the same order as the intensity of the directly excited fluorescence of the protein. Although 2PE at 566 nm excites intense protein fluorescence centred at 330 nm, the profile of the Tb3+ luminescence can be observed in the longer wavelength region of the emission spectrum, with its characteristically positioned, distinctively narrow emission bands clearly separated from the protein fluorescence. Thus, in terms of its absorption cross-section and emission profile, 2PE of Tb2–Tfr should be highly suitable for multiphoton microscopy applications, especially as the distinctive Tb3+ luminescence is emitted in a different spectral region to the background fluorescence of proteins likely to be encountered in biological environments. Indeed, background fluorescence problems could further be eliminated by using time-gated microscopy to select for the longer lifetime Tb3+ luminescence.49,50
Piszczek et al. have demonstrated multiphoton, ligand-enhanced excitation of lanthanide ions complexed to a range of fluorescent chelators, including Tb3+ complexes of the nucleotide guanosine diphosphate (GDP) and the calcium ion binding protein, troponin C.13 Their pulsed-laser system delivered light in the 770 to 800 nm region, corresponding to three-photon excitation for the Tb–GDP and Tb–troponin C complexes. Multiphoton action cross-sections were not reported, but solutions approaching 1 mM were required to achieve spectra where the Tb3+ peaks were only three times the intensity of the background noise. For the 3PE spectrum of 100 µM Tb2–Tfr, shown in Fig. 6, the major emission peak is 75 times greater in intensity than the background noise. It was reported that for 1PE, the relative intensity of Tb–GDP and Tb–troponin C luminescence was 103 times higher than that observed for aqueous Tb3+, whereas the Tb3+ luminescence enhancement factor for 1PE of Tb2–Tfr is approximately 100 times greater than this. Although the protein, troponin C, has tyrosine residues in the region of the calcium ion binding sites, none bind directly to the metal ion as in Tfr. The need for a co-ordinated ligand which can be directly excited in order to obtain high quantum efficiencies of sensitisation is clear from the comparison between the results obtained for Tb2–Tfr in this study and those obtained for troponin C by Piszczek et al.13 They also investigated a mutant of troponin C that places a single tryptophan residue close to one of the metal ion binding sites. When excited at 794 nm, the emission intensities observed for the mutant were ten times greater than those observed for the wild-type protein. This verifies that under multiphoton excitation, the protein-sensitised Tb3+ luminescence intensity still depends on the proximity of the sensitising amino acid, as expected.9 In this respect, Tfr is a particularly suitable ligand for Tb3+, as two sensitising aromatic amino acids are directly co-ordinated to each metal ion.
Beyond the high spatial resolution and penetration powers inherent in multiphoton excitation, 2PE of Tb2–Tfr has a distinct advantage over 1PE in its use of wavelengths in the visible region, suitable for microscopes with glass optical components, rather than the UV. On the other hand the excitation wavelengths, 503 and 566 nm, are still removed from the near-infrared region preferred for multiphoton microscopy. The other fluorophores shown in Table 4 are excited in the region 700 to 900 nm, allowing for deeper sample penetration and less photolytic damage; moreover, this region encompasses the optimum operating wavelengths for short pulse Ti:sapphire lasers. This study has shown that if longer wavelength excitation is desirable, detection of 3PE Tb2–Tfr luminescence is tractable with 800 nm as the excitation wavelength (Fig. 8). This 3PE signal was observable with pulse energies as low as 2 µJ. This is still a factor of 1000 greater than the energies used in a typical multiphoton microscopy experiment. However, two factors suggest that the 3PE signal reported here might also be useful in microscopy. First, the repetition rate of the sources used in multiphoton microscopy is 105 greater than used here. Second, the focal point in our experiment was approximately 10 µm in diameter. In a microscope, it is usually <1 µm. The enhanced focus will lead to a greater intensity per pulse and, consequently, a much stronger signal (because of the observed approximate dependence on I3). Both factors suggest that the 3PE signal from Tb2–Tfr will be observable with a conventional multiphoton microscope. Irrespective of the input focusing, the 3PE experiment has the added advantage of intrinsically tighter focus of the resulting image and elimination of the overlap of the excitation radiation with the most intense emission bands of Tb3+, if time gating is not an option. .51,52 Minimisation of photo-damage and photo-degradation is important in luminescent labelling studies and is, of course, one of the major advantages of multiphoton luminescence microscopy. In addition, it has been recognised that lanthanide ion luminescence is particularly resistant to photo-degradation.13 We have investigated the possibility of multiphoton photochemical degradation in our samples and found that even after many hours of irradiation, no change in optical properties was observable.
In conclusion, comparisons between the multiphoton-excited luminescence observed for Tb2–Tfr and that observed in other systems shows that the nature of the metal ion binding sites in Tb2–Tfr makes this a potential system for luminescent bio-imaging applications in terms of its absorption cross-section, emission profile, luminescence lifetime, tight focusing and resistance to photo-degradation. In order to become practicable as an intra-cellular label, however, improvements in binding affinity and selectivity of metal ion complexation in vivo will be required. A further consideration is how the Tb–protein complex will be incorporated into the cell. It is understood that lanthanide ions may interfere with vital processes in cells, particularly those involving Ca2+.53 However, it is reported that concentrations approaching 10−2 M are required before inhibition of bacterial cell growth is observed.53 Until recently, it was thought that lanthanide ions were unlikely to enter living cells, but studies providing evidence for the entry of lanthanide chelates into eukaryotic and prokaryotic cells have now been reported.31,54,55 Two approaches to the intra-cellular placement of a luminescent Tb–protein label are possible: either expression of the apo-protein inside the cell followed by passage of Tb3+ through the cell membrane or uptake of the Tb–protein complex by the cell. A protein with similar properties to Tfr, the Neisserial ferric iron binding protein (Fbp) can be over-expressed and directed to the periplasm in Escherichia coli cells.31,56 In bacterial studies, periplasmic formation of the luminescent Tb–protein complex would have advantages in that Tb3+ would only have to pass through one cell barrier and less interference is likely in the periplasmic space, as opposed to the whole cell. Investigations into the formation of intra-cellular Tb–Fbp in E. coli have been reported.31 Cellular uptake of a lanthanide–Tfr complex has also been studied, and it was reported recently that, in human erythroleukemia K562 cells, Yb2–Tfr was recognised by the transferrin receptor and taken into the cells via that mechanism.57
Acknowledgements
This work was funded by the UK research councils EPSRC and BBSRC (grants 83/B11958 and BI 11192, and a studentship to G. F. W.). Additional support from the Defence Evaluation Research Agency is gratefully acknowledged.References
- K. König, Multiphoton microscopy in life sciences, J. Microsc., 2000, 200, 83–104 CrossRef CAS.
- D. L. Andrews, Lasers in Chemistry, Springer, Berlin, 3rd edn., 1997 Search PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Kluwer Academic/Plenum Publishers, New York, 2nd edn., 1999 Search PubMed.
- R. M. Williams, W. R. Zipfel and W. W. Webb, Multiphoton microscopy in biological research, Curr. Opin. Chem. Biol., 2001, 5, 603 CrossRef CAS.
- D. J. S. Birch, Multiphoton excited fluorescence spectroscopy of biomolecular systems, Spectrochim. Acta, Part A, 2001, 57, 2313 CrossRef CAS.
- R. Y. Tsien and A. Miyawaki, Seeing the machinery of living cells, Science, 1998, 280, 1954–1958 CrossRef CAS.
- C. Xu, W. Zipfel, J. B. Shear, R. M. Williams and W. W. Webb, Multiphoton fluorescence excitation: new spectral windows for biological non-linear microscopy, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 763–10768 CAS.
763–10768 CAS. - C. G. Dos Remedios and P. D. J. Moens, Resonance energy transfer in proteins, in Resonance Energy Transfer, ed. D. L. Andrews and A. A. Demidov, Wiley, New York, 1999, pp. 1–64 Search PubMed.
- P. Allcock and D. L. Andrews, Two photon fluorescence: resonance energy transfer, J. Chem. Phys., 1998, 108, 3089 CrossRef CAS.
- P. Allcock and D. L. Andrews, Biomedical Optical Spectroscopy and Diagnostics/Therapeutic Laser Applications, ed. E. M. Sevick-Muraca, J. A. Izatt and M. N. Ediger, Trends in Optics and Photonics Vol. 22, Optical Society of America, Washington DC, 1998 Search PubMed.
- B. P. Kreuger, J. Yom, P. J. Walla and G. R. Fleming, Observation of the S-1 state of spheroidene in LH2 by two photon excitation, Chem. Phys. Lett., 1999, 310, 57 CrossRef.
- P. J. Walla, J. Yom, B. P. Krueger and G. R. Fleming, Two photon excitation spectrum of light harvesting complex II and fluorescence upconversion after one and two photon excitation, J. Phys. Chem. B, 2000, 104, 4799 CrossRef CAS.
- G. Piszczek, B. P. Maliwal, I. Gryczynski, J. Dattelbaum and J. R. Lakowicz, Multiphoton ligand-enhanced excitation of lanthanides, J. Fluor., 2001, 11, 101 CrossRef CAS.
- H. Mizuno, A. Sawano, P. Eli, H. Hama and A. Miyawaki, Red fluorescent protein from Discosoma as a fusion tag and a partner for fluorescence resonance energy transfer, Biochemistry, 2001, 40, 2502 CrossRef CAS.
- M. G. Xu, B. Crimeen, M. J. Ludford-Menting, X. S. Gan, S. M. Russell and M. Gu, Three dimensional localisation of fluorescence resonance energy transfer in living cells under two photon excitation, Scanning, 2001, 23, 9 Search PubMed.
- P. R. Selvin, Lanthanide based resonance energy transfer, IEEE J. Sel. Top. Quantum Electron., 1996, 2, 1077–1087 CrossRef CAS.
- G. S. Sittampalam, S. D. Kahl and W. P. Janzen, High throughput screening: advances in assay technologies, Curr. Opin. Chem. Biol., 1997, 1, 384–391 CrossRef CAS.
- P. R. Selvin, Principles and biophysical applications of lanthanide based probes, Annu. Rev. Biophys. Biomol. Struct., 2002, 31, 275–302 CrossRef CAS.
- W. DeW. Horrocks Jr. and M. Albin, Lanthanide ion luminescence in co-ordination chemistry, Prog. Inorg. Chem., 1984, 31, 1–104.
- W. DeW. Horrocks Jr., Luminescence spectroscopy for metalloproteins, Methods Enzymol., 1993, 226, 495–533 CrossRef.
- M. Elbanowski and B. Makowska, The lanthanides as luminescent probes of biochemical systems, J. Photochem. Photobiol., 1996, 99, 85–92 Search PubMed.
- H. Takalo, V. Mukkala and L. Merio, Luminescent Tb(III) chelates for protein labelling, effect of triplet state level, Helv. Chim. Acta, 1997, 117, 372–387 CrossRef CAS.
- M. Latva, H. Takalo, V. Mukkala, C. Matachescu, J. C. Rodriguez-Ubis and J. Kankare, Correlation between the lowest triplet state energy level of the ligand and lanthanide(III) luminescence quantum yield, J. Lumin., 1997, 75, 149–169 CrossRef CAS.
- H. Sun, H. Li and P. J. Sadler, Transferrin as a metal ion mediator, Chem. Rev., 1999, 99, 2817–2842 CrossRef CAS.
- R. T. A. MacGillivray, S. A. Moore, J. Chen, B. F. Anderson, H. Baker, A. B. Mason, R. C. Woodworth, G. D. Brayer and E. N. Baker, Crystal structures of recombinant N-lobe human transferrin, Biochemistry, 1998, 37, 7919–7928 CrossRef CAS.
- W. R. Harris, Binding constants for Nd(III) and Sm(III) with transferrin, Inorg. Chem., 1986, 25, 2041–2045 CrossRef CAS.
- O. Zak and P. Aisen, Studies on binding of Gd(III) to transferrin, Biochemistry, 1988, 27, 1075–1080 CrossRef CAS.
- W. R. Harris, B. Yang, S. Abdollahi and Y. Hamada, Steric restrictions on binding of large metal ions to transferrin, J. Inorg. Biochem., 1999, 76, 231 CrossRef CAS.
- W. R. Harris, Equilibrium constants for complexation of metal ions by serum transferrin, Adv. Exp. Med. Biol., 1989, 249, 67–93 Search PubMed.
- C. Luk, Study of metal binding sites in transferrin using lanthanide ions as fluorescent probes, Biochemistry, 1971, 10, 2838–2843 CrossRef CAS.
- G. F. White, Lanthanide Ion Binding Proteins as In Vivo Luminescent Labels, PhD Thesis, University of East Anglia, Norwich, UK, 2002 Search PubMed.
- G. F. White, I. Harvey and A. J. Thomson, unpublished results.
- G. Stein and E. Würzberg, Energy gap law in the solvent isotope effect on radiationless transitions of rare earth ions, J. Chem. Phys., 1975, 62, 208–213 CrossRef CAS.
- S. Sueda, J. Yuan and K. Matsumoto, A homogeneous DNA hybridisation system by using a new luminescence terbium chelate, Bioconjugate Chem., 2002, 13, 200 CrossRef CAS.
- M. A. Albota, C. Xu and W. W. Webb, Two photon fluorescence excitation cross sections of biomolecular probes from 690 to 960nm, Appl. Opt., 1998, 37, 7352 CAS.
- G. A. Blab, P. H. M. Lommerse, L. Cognet, G. S. Harms and T. Schmidt, Two photon excitation action cross-sections of autofluorescent proteins, Chem. Phys. Lett., 2001, 350, 71 CrossRef CAS.
- B. E. Anderson, R. D. Jones, A. A. Rehms, P. Ilich and P. R. Callis, Polarised two photon fluorescence excitation spectra of indole and benzimidazole, Chem. Phys. Lett., 1986, 125, 106 CrossRef CAS.
- Y. P. Meshalkin, Two photon absorption cross sections of aromatic amino acids and proteins, Kvantovaya Elektron., 1996, 23, 551–552 Search PubMed.
- J. J. Falke, E. E. Snyder, K. C. Thatcher and C. S. Voertler, Engineering the ion specificity of an EF-hand like Ca(II) binding site, Biochemistry, 1991, 30, 8690–8697 CrossRef CAS.
- S. K. Drake, K. L. Lee and J. K. Falke, Tuning the equilibrium ion affinity and selectivity of EF-hand Ca(II) binding, Biochemistry, 1996, 35, 6697–6705 CrossRef CAS.
- J. P. MacManus, C. W. Hogue, B. J. Marsden, M. Sikorska and A. G. Szabo, Tb(III) luminescence in synthetic peptide loops from Ca(II) binding proteins, J. Biol. Chem., 1990, 265, 10358–10366 CAS.
- I. D. Clark, J. P. MacManus, D. Banville and A. G. Szabo, Sensitised lanthanide luminescence in an engineered Ca-binding protein, Anal. Biochem., 1993, 210, 1–6 CrossRef CAS.
- M. Siedlecka, G. Goch, A. Ejchart, H. Sticht and A. Bierznski, α-helix nucleation by a calcium binding loop, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 903–908 CrossRef CAS.
- E. Snyder, B. Buoscio and J. J. Falke, Effect of size and charge on metal binding to an EF-hand like site, Biochemistry, 1990, 29, 3937–3943 CrossRef CAS.
- W. J. Meath and E. A. Power, On the importance of permanent moments in multiphoton absorption using perturbation theory, J. Phys. B: At. Mol. Phys., 1984, 17, 763–781 CrossRef CAS.
- R. Francini, U. M. Grassano, M. Tomini, S. Boiko, G. G. Tarasov and A. Scacco, Two-photon excitation spectra of divalent europium in cubic perovskite KMgF3, Phys. Rev. B, 1997, 55, 7579–7584 CrossRef CAS.
- M. C. Downer, C. D. Cordero-Montalvo and H. Crosswhite, Study of new 4f7 levels of Eu(II) in CaF2 and SrF2 using two photon absorption spectroscopy, Phys. Rev. B, 1983, 28, 4931–4943 CrossRef CAS.
- M. C. Downer and A. Bivas, Third and fourth order analysis of the intensities and polarisation dependence of two photon absorption lines of Gd(III) in LaF3 and aqueous solution, Phys. Rev. B, 1983, 28, 3677–3695 CrossRef CAS.
- T. M. Jovin and D. J. Arndt-Jovin, Luminescence digital imaging microscopy, Annu. Rev. Biophys. Biochem., 1989, 18, 271–308 Search PubMed.
- G. Vereb, E. James-Erijman, P. R. Selvin and T. M. Jovin, Temporally and spectrally resolved imaging microscopy of lanthanide chelates, Biophys. J., 1998, 74, 2210–2222 CAS.
- P. D. Higdon, P. Torok and T. Wilson, Imaging properties of high aperture multiphoton fluorescence scanning optical microscopes, J. Microsc., 1999, 193, 127–141 CrossRef.
- R. M. Williams, J. B. Shear, W. R. Zipfel, S Maiti and W. W. Webb, Biophys. J., 1999, 76, 1835–1846 CAS.
- C. H. Evans, Biochemistry of the Elements, Vol. 8: Biochemistry of the Lanthanides, series ed. E. Frieden, Plenum Press, New York, 1990 Search PubMed.
- Y. Cheng, Q. Huo, J. Lu, R. Li and K. Wang, The transport kinetics of lanthanide species in a single erythrocyte probed by confocal laser scanning microscopy, J. Biol. Inorg. Chem., 1999, 4, 447–456 CrossRef CAS.
- Y. Cheng, H. Yao, H. Lin, J Lu, R Li and K. Wang, The events relating to lanthanide ions enhanced permeability of human erythrocyte membrane: binding, conformational change, phase transition, perforation and ion transport, Chemico-Biol. Interact., 1999, 121, 267–289 Search PubMed.
- A. J. Nowalk, S. B. Tencza and T. A. Mietzner, Co-ordination of Fe(III) by Fbp of pathogenic Neisseria, Biochemistry, 1994, 33, 12769–12775 CAS.
- X. Du, T. Zhang, L. Yuan, Y. Zhao, R. Li, K. Wang, S. C. Yan, L. Zhang, H. Sun and Z. Qian, Complexation of ytterbium to human transferrin and its uptake by K562 cells, Eur. J. Biochem., 2002, 269, 6082–6090 CrossRef CAS.
| This journal is © The Royal Society of Chemistry and Owner Societies 2004 |