Fluorescence lifetime imaging (FLIM) of rhodamine 123 in living cells
Herbert
Schneckenburger
*ab,
Karl
Stock
b,
Marco
Lyttek
a,
Wolfgang S. L.
Strauss
b and
Reinhard
Sailer
b
aFachhochschule Aalen, Institut für Angewandte Forschung, Beethovenstr. 1, 73430 Aalen, Germany. E-mail: herbert.schneckenburger@fh-aalen.de
bInstitut für Lasertechnologien in der Medizin und Messtechnik an der Universität Ulm, Helmholtzstr. 12, 89081 Ulm
First published on 9th September 2003
Abstract
A novel setup for fluorescence intensity and lifetime imaging (FLIM) of living cells is reported. Time-resolving techniques are combined with total internal reflection fluorescence microscopy (TIRFM), which permits optical excitation of either plasma membranes or whole cells depending on whether the angle of incidence of the excitation light is greater or smaller than the critical angle for total internal reflection. The method is applied to BKEz-7 endothelial cells incubated with various concentrations of the well established mitochondrial marker rhodamine 123 (R123). Measurements show that only at low concentrations this dye is mainly located within the mitochondria, whereas at higher concentrations an accumulation within the plasma membrane occurs as well. Concomitantly, fluorescence quenching in the mitochondria is observed at high concentrations, probably due to aggregation of the R123 molecules. Therefore, for diagnostic applications the concentration of R123 in the incubation medium should not be above 25 µM.
Introduction
The application of time-resolving fluorescence techniques in biomedical research has been a challenge for many years. Since radiative and non-radiative transitions from the excited molecular state to the ground state are competing, the fluorescence lifetime (given as the reciprocal of the sum of all transition rates) of a molecule is sensitive to numerous parameters, e.g. degree of aggregation or conformation. Therefore, lifetime measurements are appropriate to probe the microenvironment of a fluorescent molecule. Fluorescence lifetime measurements with spatial resolution date back to the 1980s, when cells or tissue samples were scanned first in one1,2 and later in two dimensions.3 Since that time the implementation of time-resolution in laser scanning microscopy (LSM) has been improved continuously,4 and one of the most recent developments consists of a detection device, where fluorescence decay curves are measured for each pixel of a LSM image.5 Parallel developments include spatially resolved phase fluorometry6–8 as well as time-gated image intensifying camera systems. The time-resolution of image intensifiers has recently been increased from about 5 ns9,10 to the picosecond range.11 Concomitantly, detection sensitivities of image intensifying camera systems have been improved, since the detection efficiency decreases when lowering the time gate.Microscopic (lateral and axial) resolution in fluorescence lifetime imaging (FLIM) has so far been in the range of some hundreds of nanometers using either multiphoton microscopy12 or optical sectioning by a structured illumination technique.11 Very recently the axial resolution in time-resolved fluorescence microscopy has been improved using evanescent waves for selective excitation of fluorophores in close proximity to the plasma membrane. Time-gated spectroscopy upon excitation by evanescent waves was applied to single cells,13 but no fluorescence lifetime images have been reported, so far.
Rhodamine 123 (R123) is a probe of the transmembrane potential14 and is accumulated within the inner mitochondrial membrane.15 In addition to its properties of staining mitochondria, a preferential accumulation of R123 in tumour cells16 as well as some photosensitizing properties17 have been described. R123 has also been used to probe the function the respiratory chain located within the inner mitochondrial membrane.18 Following optical excitation of the coenzyme nicotinamide adenine dinucleotide in its reduced form (NADH), the non-radiative energy transfer from NADH to the R123 molecules was determined. According to the Förster mechanism19 this energy transfer is limited to intermolecular distances below 10 nm. Therefore, R123 fluorescence was related to the amount of mitochondrial NADH, which was used as a parameter to probe malfunction of enzyme complexes of the respiratory chain. A superpostion by cytoplasmic NADH or further fluorophores showing a similar emission spectrum could thus be avoided.
In addition to mitochondrial accumulation of R123, some amounts of this dye may be located in further intracellular compartments as well as in the plasma membrane.20 Therefore, the method of total internal reflection fluorescence microscopy (TIRFM)21 was used to excite R123 selectively within the plasma membrane (and adjacent cellular sites) by an evanescent electromagnetic wave, and to compare fluorescence arising from the plasma membrane with R123 fluorescence after illumination of the whole cell. Fluorescence intensity and lifetime images (FLIM) of cultivated cells incubated with various concentrations of R123 were therefore recorded using both types of illumination, to our knowledge for the first time.
Materials and methods
BKEz-7 endothelial cells from calf aorta22 were routinely cultivated in Eagle's minimum essential medium (MEM) supplemented with 10% fetal calf serum (FCS), glutamine (2 mM), and antibiotics (penicillin, streptomycin) at 37 °C and 5% CO2. 150 cells mm−2 were seeded on microscope object slides and grown for 48 h. After that period cells were incubated for 30 min in MEM (same as above, but only with 5% FCS) containing various concentrations of R123 (5, 10, 25, 50 or 100 µM) prior to rinsing with Earle's balanced salt solution (EBSS). When using an incubation period of 30 min R123 concentrations up to 25 µM were reported to be non-cytotoxic.18 For microscopic experiments an open chamber (filled with EBSS) and a 63 × /0.90 water immersion objective lens were used.A fluorescence microscope (Axioplan 1, Carl Zeiss Jena, Germany) was equipped with a novel illumination device,23 which allowed the cells to be illuminated under dark field conditions with a variable angle of incidence Θ. Total internal reflection (TIR) occurred at Θ ≥ 64.6°. Therefore, when using the angles Θ = 62° and Θ = 66° in the experiment, whole cells were illuminated in the first case, whereas the plasma membrane and adjacent cellular sites were illuminated selectively in the second case (with a penetration depth d ≈ 160 nm of the evanescent wave). In all cases the electric field vector was polarized perpendicular to the plane of incidence. As a picosecond light source a modelocked argon ion laser (Innova 90, Coherent, Palo Alto, USA) was used at a wavelength of 476 nm, a pulse duration of 200 ps and a repetition rate of 99 MHz. The repetition rate was reduced to 2.5 MHz by a pulse picker (Pockels cell, LM 0202, LINOS Photonics, Planegg, Germany). A monomode quartz fiber with collimation optics at its entrance and exit (KineFLEX-P-3; Point Source, Southampton, UK) was used to couple excitation light to the illumination device, permitting a small divergence angle of ±0.25° in the plane of the sample. Light pulses with an energy of 40 pJ each (corresponding to an average power of 100 µW) were used to irradiate an elliptic object field with a major axis of about 1 mm and a minor axis of 400 µm. No phototoxicity and almost no photobleaching were observed under these conditions. The electrical output of the modelocker was used to trigger the Pockels cell as well as the time gate of the image intensifier (Picostar HR 12, LaVision, Göttingen) which was coupled to a cooled ICCD camera with 640 × 480 pixels. Time gates between 200 ps and 1000 ps as well as variable delay times with respect to the trigger pulse could be adjusted. The whole setup is depicted in Fig. 1.
 | ||
| Fig. 1 Experimental setup for fluorescence lifetime imaging (FLIM) with variable-angle excitation. | ||
Fluorescence decay kinetics and images were recorded from object fields of 220 × 160 µm, which typically contained 5–10 individual cells in each case. Since the fluorescence maximum of R123 was around 530 nm, a long pass filter for 515 nm was used to avoid the detection of scattered light as well as autofluorescence from the cells. Fluorescence decay kinetics were measured by shifting the time gate in intervals of 200 ps over a scale of 10–20 ns (dwell time: 1 s for each interval), and lifetimes were obtained from mono- or biexponential curve fitting using a least-square fitting algorithm (without deconvolution). For each R123 concentration between 7 and 14 fluorescence decay curves were recorded, and the mean values as well as the standard deviations of fluorescence lifetimes were calculated. In addition, for biexponential decays, which are described by the fluorescence intensity IF(t) = A1e−t/τ1 + A2e−t/τ2, the relative intensity of the component i (i = 1, 2) with fluorescence lifetime τi and amplitude Ai was calculated according to the formula:
| Ii = Aiτi/(A1τ1 + A2τ2) | (1) |
Fluorescence images were recorded within the time intervals of 0.5–1.0 ns (A) and 4.5–5.0 ns (B) after the pulse maximum (see Fig. 2) The acquisition time of each image was 1 s at Θ = 62° and 10 s at Θ = 66°. In addition to the fluorescence intensities IA and IB, the effective fluorescence lifetime τeff could be displayed as an image. The value of τeff was calculated for each pixel from the intensities IA and IB, and the time shift Δt = 4 ns between the intervals A and B using eqn. (2):
| τeff = Δt/ln(IA/IB) | (2) |
 | ||
| Fig. 2 Fluorescence decay curve of BKEz-7 endothelial cells incubated with R123 (50 µM, 30 min) obtained from an image of 220 × 160 µm. Excitation wavelength: λex = 476 nm, detection range: λd ≥ 515 nm, angles of incidence: Θ = 62° (whole cell illumination; upper curve) or Θ = 66° (TIR illumination; lower curve). Fluorescence intensities IA and IB (measured within the time gates A and B) as well as the time shift Δt used for fluorescence lifetime imaging are indicated. | ||
Only when the fluorescence decay curve was monoexponential, τeff corresponded to the real fluorescence lifetime (as measured by decay kinetics); if this curve was biexponential, τeff depended on both lifetimes. For all fluorescence images the background (recorded without laser illumination) was subtracted.
In an additional experiment, the fluorescence intensity IF within a certain time interval close to the maximum of the fluorescence decay curve (width: 1 ns) was determined as a function of the angle Θ of light incidence between 65° and 72° in steps of 1°. As previously shown,23 the intensity of a fluorescence marker IF excited by an evanescent electromagnetic wave can be described by eqn. (3):
| IF(Θ) = FcT(Θ)te−Δ/d(Θ) | (3) |
| IF(Θ) = FcT(Θ)d(Θ)e−Δ/d(Θ) | (4) |
![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) sin2Θ
−
n22)−1/2 the penetration depth of the evanescent wave and T(Θ)
= 4cos2Θ/
[1 −
(n2/n1)2] the transmission factor through the cell/substrate interface, if the electric field vector is polarized perpendicular to the plane of incidence.24 For fitting the experimental data according to a two-layer model (with refractive indices n1
= 1.53 for the glass substrate and n2
= 1.38 for the cytoplasm23), eqns. (3) and (4) were used, and Δ was calculated by a least-square fitting algorithm. Variable-angle experiments were performed using R123 concentrations of 10 or 50 µM.
sin2Θ
−
n22)−1/2 the penetration depth of the evanescent wave and T(Θ)
= 4cos2Θ/
[1 −
(n2/n1)2] the transmission factor through the cell/substrate interface, if the electric field vector is polarized perpendicular to the plane of incidence.24 For fitting the experimental data according to a two-layer model (with refractive indices n1
= 1.53 for the glass substrate and n2
= 1.38 for the cytoplasm23), eqns. (3) and (4) were used, and Δ was calculated by a least-square fitting algorithm. Variable-angle experiments were performed using R123 concentrations of 10 or 50 µM.
Results
Fluorescence decay curves of BKEZ-7 endothelial cells incubated with 50 µM R123 are depicted in Fig. 2. Either whole cells (Θ = 62°) or only plasma membranes (Θ = 66°) were illuminated. The time gates A and B (with intensities IA and IB) as well as the time shift Δt = 4 ns are also indicated. During illumination of whole cells, decay curves were monoexponential with a fluorescence lifetime τ = (2.7 ± 0.3) ns, if R123 concentrations of 5 or 10 µM were used. For high R123 concentrations of 50 or 100 µM, decay curves were biexponential with τ1 = (0.55 ± 0.1) ns and τ2 = (2.7 ± 0.3) ns. Relative fluorescence intensites were I1 = 0.4 ± 0.2 and I2 = 0.6 ± 0.2 for both concentrations. For a R123 concentration of 25 µM only part of the decay profiles could be fitted by two exponentials whereas all other profiles were monoexponential with the decay time τ2. In contrast, when the plasma membrane was illuminated selectively, the decay profiles were always monoexponential with τ = (2.8 ± 0.2) ns at R123 concentrations between 10 and 100 µM. However, at a R123 concentration of 5 µM the fluorescence intensity at Θ = 66° was very low, and decay profiles could not be evaluated reliably. Differences between whole cell illumination (biexponential decay) and TIR illumination (monoexponential decay) can be deduced from Fig. 2.Fig. 3 shows the patterns of fluorescence intensity (measured in the interval A; top) and effective fluorescence lifetime (bottom) of BKEz-7 endothelial cells incubated with 10 µM R123 upon illumination of whole cells. The intensity image shows dark cell nuclei and brightly fluorescent mitochondria, whereas the lifetime image shows an almost homogenous distribution of the effective fluorescence lifetimes all over the cells. Upon selective illumination of the plasma membrane and its adjacent cellular parts, a rather diffuse fluorescence pattern and a few bright spots can be deduced from the intensity image (Fig. 4, top). The bright spots probably arise from parts of those mitochondria, which are in close proximity to the plasma membrane. Again, fluorescence lifetimes (Fig. 4, bottom) were rather homogenous all over the cells with a large noise level resulting from the quotient of low intensities which were used to calculate the effective lifetime according to eqn. (2).
 | ||
| Fig. 3 Fluorescence intensity IA (top) and fluorescence lifetime τeff (bottom; scale: 0–8 ns) of BKEz-7 endothelial cells incubated with 10 µM R123 measured at Θ = 62° (λd ≥ 515 nm; image size: 220 × 160 µm). | ||
 | ||
| Fig. 4 Fluorescence intensity IA (top) and fluorescence lifetime τeff (bottom; scale: 0–8 ns) of BKEz-7 endothelial cells incubated with 10 µM R123 measured at Θ = 66° (λd ≥ 515 nm; image size: 220 × 160 µm). Same object field as in Fig. 3. | ||
After incubation of BKEz-7 cells with 50 µM R123, illumination of the whole cells results in brightly fluorescent mitochondria superimposed by some diffuse fluorescence distributed all over the cells (Fig. 5, top). The corresponding lifetime image (Fig. 5, bottom) shows a homogenous distribution of fluorescence lifetimes in the cytoplasm (and the cell nucleus), but a lower effective lifetime within the mitochondria. This can be deduced from the dark spots in the lifetime image. Upon selective illumination of the plasma membrane (and adjacent cellular sites), fluorescence was distributed all over the cell surface and was considerably higher than after incubation of 10 µM R123 (Fig. 6, top). Again, bright fluorescent spots arising from mitochondria in close proximity to the plasma membrane were identified. The fluorescence lifetime image in this case was homogenous all over the cell surface (Fig. 6, bottom).
 | ||
| Fig. 5 Fluorescence intensity IA (top) and fluorescence lifetime τeff (bottom; scale: 0–8 ns) of BKEz-7 endothelial cells incubated with 50 µM R123 measured at Θ = 62° (λd ≥ 515 nm; image size: 220 × 160 µm). | ||
 | ||
| Fig. 6 Fluorescence intensity IA (top) and fluorescence lifetime τeff (bottom; scale: 0–8 ns) of BKEz-7 endothelial cells incubated with 50 µM R123 measured at Θ = 66° (λd ≥ 515 nm; image size: 220 × 160 µm). Same object field as in Fig. 5. | ||
In addition, the fluorescence intensity excited by the evanescent electromagnetic wave was integrated over images recorded at variable angles of incidence 65 ≤ Θ ≤ 72°. The result for a sample of BKEz-7 cells incubated with 50 µM R123 is depicted in Fig. 7. This Figure also shows the fitted curves for a fluorescence marker of the plasma membrane according to eqn. (3) and for a marker of the cytoplasm according to eqn. (4). Only eqn. (3) provides a reasonable fit with an average distance between the plasma membrane and the glass substrate of Δ = 90 nm. For cells incubated with 10 µM R123 the angular dependence of the fluorescence signal was similar for Θ ≥ 66°, but the fluorescence intensity was considerably lower than for cells incubated with 50 µM R123 (data not shown).
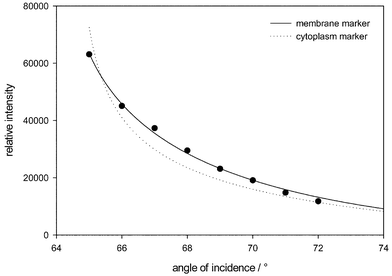 | ||
| Fig. 7 Angular dependence of fluorescence intensity of BKEz-7 endothelial cells incubated with R123 (50 µM) and fitted curves for a marker of either the plasma membrane (according to eqn. (3)) or the cytoplasm (according to eqn. (4)). | ||
Discussion
Fluorescence lifetime imaging revealed to be a valuable method to probe both, the cellular location and the microenvironment of fluorescent molecules. Precision of this method depends on time resolution as well as on the signal/noise ratio of the detection device. After background subtraction the noise level within individual pixels varied between 0 and 20 counts in comparison with measured signals within the time gate A around 4000 counts (whole cell illumination), 400 counts (TIR illumination) or 40 counts (extremely low value in TIR images at low R123 concentrations). Corresponding signals within the time gate B were about 1000, 100 or 10 counts. Without any background a fluorescence lifetime τeff = 2.88 ns is calculated according to eqn. (2) when using the values mentioned above. A maximum background of 20 counts would result in an effective fluorescence lifetime of 2.92, 3.19 or 5.77 ns for the three cases. In other words, the influence of the background on effective fluorescence lifetime is rather low for most of the experiments reported above. However, at very low fluorescence intensities an increase of the mean value and strong fluctuations of τeff are observed, e.g. in the plasma membrane (at low R123 amounts, see Fig. 4) or at cell edges (Fig. 5).As shown above, R123 is preferentially accumulated within the mitochondria of BKEz-7 endothelial cells after incubation with rather low concentrations (5–10 µM). At higher concentrations R123 is increasingly found in further intracellular compartments as well as within or close to the plasma membrane. Location of this dye in further intracellular compartments is deduced from measurements by laser scanning microscopy showing pronounced mitochondrial fluorescence in addition to some diffuse fluorescence all over the cytoplasm (data not shown). Location within the plasma membrane or its close vicinity can be deduced from the angular dependence of fluorescence intensity when using an evanescent electromagnetic wave for excitation. Fluorescence lifetimes are distributed homogenously over the plasma membrane with a time constant around 2.8 ns. The lifetime remains unchanged, when R123 concentrations are increased up to 100 µM. Fluorescence lifetimes measured inside the cells are about the same, but at concentrations above 25 µM, an additional short-lived component (τ ≈ 0.6 ns) is detected. Short-lived fluorescence mainly arises from the mitochondria, as shown by the lifetime image in Fig. 5. Since the fluorescence lifetime τ corresponds to the reciprocal rate of radiative (kr) and non-radiative (knr) transitions from the excited molecular state to the ground state according to eqn. (5):
| τ = (kr + knr)−1 | (5) |
Quenching of R123 fluorescence in mitochondria and accumulation of R123 in further cell membranes (in particular the plasma membrane) at high concentration limits the application of this dye for in vitro diagnostics. For example, for measurements of mitochondrial malfunction using non-radiative energy transfer from the mitochondrial coenzyme NADH to R123 molecules,18,25 the concentration of R123 in the incubation medium should not be above 25 µM.
Selective fluorescence lifetime imaging (FLIM) of the plasma membrane of living cells became possible by a combination of FLIM and total internal fluorescence microscopy (TIRFM). In the near future this combination may become promising for numerous studies, e.g. for measuring dynamics of membrane lipids26 or proteins,27 for the detection of membrane-proximal ion fluxes,28 for imaging of endocytosis or exocytosis,29,30 as well as for the investigation of photosensitizers (with tumour-localizing properties) in close proximity to the plasma membrane.31,32
Acknowledgements
The authors thank C. Hintze, M. Wagner and M. Kretzschmar for technical assistance. The project was supported by the Bundesministerium für Bildung und Forschung (BMBF), grants no. 1706698 and 13N7514.References
- H. Schneckenburger, F. Pauker, E. Unsöld and D. Jocham, Intracellular distribution and retention of the fluorescent components of Photofrin II, Photobiochem. Photobiophys., 1985, 10, 61–67 Search PubMed.
- I. Bugiel, K. König and H. Wabnitz, Investigation of cells by fluorescence laser scanning microscopy with subnanosecond resolution, Lasers Life Sci., 1989, 3, 47–53 Search PubMed.
- E. P. Buurman, R. Sanders, A. Draijer, H. C. Gerritsen, J. J. F. van Veen, P. M. Houpt and Y. K. Levine, Fluorescence lifetime imaging using a confocal laser scanning microscope, Scanning, 1992, 14, 155–159 Search PubMed.
- H. C. Gerritsen, M. A. Asselbergs, A. V. Agronskaia and W. G. van Sark, Fluorescence lifetime imaging in scanning microscopes: acquisition, speed, photon economy and lifetime resolution, J. Microsc., 2002, 206, 218–224 CrossRef CAS.
- W. Becker, A. Bergmann, C. Biskup, L. Kelbaukas, T. Zimmer and N. Klöcker, K. Benndorf, High resolution TCSPC lifetime imaging, in Multiphoton Microscopy in the Biomedical Sciences III, eds. A. Periasamy and P. T. C. So, Proc. SPIE, Vol. 4963, Bellingham, USA, 2003, in press Search PubMed.
- J. R. Lakowicz and K. Berndt, Lifetime-selective fluorescence imaging using an rf phase-sensitive camera, Rev. Sci. Instrum., 1991, 62, 1727–1734 CrossRef CAS.
- T. W. J. Gadella, T. M. Jovin and R. M. Clegg, Fluorescence lifetime imaging microscopy (FLIM): Spatial resolution of microstructures on the nanosecond time scale, Biophys. Chem., 1993, 48, 221–239 CrossRef CAS.
- A. Squire, P. J. Verveer and P. I. H. Bastiaens, Multiple frequency fluorescence lifetime imaging microscopy, J. Microsc., 2000, 197, 136–149 CrossRef.
- M. Kohl, J. Neukammer, U. Sukowski, H. Rinneberg, D. Wöhrle, H.-J. Sinn and A. Friedrich, Delayed observation of laser-induced fluorescence for imaging of tumours, Appl. Phys. B, 1993, 56, 131–138.
- H. Schneckenburger, K. König, T. Dienersberger and R. Hahn, Time-gated microscopic imaging and spectroscopy in medical diagnosis and photobiology, Opt. Eng., 1994, 33, 2600–2606.
- M. J. Cole, J. Siegel, S. E. Webb, R. Jones, K. Dowling, M. J. Dayel, D. Parsons-Karavassilis, P. M. French, M. J. Lever, L. O. Sucharov, M. A. Neil, R. Juskaitis and T. Wilson, Time-domain whole-field fluorescence lifetime imaging with optical sectioning, J. Microsc., 2001, 203, 246–257 CrossRef CAS.
- M. Straub and S. W. Hell, Fluorescence lifetime three-dimensional microscopy with picosecond precision using a multifocal multiphoton microscope, Appl. Phys. Lett., 1998, 73, 1769–1771 CrossRef CAS.
- H. Schneckenburger, K. Stock, W. S. L. Strauss, J. Eickholz and R. Sailer, Time-gated total internal reflection fluorescence spectroscopy (TG-TIRFS): application to the membrane marker laurdan, J. Microsc., 2003, 211, 30–36 CrossRef CAS.
- R. K. Emaus, R. Grunwalg and J. LeMasters, Rhodamine 123 as a probe of transmembrane potential in isolated rat liver mitochondria: spectral and metabiolic properties, Biochim. Biophys. Acta, 1986, 850, 436–448 CrossRef CAS.
- L. V. Johnson, M. L. Walsh and L. B. Chen, Localization of mitochondria in living cells with rhodamine 123, Proc. Natl. Sci. USA, 1980, 77, 990–994 CAS.
- K. Nadakavukaren, J. Nadakavukaren and L. B. Chen, Increased rhodamine 123 uptake by carcinoma cells, Cancer Res., 1985, 45, 6093–6099 CAS.
- P. Moliè, R. Santus, M. Bazin, E. Kohen, V. Carillet, F. Bon, J. Rainasse and L. Dubertret, Is Rhodamine 123 a photosensitizer?, Photochem. Photobiol., 1990, 52, 703–710 CAS.
- H. Schneckenburger, M. H. Gschwend, W. S. L. Strauss, R. Sailer, M. Kron, U. Steeb and R. Steiner, Energy transfer spectroscopy for measuring mitochondrial metabolism in living cells, Photochem. Photobiol., 1997, 66, 33–41.
- T. Förster, Zwischenmolekularer Übergang von Elektronenanregungsenergie, Z. Elektrochem., 1960, 64, 157–164 Search PubMed.
- J. L. Weaver, P. S. Pine, A. Aszalos, P. V. Schoenlein, S. J. Currier, R. Padmanabhan and M. M. Gottesman, Laser-scanning and confocal microscopy of daunorubicin, doxorubicin and rhodamine 123 in multidrug-resistant cells, Exp. Cell Res., 1991, 196, 323–329 CAS.
- D. Axelrod, Cell-substrate contacts illuminated by total internal reflection fluorescence, J. Cell Biol., 1981, 89, 141–145 CAS.
- W. Halle, W.-E. Siems, K. D. Jentzsch, E. Teuscher and E. Göres, Die in vitro kultivierte Aorten-Endothelzelle in der Wirkstofforschung – Zellphysiologische Charakterisierung und Einsatzmöglichkeiten der Zellinie BKEz-7, Pharmazie, 1984, 39, 77–81 Search PubMed.
- K. Stock, R. Sailer, W. S. L. Strauss, M. Lyttek, R. Steiner and H. Schneckenburger, Variable-angle total internal reflection fluorescence microscopy (VA-TIRFM): realization and application of a compact illumination device, J. Microsc., 2003, 211, 19–29 CrossRef CAS.
- W. M. Reichert and G. A. Truskey, Total internal reflection fluorescence (TIRF) microscopy (I) Modelling cell contact region fluorescence, J. Cell Sci., 1990, 96, 219–230 Search PubMed.
- M. H. Gschwend, R. Rüdel, W. S. L. Strauss, R. Sailer, H. Brinkmeier and H. Schneckenburger, Optical detection of mitochondrial NADH content in human myotubes, Cell. Mol. Biol., 2001, 47, OL95–OL104 Search PubMed.
- T. Parasassi, E. K. Krasnowska, L. Bagatolli and E. Gratton, Laurdan and prodan as polarity-sensitive fluorescent membrane probes, J. Fluoresc., 1998, 4, 65–373.
- S. E. Sund and D. Axelrod, Actin dynamics at the living cell submembrane imaged by total internal reflection fluorescence photobleaching, Biophys. J., 2000, 79, 1655–1669 CAS.
- G. M. Omann and D. Axelrod, Membrane-proximal calcium transients in stimulated neutrophils detected by total internal reflection fluorescence, Biophys. J., 1996, 71, 2885–2891 CAS.
- W. J. Betz, F. Mao and C. B. Smith, Imaging exocytosis and endocytosis, Curr. Opin. Neurobiol., 1996, 6, 365–371 CrossRef CAS.
- M. Oheim, D. Loerke, W. Stühmer and R. H. Chow, The last few milliseconds in the life of a secretory granule, Eur. J. Biophys., 1998, 27, 83–98 Search PubMed.
- W. S. L. Strauss, R. Sailer, M. H. Gschwend, H. Emmert, R. Steiner and H. Schneckenburger, Selective examination of plasma associated photosensitizers using total internal reflection fluorescence spectroscopy (TIRFS) – correlation between photobleaching and photodynamic efficacy of protoporphyrin IX, Photochem. Photobiol., 1998, 67, 363–369 CAS.
- R. Sailer, W. S. L. Strauss, H. Emmert, K. Stock, R. Steiner and H. Schneckenburger, Plasma membrane associated location of sulfonated meso-tetraphenylporphyrins of different hydrophilicity probed by total internal reflection fluorescence spectroscopy, Photochem. Photobiol., 2000, 71, 460–465 CAS.
| This journal is © The Royal Society of Chemistry and Owner Societies 2004 |