A novel and facile synthesis of dienals and substituted 2H-pyrans via the Vilsmeier reaction of α-oxo-ketenedithioacetals
Yingchun
Liu
,
Dewen
Dong
*,
Qun
Liu
,
Yimei
Qi
and
Zuo
Wang
Department of Chemistry, Northeast Normal University, Changchun, 130024, P. R. China. E-mail: dewend@yahoo.com
First published on 6th November 2003
Abstract
A novel and facile synthesis of dienals (3a, 3b) and substituted 2H-pyrans (4c, 4d) from a series of α-oxo ketenedithioacetals containing a methyl group adjacent to the carbonyl group (1a–d) via the Vilsmeier reaction has been developed and a mechanism for the reactions has been proposed.
Over the last decades, the Vilsmeier reaction, associated with its mild reaction conditions, commercial viability of the reagents and improved understanding of the reaction mechanism, has proven to be a versatile pathway to the synthesis of various heterocyclic compounds, such as quinolines, indoles, quinazolines, pyridines, and naphthyridines.1–5 On the other hand, α-oxo ketenedithioacetals as the organic synthetic intermediates have been widely used in the formation of heterocycles, aromatic compounds and various valuable reactive intermediates.6–10 The α-oxo ketenedithioacetals owe their potential synthetic applications to their varied intrinsic chemical properties. The presence of the carbonyl functionality and its position in conjugation with the double bond carrying the bis(alkylthio) groups at the β-position places α-oxo ketenedithioacetals among the versatile 1, 3-electrophilic 3-carbon equivalents.6 Meanwhile, we note that the bis(alkylthio) groups as electron-donating groups, may activate at least to a certain extent, the carbonyl group of the α-oxo ketenedithioacetals. Such activation might drive the α-oxo ketenedithioacetals to react with the Vilsmeier reagent, and hence develop new strategies towards the synthesis of heterocycles via the cyclization potential of the resulting halomethyleniminium salts. To the best of our knowledge, the novelty of the process lying in the Vilsmeier reaction of α-oxo ketenedithioacetals is unprecedented, although there are a few reports on the Vilsmeier reactions of α-hydroxy ketenedithioacetals12 and α-oxoketene-N,S-acetals.13 As a continuation of our interest in the chemistry of α-oxo ketenedithioacetals,9,10 we herein wish to report a novel synthetic strategy of dienals and 2H-pyrans directly from α-oxo-ketenedithioacetals via the Vilsmeier reaction.
In this communication, a series of α-oxo ketenedithioacetals 1a–1d (Scheme 1) containing a methyl group adjacent to the carbonyl group was prepared in very high yields (up to 99%) according to our earlier reported procedure.11 The Vilsmeier reactions of 1a–1d were investigated using varied conditions (Schemes 2 and 3), some results are listed in Tables 1 and 2.
 | ||
| Scheme 1 | ||
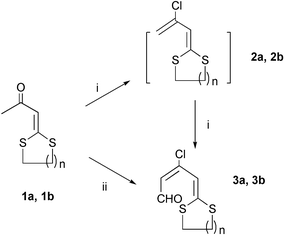 | ||
| Scheme 2 Reagents and conditions: (i) POCl3–DMF (1 equiv.), r.t., 6 h; (ii) POCl3–DMF (2 equiv.), r.t., 12 h. | ||
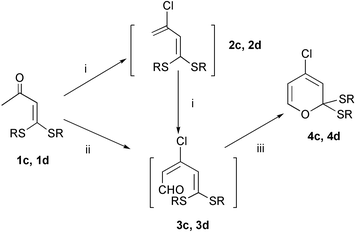 | ||
| Scheme 3 Reagents and conditions: (i) POCl3–DMF (1 equiv.), r.t., 6 h.; (ii) POCl3–DMF (2 equiv.), r.t., 12 h; (iii) diethyl ether, r.t., 48 h. | ||
The initial studies were performed on the reactions of acyclic α-oxo ketenedithioacetals, 1a and 1b, with Vilsmeier reagent (1 equiv.) at 0 °C, respectively. Halogenation products 2a and 2b were detected, however, they were not stable. The addition of another one equivalent of Vilsmeier reagent to the halogenated reaction mixture subsequently resulted in the formation of haloformylation product, dienal (3a or 3b), in very high yield. According to the 1H NMR spectra, the haloformylation product is a mixture of isomers with the trans-isomer as the predominant one (Table 1†). One-pot conversion to dienal 3a or 3b was successfully achieved when 2 equivalents of the Vilsmeier reagent was employed. The above results indicate that the bis(alkylthio) group of the α-oxo ketenedithioacetals can activate the carbonyl group and lead to the commencement of the Vilsmeier reaction (i.e. the halogenation reaction and the haloformylation reaction). Moreover, the haloformylation reaction exhibits stereoselectivity.
In an extension of this reaction, we next turned our attention to the Vilsmeier reaction of cyclic α-oxo ketenedithioacetals, 1c and 1d. The reactions of 1c and 1d with the Vilsmeier reagent (1 equiv.) were carried out at 0 °C, respectively. Just like compounds 2a, and 2b, halogenation products 2c and 2d were detected and unstable. With the addition of another one equivalent of the Vilsmeier reagent to the halogenated mixture, haloformylation products 3c and 3d were formed and detected, but it was found they were not stable, either. Kept in diethyl ether at room temperature for about 48 h, 3c and 3d were further converted to the corresponding 2H-pyrans 4c and 4d. One-step conversion to 4c and 4d was successfully achieved by the treatment of 1c and 1d with 2 equivalents Vilsmeier reagents, respectively.‡ It was noted that the conversion could be speeded up with increasing the reaction temperature. Obviously, the cyclization gives the evidence that the haloformylation is a stereo-selective reaction and the cyclization might follow a 6π-electrocyclic ring-closure mechanism.1d It is worthy of note that 3c and 3d could not undergo the reaction to afford 2H-pyrans under similar conditions.
In general, the carbonyl and the β-carbon atoms in α-oxo ketenedithioacetals can be regarded as hard and soft electrophilic centers. Therefore, many regioselective reagents can be selected either from hard nucleophiles undergoing 1,2-addition or from soft nucleophiles adding preferentially in a 1,4-fashion.6b However, in our previous work on the addition of Grignard reagents to α-oxo ketenedithioacetals with cyclic alkyldithio groups (e.g. S(CH2)2S and S(CH2)3S) rather than acyclic groups (e.g. SCH3), only 1,2-addition products were formed.9 We attributed this to the steric hindrance effect of the rigid cyclic dithioacetal moiety. The present work further demonstrates that there is great difference between the α-oxo ketenedithioacetals with cyclic alkyldithio groups and those with acyclic alkyldithio groups from the synthetic intermediate point of view. The reason for this difference is complicated and worthy of further investigation.
A possible mechanism for the Vilsmeier reactions to yield dienals and 2H-pyrans from α-oxo ketenedithioacetals is depicted in Scheme 4.
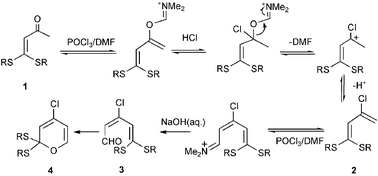 | ||
| Scheme 4 | ||
In summary, the reactions between Vilsmeier reagents and a series of α-oxo ketenedithioacetals containing a methyl group adjacent to the carbonyl group (1a–d) were investigated. A novel and convenient route to dienals and substituted 2H-pyrans directly from α-oxo ketenedithioacetal via the Vilsmeier reaction has been developed. The potential applications and extension of the scope of the methodology are currently under investigation in our laboratory.
This work was supported by the National Natural Science Foundation of China (20272008).
Notes and references
- For reviews on the Vilsmeier reaction and its synthetic applications see: (a) D. Burn, Chem. Ind.(London), 1973, 870 Search PubMed; (b) S. Seshadri, J. Sci. Ind. Res., 1973, 32, 128 Search PubMed; (c) J. C. Tebby and S. E. Willetts, Phosphorus Sulfur, 1987, 30, 293 Search PubMed; (d) C. M. Marson, Tetrahedron, 1992, 48(18), 3659 CrossRef CAS.
- (a) Z. Arnold and J. Zemlicka, Collect. Czech. Chem. Commun., 1959, 24, 2385 CAS; (b) Z. Arnold and J. Zemlicka, Proc. Chem. Soc., 1958, 227 RSC.
- (a) J. M. Vattoly and T. P. Paramasivan, Tetrahedron Lett., 1997, 6889; (b) C. Ian and L. C. Jose, Tetrahedron Lett., 1999, 4069.
- (a) V. J. Majo and P. T. Perumal, Tetrahedron Lett., 1996, 37, 5015 CrossRef CAS; (b) A. Shanmugam, S. Srinivasan and B. Krishna, Tetrahedron, 2001, 57, 3465 CrossRef.
- (a) O. Meth-Cohn and D. Taylor, J. Chem. Soc., Chem Commun., 1995, 1463 RSC; (b) O. Meth-Cohn and S. Goon, J. Chem. Soc., Perkin Trans. 1, 1997, 85 RSC.
- For reviews on the synthesis and applications of α-oxo ketenedithioacetals, see: (a) R. K. Dieter, Tetrahedron, 1986, 42, 3029 CrossRef CAS; (b) H. Junjappa, H. Ila and C. V. Asokan, Tetrahedron, 1990, 46, 5423 CrossRef CAS.
- (a) O. Barun, H. Ila, H. Junjappa and O. M. Sign, J. Org. Chem, 2000, 65, 1583 CrossRef CAS; (b) M. V. B. Rao, U. K. S. Kumar, H. Ila and H. Junjappa, Tetrahedron, 1999, 55, 11563 CrossRef; (c) J. R. Suresh, U. K. S. Kumar, H. Ila and H. Junjappa, Tetrahedron, 2001, 57, 781 CrossRef CAS; (d) K. Kishore, K. R. Reddy, J. R. Suresh, H. Ila and H. Junjappa, Tetrahedron, 1999, 55, 7645 CrossRef CAS; (e) J. R. Suresh, O. Barun, H. Ila and H. Junjappa, Tetrahedron, 2000, 56, 8153 CrossRef CAS.
- M. X. Wang, Y. Liu and Z. T. Huang, Tetrahedron Lett., 2001, 42, 2553 CrossRef CAS.
- (a) Q. Liu, Z. Zhu, Z. Yang, Y. Hu, F. Jing and Y. Xiao, Chem. J. Chin. Univ., 1993, 14, 1538 Search PubMed; (b) Q. Liu, D. Dong, Z. Yang, Y. Hu and C. Zhang, Chem. J. Chin. Univ., 1994, 15, 383 Search PubMed; (c) D. Dong, Z. Xie, Q. Liu, Z. Yang and J. Liu, Chem. J. Chin. Univ., 1997, 18(1), 68 Search PubMed.
- (a) S. Gill, P. J. Kocienski, A. Kohlor, A. Pontiroli and Q. Liu, Chem. Commun., 1996, 1743 RSC; (b) P. J. Kocienski, P. Alessandro and Q. Liu, J. Chem. Soc., Perkin Trans. 1, 2001, 2356 RSC.
- M. Wang, L. Ai, J. Zhang, Q. Liu and L. Gao, Chin. J. Chem., 2002, 20, 1591 Search PubMed.
- A. D. Thomas and C. V. Asokan, Tetrahedon Lett., 2002, 43, 2273 CrossRef CAS.
- P. K. Mahata, C. Venkatesh, U. K. Syam Kumar, H. Ila and H. Junjappa, J. Org. Chem., 2003, 68, 3966 CAS.
Footnotes |
| † Typical procedure for 3: The Vilsmeier reagent was prepared by adding POCl3 (10 mmol) dropwise to ice cold dry N,N-dimethylformamide (DMF, 10 mL) under stirring. The mixture was then stirred for 10–15 min at 0 °C. To the above Vilsmeier reagent was added 1a (5 mmol) as a solution in DMF (5 mL). Then the mixture was allowed to warm to room temperature and was stirred for 12–15 h. After the starting material was consumed (monitored by TLC), the reaction mixture was poured onto crushed ice (10 g) with stirring, followed by basification with cold aqueous NaOH (0.5 M) to adjust the pH value of the solution to 9. The mixture was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with brine (3 × 20 mL), dried over anhydrous MgSO4, filtered and concentrated under reduced pressure to yield the crude product which was purified by chromatography over silica gel using diethyl ether–hexane (1 : 80) as eluent.Analytical data for 3a: 1H NMR (400 MHz, CDCl3, 25 °C): (1) trans-isomer: 3.40 (2H, m, –SCH2), 3.60 (2H, m, –SCH2), 6.18 (1H, d, J = 8 Hz, –H), 6.37 (1H, s, –H), 10.03 (1H, d, J = 8 Hz, –CHO); (2) cis-isomer: 3.54 (2H, m, –SCH2), 3.55 (2H, m, –SCH2), 6.03 (1H, d, J = 8 Hz, –H), 7.28 (1H, s, = –H), 9.73 (1H, d, J = 8 Hz, –CHO); IR: 2933, 2864, 1640, 1557, 1177cm−1; MS m/z [(M − 1)+]: 206Analytical data for 3b: 1H NMR (400 MHz, CDCl3, 25 °C): (1) trans-isomer: 2.22 (2H, m, –CH2), 3.03 (4H, t, –SCH2), 6.17 (1H, d, J = 8 Hz, –H), 6.37 (1H, s, –H), 10.04 (1H, d, J = 8 Hz, –CHO); (2) cis-isomer: 2.22 (2H, m, –CH2), 3.03 (4H, t, –SCH2), 6.22 (1H, s, –H), 6.70 (1H, d, J = 8 Hz, –H), 9.59 (1H, d, J = 8 Hz, –CHO); IR: 2980, 2833, 1744, 1651, 1550, 1131cm−1; MS m/z [(M − 1)+]: 220.. |
| ‡ Typical procedure for 4: following the same procedure described above, the Vilsmeier reaction of 1c was carried out. The resulting reaction mixture containing 3c was worked up. The dried organic extracts were stirred at room temperature for 48 h, then concentrated under reduced pressure. The crude product was purified by chromatography over silica gel using diethyl ether–hexane (1 : 80) as eluent to yield 4c as a yellow solid.Analytical data for 4c: 1H NMR(400 Hz, CDCl3, 25 °C): 2.36 (3H, s, –SCH3), 2.42 (3H, s, –SCH3), 6.15 (1H, s, –H), 7.43 (1H, d, J = 14 Hz, = –H), 7.50 (1H, d, J = 14 Hz, –H); IR: 3068, 2921, 1656, 1572, 1541, 1428, 1043 cm−1; MS m/z [(M − 1)+]: 208Analytical data for 4d: 1H NMR (400 Hz, CDCl3, 25 °C): 4.10 (2H, s, –SCH2), 4.18 (2H, s, –SCH2), 6.11(1H, s, –H), 7.36 (11H, m, –H, –PhH), 7.68 (1H, d, J = 16 Hz, –H); IR: 3060, 1647, 1563, 1534, 1032, 698 cm−1; MS m/z [(M − 1)+]: 360.. |
| This journal is © The Royal Society of Chemistry 2004 |