Synthesis of an S-linked glycopeptide analog derived from human Tamm–Horsfall glycoprotein†
Xiangming
Zhu
,
Tobias
Haag
and
Richard R.
Schmidt
*
Fachbereich Chemie, Universität Konstanz, Fach M 725, D–78457 Konstanz, Germany. E-mail: Richard.Schmidt@uni-konstanz.de; Fax: (+49) 7531 883135
First published on 18th November 2003
Abstract
Direct base catalyzed S-glycosylation of a cysteine and a homocysteine containing peptide with O-acetyl protected bromides in DMF–water solution furnished two glycopeptide fragments. The two glycopeptide fragments were linked to the target glycopeptide with two S-glycosyl residues mimicking a part of Tamm–Horsfall glycoprotein.
Glycosylated proteins are ubiquitous components of extracellular matrices and cellular surfaces where their oligosaccharide moieties are implicated in a wide range of cell–cell and cell–matrix recognition events.1 Abnormalities in posttranslational glycosylation are often intimately associated with cell transformation, malignancy and other pathological conditions, including immunodeficiency syndromes, cancer and inflammation.2 However, homogenous glycopeptides for biological investigation are difficult to obtain from biological sources due to the significant levels of microheterogeneity in naturally produced glycopeptides.3 Therefore, the synthesis of structurally defined glycopeptides for the study of glycoprotein binding interactions, cell–cell adhesion and receptor binding specificity is of significant current interest.4
Naturally occurring glycopeptides most commonly incorporate an O-glycosidic or an N-glycosidic linkage between the carbohydrate moiety and the side-chain of an appropriate amino acid residue.4a However, the use of these substances as therapeutics is hampered in many cases due to their inherent in vivo instability, i.e. glycosidase involved cleavage of the glycan from the peptide backbone. This has stimulated the development of catabolically stable glycopeptide mimetics as fundamental tools for biological research and as potential agents for therapeutic intervention.5 As a result, S-linked glycopeptides which contain sulfur in the glycosidic linkage have attracted much attention,6,7 because of their enhanced chemical stability and enzymatic resistance.8 In this context, we have initiated a program to synthesize S-linked glycopeptides6 that may prove useful in biological studies and as potential therapeutic agents.
Herein we wish to report the synthesis of an S-linked glycopeptide analog derived from Tamm–Horsfall glycoprotein (THP),9 the most abundant glycoprotein present in normal human urine. Over the past 50 years, considerable investigations have been centered on THP, owing to its various biological activities.10 THP was shown to block hemagglutination induced by influenza, mumps, and New Castle disease viruses. THP has been assumed essential for protecting the kidneys from bacterial infections, also it has been implicated as a potential component necessary for maintenance of the electrolyte balance in the nephron. In addition, THP may also play a significant role in several pathological conditions involving the kidney, including acute renal failure, urinary tract infection, stone formation and interstitial nephritis. However, the biological role of THP, although extensively studied, remains unclear.11 Structurally, THP, built up from a polypeptide backbone of 616 amino acids, contains approximately 30% carbohydrate (w/w), comprising seven N-glycosidically bound sugar chains,9 which are specific ligands for lectins and cytokines and are probably responsible for most of THP functions.12
In order to contribute to the unraveling of the biological function of human THP, a small S-linked glycopeptide carrying two sugar residues 1 was designed (Fig. 1) and synthesized, which corresponds to the sequence Ala484–Ala490 in THP, containing Thr485 as potential O-glycosylation site and Asn489 as N-glycosylation site.
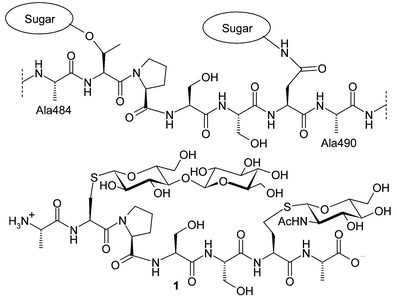 | ||
| Fig. 1 Structures of THP fragment Ala484–Ala490 and target molecule 1. | ||
As is evident from Fig. 1, Thr485 and Asn489 in THP were replaced by the corresponding cysteine (Cys) and homocysteine (Hcy) residues in 1, respectively, which was based on the viewpoint that the distance between sugar residue and cysteine is similar to O-glycan linkage to threonine and ligation of sugar residue to homocysteine mimics N-glycan linkage to asparagine. Thus, a catabolically stable S-linked glycopeptide was then resultant from the above substitution, providing the opportunity to probe biological properties of glycopeptides at the molecular level.
Very recently, we reported a new protocol for the synthesis of S-linked glycopeptides in aqueous solution by direct S-glycosylation of cysteine or homocysteine containing peptides with O-acetyl protected glycosyl halides,6a wherein chemical ligation techniques13 were utilized to prepare the required peptides. This procedure is a significant advance in the convergent assembly of S-linked glycopeptides because it demonstrates that under mild basic, aqueous conditions peptides can be S-glycosylated effectively. Based on this approach, the corresponding cysteine and homocysteine containing peptides should be firstly prepared in order to construct the target structure 1. For this purpose, the coupling reaction between commercially available N-Boc protected cystine (Boc-Cys-OH)2 and H-Pro-OBzl·HCl was performed in DMF using PyBOP as condensing agent,14 as shown in Scheme 1, leading to the desired peptide 2 in almost quantitative yield, which was then treated with 20% TFA in CH2Cl2 to give the free amine 3. The disulfide bond of 3 was subsequently cleaved by DTT15 in Tris buffer (pH 8) at room temperature, and ensuing addition of the alanine thioester 416 to the reaction mixture gave rise to the desired tripeptide Boc-Ala-Cys-Pro-OBzl 5 by chemical ligation strategy in 68% overall yield. At the same time, for the sake of mimicking Asn489 in THP, homocysteine containing dipeptide 8 (Scheme 1) was prepared in excellent yield by coupling of N-Boc protected homocystine 66a with H-Ala-OBzl·HCl in the presence of PyBOP and DIPEA in DMF followed by reduction of its disulfide bond with PBu317 in CHCl3–MeOH–H2O.
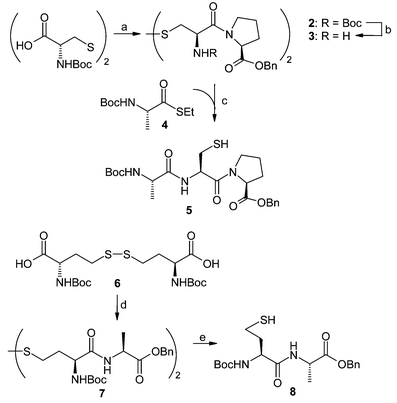 | ||
| Scheme 1 Reagents and conditions: (a) H-Pro-OBzl·HCl, PyBOP, DIPEA, DMF, quant.; (b) TFA, CH2Cl2; (c) DTT, Tris buffer (pH 8.0), guanidinium hydrochloride, 68% from 3; (d) H-Ala-OBzl·HCl, PyBOP, DIPEA, DMF, quant.; (e) PBu3, CHCl3–MeOH–H2O, 98%. | ||
With the requisite peptides 5 and 8 in hand, introduction of the sugars by their sulfhydryl groups was then investigated based on our previous work.6a In practice, tripeptide 5 was treated with lactosyl bromide 918 in the presence of Na2CO3 in DMF–H2O (Scheme 2) and, as expected, the desired glycotripeptide 10 was smoothly produced in 65% yield. Similarly, glycosylation of dipeptide 8 with N-Troc protected 2-amino glucosyl bromide 1618 also proceeded smoothly by the action of Na2CO3, providing the desired glycodipeptide 17 in very high yield (90%). To access the target molecule 1, the peptide chain of 10 was then elongated at the C-terminus as shown in Scheme 2. Debenzylation of 10 by catalytic hydrogenation gave the acid 11 in almost quantitative yield, which was subsequently coupled with H-Ser-OBzl·HCl by the action of PyBOP to produce the desired glycotetrapeptide 12 in high yield (86%).
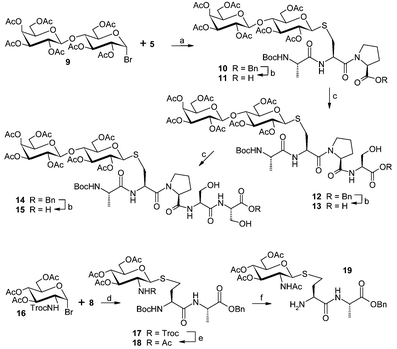 | ||
| Scheme 2 Reagents and conditions: (a) Na2CO3, DMF–H2O, 65%; (b) H2, 10% Pd/C, EtOAc–EtOH, 94% for 11, 85% for 13, 89% for 15; (c) H-Ser-OBzl·HCl, PyBOP, DIPEA, DMF, 88% for 12, 85% for 14; (d) Na2CO3, TBAHS, EtOAc–H2O, 90%; (e) Zn, Ac2O, Et3N, ultrasonic bath, 3 h, 87%; (f) TFA, TESH, CH2Cl2. | ||
The removal of the benzyl group from 12 was then conducted again by catalytic hydrogenation to give rise to the free acid 13, which was converted into glycopentapeptide 15 in two steps by treatment with H-Ser-OBzl·HCl in the presence of PyBOP and DIPEA in DMF followed by debenzylation with catalytic hydrogenation (Scheme 2). On the other hand, using the procedure developed recently,19 the N-Troc protecting group in glycodipeptide 17 was selectively transformed to an N-Ac group in compound 1819 in the presence of N-Boc protecting group by treatment with Zn–Ac2O–Et3N. Then 18 was exposed to 20% TFA in CH2Cl2 to lead smoothly to the required building block 19, which was used directly in the next reaction.
Coupling between acid 15 and amine 19 was conducted in the presence of PyBOP and DIPEA in DMF, as shown in Scheme 3, and fortunately, S-linked glycopetide 20 was produced in pleasing yield (69%). Subsequently, deprotection of 20 to convert into final product 1 was achieved in three steps: firstly, debenzylation by catalytic hydrogenation; secondly, deacetylation by treatment with 6 mM NaOMe in MeOH;20 finally, removal of the Boc protecting group by the action of TFA in the presence of triethylsilane and water. The synthetic target 1 was purified by reversed-phase HPLC and characterized by MALDI-MS and 600 MHz NMR.21
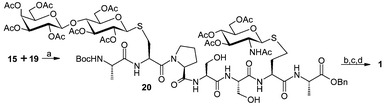 | ||
| Scheme 3 Reagents and conditions: (a) PyBOP, DIPEA, DMF, 69% from 18; (b) H2, 10% Pd/C, EtOAc–EtOH–HOAc; (c) 6 mM NaOMe, MeOH; (d) TFA, TESH, H2O, 0 °C, then RP–HPLC, 37% (3 steps). | ||
In conclusion, as an extension of our previous project, in this report, an S-linked glycopeptide (1) carrying two sugar moieties was synthesized in solution phase, which mimics the peptide sequence Ala484–Ala490 in the Tamm–Horsfall protein (THP) by replacing of the glycosylation sites Thr485 and Asn489 in THP with Cys and Hcy respectively. In view of the enhanced catabolic stability of S-linked glycopeptides, the resultant structure of 1 is of particular interest for studying its biological properties, also because in terms of distance between the sugar and the peptide backbone ligation of sugar to Cys mimics O-glycan linkage to Thr and linkage to Hcy mimics N-glycan linkage to Asn, respectively.
Notes and references
- (a) A. Varki, Glycobiology, 1993, 3, 97 CAS; (b) H. Lis and N. Sharon, Eur. J. Biochem., 1993, 218, 1 CAS; (c) R. A. Dwek, Chem. Rev., 1996, 96, 683 CrossRef CAS.
- S. Tsuboi and M. Fukuda, BioEssays, 2001, 23, 46 CrossRef CAS.
- R. S. Rush, P. L. Derby, D. M. Smith, C. Merry, G. Rogers, M. F. Rohde and V. Katta, Anal. Chem., 1995, 67, 1442 CrossRef CAS.
- For some recent reviews on glycopeptide synthesis, see: (a) A. M. Jansson, P. M. S. Hilaire and M. Meldal, in Synthesis of peptides and peptidomemetics, M. Goodman, A. Felix, L. Moroder and C. Toniolo, Eds., Houben-Weyl, Stuttgart, 2003, 235–332 Search PubMed; (b) B. G. Davis, Chem. Rev., 2002, 102, 579 CrossRef CAS; (c) V. Wittmann, in Glycoscience: Glycoproteins, B. O. Fraser-Reid, K. Tatsuta and J. Thiem, Eds., Springer-Verlag, Berlin, 2001, 2253–2352 Search PubMed; (d) H. Herzner, T. Reipen, M. Schultz and H. Kunz, Chem. Rev., 2000, 100, 4495 CrossRef CAS; (e) G. Arsequell and G. Valencia, Tetrahedron: Asymmetry, 1999, 10, 3045 CrossRef CAS; (f) C. M. Taylor, Tetrahedron, 1998, 54, 11317 CrossRef CAS.
- L. A. Marcaurelle and C. R. Bertozzi, Chem. Eur. J., 1999, 5, 1384 CrossRef CAS.
- (a) X. Zhu, K. Pachamuthu and R. R. Schmidt, J. Org. Chem., 2003, 68, 5641 CrossRef CAS; (b) X. Zhu and R. R. Schmidt, Tetrahedron Lett., 2003, 44, 6063 CrossRef CAS; (c) X. Zhu, R. R. Schmidt, Chem. Eur. J., in press Search PubMed.
- For more examples of S-glycopeptide synthesis, see ref. 4 cited in ref. 6b.
- (a) D. Horton and J. D. Wander, Carbohydrates: Chemistry and Biochemistry, Vol. 4B, W. W. Pigman and D. Horton, Eds., Academic Press, New York, 1990, 799–842 Search PubMed; (b) T. Eisele and R. R. Schmidt, Liebigs Ann., 1997, 865–872, 1303 Search PubMed; (c) M. J. Kiefel, R. J. Thomson, M. Radovanovic and M. V. Itzstein, J. Carbohydr. Chem., 1999, 18, 937 CAS; (d) H. Driguez, ChemBioChem, 2001, 2, 311 CrossRef CAS.
- J. J. M. van Rooijen, A. F. Voskamp, J. P. Kamerling and J. F. G. Vliegenthart, Glycobiology, 1999, 9, 21 CrossRef CAS.
- R. L. Easton, M. S. Patankar, G. F. Clark, H. R. Morris and A. Dell, J. Biol. Chem., 2000, 275, 21928 CrossRef CAS and references cited therein..
- T. Olczak, A. Kubicz, F. Kokot and J. Dulawa, Eur. J. Clin. Invest., 1998, 28, 475 CrossRef CAS.
- (a) M. Olczak and T. Olczak, Clin. Chim. Acta, 1999, 282, 35 CrossRef CAS and references cited therein; (b) T. Olczak, M. Olczak and A. Kubicz, Glycoconjugate J., 1999, 16, 481 CrossRef CAS.
- (a) K. Pachamuthu and R. R. Schmidt, Synlett, 2003, 659 CAS; (b) M. Schnolzer and S. B. H. Kent, Science, 1992, 256, 221 CAS; (c) J. P. Tam, Y. A. Lu, C. F. Liu and J. Shao, Proc. Natl. Acad. Sci. USA, 1995, 92, 12485 CAS.
- J. Coste, D. Le-Nguyen and B. Castro, Tetrahedron Lett., 1990, 31, 205 Search PubMed.
- W. L. Zahler and W. W. Cleland, J. Biol. Chem., 1968, 243, 716 CAS.
- H. Tokuyama, S. Yokoshima, S. C. Lin, L. Li and T. Fukuyama, Synthesis, 2002, 1121 CrossRef CAS.
- M. Iwai, I. Kohda, C. Fukaya, Y. Naito, K. Yokoyama, H. Nakajima, K. Tsujikawa, M. Okabe and T. Mimura, Chem. Pharm. Bull., 1987, 35, 4616 CAS.
- S. A. Mitchett, M. R. Pratt, V. J. Hruby and R. Polt, J. Org. Chem., 2001, 66, 2327 CrossRef.
- X. Zhu and R. R. Schmidt, Synthesis, 2003, 1262 CAS.
- P. Sjölin, M. Elofsson and J. Kihlberg, J. Org. Chem., 1996, 61, 560 CrossRef.
- Analytical data for compound 1: 1H NMR (600 MHz, HMQC, COSY, ROESY, DMSO-d6) δ 8.72–7.77 (NH), 5.43–4.82 (OH), 4.70 (m, 1H, Cysα), 4.41 (d, J = 9.6 Hz, 1H, HGlc-1), 4.35 (m, 3H, Proα, Hcyα, HGlcNAc-1), 4.29 (m, 1H, Serα), 4.25 (m, 1H, Serα), 4.20 (d, J = 6.9 Hz, 1H, HGal-1), 4.09 (m, 1H, Alaα), 3.82 (m, 1H, Alaα), 3.75 (dd br, J ≈ 11.5 Hz, 2H, Serβ), 3.68–3.27 (overlapped with water peak, 19H, Serβ, Proδ, HGal-4, HGal-2, HGal-5, 2 HGal-6, HGlc-3, HGlc-4, HGlc-5, 2 HGlc-6, HGlcNAc-3, HGlcNAc-4, 2 HGlcNAc-6), 3.10 (m, 5H, Hcyγ), HGlc-2, HGal-3, HGlcNAc-5), 2.71 (s-like, 2H, Cysβ), 2.65 (m, 1H, Proγ), 2.42 (overlapped m, 1H, Proγ), 1.98 and 1.73 (m, 2H, Proβ), 1.83 (m, 2H, Hcyβ), 1.79 (s, 3H, Ac), 1.30 (d, J = 6.9 Hz, 3H, Alaβ), 1.25 (d, J = 7.3 Hz, 3H, Alaβ). C44H74N8O25S2 (1179.2); MALDI-MS m/z 1200 [M − 1 + Na+], 1216 [M − 1 + K+].
Footnote |
| † This work was supported by the Deutsche Forschungsgemeinschaft, the Fonds der Chemischen Industrie, and the European Community (Grant No. HRPN-CT-2000-0001/GLYCOTRAIN). |
| This journal is © The Royal Society of Chemistry 2004 |