Rapid methylation of terminal acetylenes by the Stille coupling of methyl iodide with alkynyltributylstannanes: a general protocol potentially useful for the synthesis of short-lived 11CH3-labeled PET tracers with a 1-propynyl group†
Takamitsu
Hosoya
ab,
Masahiro
Wakao
a,
Yurie
Kondo
b,
Hisashi
Doi
a and
Masaaki
Suzuki
*ab
aDivision of Regeneration and Advanced Medical Science, Gifu University Graduate School of Medicine, Yanagido 1-1, Gifu 501-1193, Japan. E-mail: suzukims@biomol.gifu-u.ac.jp
bDepartment of Biomolecular Science, Faculty of Engineering, Gifu University, Yanagido 1-1, Gifu 501-1193, Japan
First published on 11th November 2003
Abstract
The Pd(0)-mediated rapid coupling (trapping) reaction of methyl iodide with an excess amount of alkynyltributylstannane has been developed with the aim to incorporate a short-lived 11C-labeled methyl group into biologically active organic compounds with a 1-propynyl structural unit.
Positron emission tomography (PET) is a particularly powerful, noninvasive method for the molecular imaging of living systems such as the brain, heart, and other active organs.1 In the PET study, a specific organic PET tracer is efficiently used as a molecular probe to monitor the biochemical processes and localization of a target molecule involved in important biofunctions and related phenomena. The need for the development of new PET tracers has grown with the increase in the use of this technique in in vivo biochemistry and medicine.1 In light of this need, we have recently developed the rapid Stille cross-coupling (5 min, 60 °C in DMF) of methyl iodide with aryltributylstannanes possessing sp3 and sp2 carbons, respectively, at the reaction centers2 and applied it successfully to the synthesis of a short-lived, 11C-incorporated PET tracer (t1/2 = 20.3 min), 15R-[11C]TIC methyl ester,3 an efficient prostaglandin probe, using 11CH3I,4 a synthetically established precursor, to image a novel prostacyclin (PGI2) receptor (IP2) present in the central nervous system in living monkey and human brains.3,5
Our next interest has been directed at the rapid introduction of a 11CH3 group to an alkyne terminal to develop new PET tracers of biologically significant molecules with a 1-propynyl structure. Examples of molecules with this structure include iloprost (1)
(a stable PGI2 analogue specific for the PGI2 receptor, IP1,3 in peripheral systems used as potential therapeutic agent),6,7 beraprost (a PGI2 analogue possessing the same ω side-chain as 1),8 2-(1-propnyl)estradiol (a tubulin polymerization inhibitor),9 RU 28362 (a glucocorticoid receptor agonist),10 E3620 (a 5-HT3 receptor antagonist/5-HT4 receptor agonist),11 2′-deoxy-5-(1-propynyl)uridine (an antiviral agent),12 and several others. Among the numerous methylation methods available,13 the Stille coupling14 using an alkynyltributylstannane as a trapping substrate of methyl iodide is ideal because: (1) the usual reaction conditions are compatible with a broad range of functional groups,13
(2) alkynylstannanes are tolerant enough of various reaction, workup, and chromatographic conditions to be transformed intact to the actual stage of the coupling reaction,15,16 and (3) the extremely low polarity of tributyl-substituted alkynylstannanes makes for easy chromatographic isolation of the desired 11C-incorporated coupling product from the remaining excess amount of tin substrate after the reaction.13,16 Although innumerable reports have described,14,17 the reports of Stille reactions between sp3/sp carbons are quite limited,18,19 and, to our surprise, Stille reactions including methyl iodide remain unexplored. Described herein is a general protocol which provides a firm chemical basis for the synthesis of a 11CH3-incorporated PET tracer with a 1-propynyl moiety in the structure.

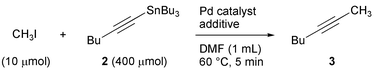
Non-functional tributyl-1-hexynylstannane (2) was used as a trapping agent to establish the standard reaction conditions. The reaction conditions were fixed at 60 °C in DMF2a for 5 min with the use of a large excess (40-fold) of the stannyl substrate, keeping the actual PET tracer synthesis in mind.13 Initially, some Pd(0)-complexes often utilized in the Stille reaction14 (Table 1, entries 1–4), and our conditions previously established in the rapid methylation of aryl substrates2 (entry 6) were investigated, resulting in the desired 2-heptyne (3) in yields between 11 and 61%. In the course of further studies, we noted that the steric and electronic properties of phosphine ligands influenced the yield of the coupling product. Among the ligands examined, tri-tert-butylphosphine (PtBu3)20 facilitated the reaction the most, resulting in a 72% yield (entry 9). The reaction was further improved to give an 81% yield (entry 10) by the direct use of bis(tri-tert-butylphosphine)palladium(0) (Pd(PtBu3)2), a commercially available complex,21 instead of generating it in situ by mixing Pd2(dba)3 and PtBu3. Further, the addition of CsF or KF facilitated an even higher reaction yield (entries 14 and 15, respectively), while the addition of LiCl or TBAF retarded the reaction (entries 12 and 13, respectively).22 Thus, as can be seen in Table 1, the highest yield (95%) was generated by the reaction in the presence of KF (entry 15).‡ Interestingly, the addition of CsF to the Pd2(dba)3/PMe(tBu)2 system19,20 led to an unexpectedly poor result (entry 16).
| Entry | Pd catalyst (µmol) | Additive(s) (µmol) | Yield of 3a (%) |
|---|---|---|---|
| a Determined by GLC analysis based on CH3I consumption. | |||
| 1 | Pd(PPh3)4 (10) | — | 27 |
| 2 | Pd2(dba)3 (5) | PPh3 (20) | 11 |
| 3 | Pd2(dba)3 (5) | P(2-furyl)3 (20) | 30 |
| 4 | Pd2(dba)3 (5) | AsPh3 (20) | 61 |
| 5 | Pd2(dba)3 (5) | P(o-tolyl)3 (20) | 20 |
| 6 | Pd2(dba)3 (5) | P(o-tolyl)3 (20), K2CO3 (20), CuCl (20) | 43 |
| 7 | Pd2(dba)3 (5) | P(cyclohexyl)3 (20) | 49 |
| 8 | Pd2(dba)3 (5) | PMe(tBu)2 (20) | 67 |
| 9 | Pd2(dba)3 (5) | PtBu3 (20) | 72 |
| 10 | Pd(PtBu3)2 (10) | — | 81 |
| 11 | Pd(PtBu3)2 (10) | K2CO3 (20), CuCl (20) | 45 |
| 12 | Pd(PtBu3)2 (10) | LiCl (20) | 27 |
| 13 | Pd(PtBu3)2 (10) | TBAF (20) | 15 |
| 14 | Pd(PtBu3)2 (10) | CsF (20) | 92 |
| 15 | Pd(PtBu3)2 (10) | KF (20) | 95 |
| 16 | Pd2(dba)3 (5) | PMe(tBu)2 (20), CsF (20) | 10 |
The reaction of methyl iodide and a stoichiometric amount of 2 at 60 °C for 5 min also proceeded under catalytic conditions (CH3I/2/Pd(PtBu3)2/KF = 1/1/0.1/1 and 1/1/0.02/1) to give 96 and 91% of 3, respectively, strengthening the utility of the coupling reaction from the synthetic point of view.23
The reaction most likely proceeds by the mechanism proposed in eqns. (1)–(4), where M = Bu3Sn or Bu3SnF−. First, methyl iodide undergoes oxidative addition with Pd(PtBu3)2 to generate methyl–Pd(II) iodide 4 or 4′ (eqn. (1)).24,25 These Pd(II) complexes may then react directly with alkynylstannane 5 or more favorably with the hypervalent stannate 6, formed in situ by the addition of a fluoride ion source (eqn. (2)), to afford the (alkynyl)(methyl)Pd(II) complex 7 (eqn. (3)). Finally, the desired methylalkyne 8 is formed by reductive elimination of the Pd(II) complex 7 (eqn. (4)).13f Here, we assume that the bulkiness (cone angle 182°)26 and the strong σ-electron-donating ability (pKa = 11.4)27 of PtBu3 would contribute to the generation of highly reactive, coordinatively unsaturated Pd(0) and Pd(II) intermediates, and also to the stabilization of the palladium complexes to avoid metal precipitation at high temperatures throughout the reaction cycle. We further assume that the extraordinarily high basicity of PtBu3 would also serve not only to increase the nucleophilicity for the oxidative addition step (eqn. (1)), but also to facilitate the dissociation of iodide to stabilize the transition state structure for the substitution step (eqn. (3)). In addition, the effect of fluoride ion can be explained by its facilitation of the formation of the hypervalent stannyl complex 6, whose nucleophilic C–Sn bond is more polar than that in 5 (eqn. (2)).20,28 The choice of a proper counter cation in the formation of the stannate complex 6 is also important to promote the reaction effectively, although the role of the cationic species remains unclear.
 | (1) |
 | (2) |
 | (3) |
 | (4) |
The best protocol (entry 15 in Table 1)‡ allows for the controlled methylation of a variety of alkynyltributylstannanes consisting of oxygen-based functional groups such as ethers, hydroxy groups, and ester groups. Compounds 9–14 are examples in our studies that were generated in 80, 89, 91, 88, 96 and 82% yields, respectively.29 The reaction was also applicable to the stannyl precursors 15 and 16, which are the substrates with steroid and deoxyribonucleoside frameworks, giving methylated compounds 17 and 1812 in 87 and 74%29 yields, respectively.
Finally, we demonstrated the trapping protocol of methyl iodide with a five-fold amount of stannyl precursor 19 leading to iloprost methyl ester (20) in 81% yield (based on the consumption of CH3I)
(Scheme 1).§ This ester 20 undergoes rapid enzymatic hydrolysis in the vital system to generate the free acid 1 as the bioactive form,3 and, therefore, the corresponding [11C]iloprost methyl ester can be used as a PET tracer.30

 | ||
| Scheme 1 | ||
Thus, we have succeeded in elaborating the rapid methylation of terminal acetylenes under mild conditions, which is potentially useful for the synthesis of short-lived 11CH3-labeled PET tracers to promote in vivo molecular science for medical purposes. The synthesis of actual PET tracers and their use for molecular imaging will be reported in due course.
Acknowledgements
This work was supported in part by a Grant-in-Aid for Creative Scientific Research (No. 13NP0401) of the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan. M. W. and H. D. are thankful for JSPS fellowships.Notes and references
- (a) J. S. Fowler, A. P. Wolf, J. R. Barrio, J. C. Mazziotta and M. E. Phelps in Positron Emission Tomography and Autoradiography, ed. M. E. Phelps, J. C. Mazziotta and H. R. Schelbert, Raven Press, New York, 1986, ch. 9–11 Search PubMed; (b) B. Långström and R. F. Dannals in Principle of Nuclear Medicine, ed. H. N. Wagner, Z. Szabo and J. W. Buchanan, W. B. Saunders, Philadelphia, PA, 2nd edn., 1995, section 1, ch. 11 Search PubMed; (c) J. S. Fowler and A. P. Wolf, Acc. Chem. Res., 1997, 30, 181–188 CrossRef CAS.
- (a) M. Suzuki, H. Doi, M. Björkman, Y. Andersson, B. Långström, Y. Watanabe and R. Noyori, Chem. Eur. J., 1997, 3, 2039–2042 CrossRef CAS; (b) M. Suzuki, H. Doi, K. Kato, M. Björkman, B. Långström, Y. Watanabe and R. Noyori, Tetrahedron, 2000, 56, 8263–8273 CrossRef CAS.
- (a) M. Suzuki, K. Kato, R. Noyori, Yu. Watanabe, H. Takechi, K. Matsumura, B. Långström and Y. Watanabe, Angew. Chem., Int. Ed. Engl., 1996, 35, 334–336 CAS; (b) M. Björkman, Y. Andersson, H. Doi, K. Kato, M. Suzuki, R. Noyori, Y. Watanabe and B. Långström, Acta Chem. Scand., 1998, 52, 635–640; (c) M. Suzuki, R. Noyori, B. Långström and Y. Watanabe, Bull. Chem. Soc. Jpn., 2000, 73, 1053–1070 CrossRef CAS; (d) R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008–2022 CrossRef CAS.
- (a) R. A. Ferrieri and A. P. Wolf, Radiochim. Acta, 1983, 34, 69–83 CAS; (b) B. Långström, G. Antoni, P. Gullberg, C. Halldin, P. Malmborg, K. Någren, A. Rimland and H. Svärd, J. Nucl. Med., 1987, 28, 1037–1040 Search PubMed.
- For application of rapid methylation to the synthesis of 11C-labeled PGF2α analogue: (a) M. Björkman, H. Doi, B. Resul, M. Suzuki, R. Noyori, Y. Watanabe and B. Långström, J. Labelled Compd. Radiopharm., 2000, 43, 1327–1334 CrossRef CAS ; [p-11C-methyl]MADAM; (b) J. Tarkiainen, J. Vercouillie, P. Emond, J. Sandell, J. Hiltunen, Y. Frangin, D. Guilloteau and C. Halldin, J. Labelled Compd. Radiopharm., 2001, 44, 1013–1023 CrossRef ; [11C]toluene; (c) M. Gerasimov, J. Logan, R. A. Ferrieri, R. D. Muller, D. Alexoff and S. L. Dewey, Nucl. Med. Biol., 2002, 29, 607–612 CrossRef CAS , and references therein; 5-[11C]methyl-6-nitroquipazine; (d) J. Sandell, M. Yu, P. Emond, L. Garreau, S. Chalon, K. Någren, D. Guilloteau and C. Halldin, Bioorg. Med. Chem. Lett., 2002, 12, 3611–3613 CrossRef CAS and references therein; 4-[11C]methylmetaraminol; (e) O. Langer, T. Forngren, J. Sandell, F. Dollé, B. Långström, K. Någren and C. Halldin, J. Labelled Compd. Radiopharm., 2003, 46, 55–65 CrossRef CAS ; 11C-labeled citalopram analogue; (f) J. Madsen, P. Merachtsaki, P. Davoodpour, M. Bergström, B. Långström, K. Andersen, C. Thomsen, L. Martiny and G. M. Knudsen, Bioorg. Med. Chem., 2003, 11, 3447–3456 Search PubMed.
- (a) W. Skuballa and H. Vorbrüggen, Angew. Chem., Int. Ed. Engl., 1981, 20, 1046–1048 CrossRef; (b) W. Skuballa and H. Vorbrüggen, Adv. Prostaglandin Thromboxane Leukot. Res., 1983, 11, 299–305 Search PubMed.
- (a) P. W. Collins and S. W. Djuric, Chem. Rev., 1993, 93, 1533–1564 CrossRef CAS; (b) B. Buchman, in Prostaglandins, Leukotrienes and Other Eicosanoids, ed. F. Marks and G. Fürstenberger, Wiley–VCH, Weinheim, 1999, pp. 331–374 Search PubMed; (c) H. Wise and R. L. Jones, Prostacyclin and Its Receptors, Kluwer Academic/Plenum Publishers, New York, 2000 Search PubMed.
- H. Wakita, H. Yoshiwara, H. Nishiyama and H. Nagase, Heterocycles, 2000, 53, 1085–1110 CAS , and references therein.
- M. Cushman, A. K. Mohanakrishnan, M. Hollinshead and E. Hamel, J. Med. Chem., 2002, 45, 4748–4754 CrossRef CAS.
- G. Teutsch, M. Gaillard-Moguilewsky, G. Lemoine, F. Nique and D. Philibert, Biochem. Soc. Trans., 1991, 19, 901–908 CAS.
- S. Miyazawa, S. Hibi, H. Yoshimura, T. Mori, Y. Hoshino, M. Nagai, K. Kikuchi, H. Shibata, K. Hirota, T. Yamanaka, I. Yamatsu and M. Mizuno, Jpn. Pat., JP H06-128254, 1994 Search PubMed.
- E. De Clercq, J. Descamps, J. Balzarini, J. Giziewicz, P. J. Barr and M. J. Robins, J. Med. Chem., 1983, 26, 661–666 CrossRef CAS.
- The reaction is usually conducted using >10−6 g of 11CH3I and 10−3 g of a trapping substrate. Sonogashira coupling is not feasible because of the formation of undesired tetraalkylammonium salts by methyl iodide and tertiary amines. The method using alkynylmetalloids is too drastic for substrates with various base-sensitive functions. Both of the reactions leave a large amount of unreacted acetylenic compounds after the reaction under PET conditions, making the isolation of the desired methylated compound difficult. For other methylation reactions of a terminal alkyne: (a) R. C. Larock and S. S. Hershberger, J. Organomet. Chem., 1982, 225, 31–41 CrossRef CAS; (b) J. A. Sikorski, N. G. Bhat, T. E. Cole, K. K. Wang and H. C. Brown, J. Org. Chem., 1986, 51, 4521–4525 CrossRef CAS; (c) C. J. Elsevier and H. H. Mooiweer, J. Org. Chem., 1987, 52, 1536–1539 CrossRef CAS; (d) A. Pelter and R. A. Drake, Tetrahedron Lett., 1988, 29, 4181–4184 CrossRef CAS; (e) J. A. Soderquist and B. Santiago, Tetrahedron Lett., 1990, 31, 5541–5542 CrossRef CAS; (f) Y. J. Kim, K. Osakada and A. Yamamoto, J. Organomet. Chem., 1993, 452, 247–250 CrossRef CAS.
- (a) J. K. Stille, Angew. Chem., Int. Ed. Engl., 1986, 25, 508–524 CrossRef; (b) V. Farina, V. Krishnamurthy and W. J. Scott, Org. React., 1997, 50, 1–657 CAS.
- It was found that alkynylboronic acids or alkynylboronates used for the Suzuki reaction are too unstable to isolate or hard to purify by usual chromatographic techniques. See: (a) H. C. Brown, N. G. Bhat and M. Srebnik, Tetrahedron Lett., 1988, 29, 2631–2634 CrossRef CAS; (b) H. Fujishima, E. Takada, S. Hara and A. Suzuki, Chem. Lett., 1992, 695–698 CAS.
- For reviews on organostannanes: A. G. Davies, Organotin Chemistry, VCH, Weinheim, 1997 Search PubMed.
- For review on Pd-catalyzed alkynylation: (a) E. Negishi and L. Anastasia, Chem. Rev., 2003, 103, 1979–2018 CrossRef ; for recent reviews on Pd catalyzed cross-coupling reactions; (b) Handbook of Organopalladium Chemistry for Organic Synthesis, ed. E. Negishi, John Wiley & Sons, New York, 2002 Search PubMed.
- For the Stille reaction with allyllic, benzylic, propargylic electrophiles, and perfluoroalkyl halides: (a) pp. 474–516 in ref. 14b.; (b) S. Matsubara, M. Mitani and K. Utimoto, Tetrahedron Lett., 1987, 28, 5857–5860 CrossRef CAS.
- Menzel and Fu reported efficient Stille-type cross-coupling of alkylbromides with alkenylstannnannes (coupling between sp3/sp2 carbons). They mentioned, however, alkynylstannannes are not suitable substrates: K. Menzel and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 3718–3719 Search PubMed.
- A. F. Littke, L. Schwarz and G. C. Fu, J. Am. Chem. Soc., 2002, 124, 6343–6348 CrossRef CAS , and references therein.
- T. Yoshida and S. Otsuka, Inorg. Synth., 1990, 28, 113–119 CAS ; commercially available from Strem Chemicals (MA, USA).
- The effects of other additives: TASF (20 µmol, 75%), CaF2 (10 µmol, 88%), KF and 18-crown-6 (20 µmol each, 91%).
- The reaction was conducted using 10 µmol of methyl iodide at 10 mM concentration.
- (a) J. A. Casares, P. Espinet and G. Salas, Chem. Eur. J., 2002, 8, 4844–4853 CrossRef; (b) A. Ricci, F. Angelucci, M. Bassetti and C. LoSterzo, J. Am. Chem. Soc., 2002, 124, 1060–1071 CrossRef CAS; (c) C. Amatore, A. A. Bahsoun, A. Jutand, G. Meyer, A. N. Ntepe and L. Ricard, J. Am. Chem. Soc., 2003, 125, 4212–4222 CrossRef CAS , and references therein.
- (a) J. P. Stambuli, M. Bühl and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 9346–9347 CrossRef CAS; (b) J. H. Kirchhoff, M. R. Netherton, I. D. Hills and G. C. Fu, J. Am. Chem. Soc., 2002, 124, 13662–13663 CrossRef CAS.
- Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- T. Allman and R. G. Goel, Can. J. Chem., 1982, 60, 716–722 CAS.
- A. García Martínez, J. Osío Barcina, M. R. Colorado Heras and Á. De Frensno Cerezo, Organometallics, 2001, 20, 1020–1023 CrossRef CAS.
- In these cases, the stepwise procedure, which consists of the addition of an alkynylstannane after mixing the palladium complex and methyl iodide in DMF for 1 min at 60 °C, was employed. The total synthetic time was fixed to 5 min. See also ref. 2b. Yields were determined by GLC or HPLC analysis based on CH3I consumption. In the actual PET tracer synthesis, the reaction mixture is subjected directly to reversed-phase HPLC purification (CH3CN–H2O solvent system) to afford the desired radiolabeled compound in nanomole scale, which is used immediately.
- Successful synthesis of [11C]iloprost methyl ester will be reported independently.
Footnotes |
| † Electronic supplementary information (ESI) available: general experimental remarks and synthetic methods and characterization of tributylalkynylstannanes and the corresponding methylacetylenes. See http://www.rsc.org/suppdata/ob/b3/b311532a/ |
| ‡ Trapping reaction of methyl iodide with an excess amount of tributyl-1-hexynylstannane (2) leading to 2-heptyne (3): In a dry Schlenk tube (10 mL), bis(tri-tert-butylphosphine)palladium(0) (5.1 mg, 10 µmol) and potassium fluoride (1.2 mg, 20 µmol) were placed under Ar. After the addition of DMF (950 µL), the mixture was stirred for 5 min at room temperature followed by successive additions of a methyl iodide solution (50 µL, 0.20 M, 10 µmol) in DMF and a stannane 2 (148 mg, 400 µmol). After stirring at 60 °C for 5 min, the mixture was rapidly cooled in an ice bath, followed by the addition of n-nonane (50 µL, 0.10 M diethyl ether solution, 5.0 µmol) as an internal standard, and diethyl ether (1 mL). The mixture was loaded onto a short column of silica-gel (1.0 g) and then eluted with diethyl ether (ca. 1 mL). The combined eluates were analyzed by GLC (Shimadzu GC-14B instrument equipped with a flame ionization detector; capillary column: GL Science TC-5, 60 m × 0.25 mm i.d., df = 0.25 µm; carrier gas: He; flow rate: 0.5 mL min−1; injector temperature: 280 °C; detector temperature: 280 °C; column temperature: initial 70 °C, final 100 °C; temperature rising rate: 5 °C min−1, from 10 to 16 min): yield of 2-heptyne (3): 95% based on starting CH3I; retention time: 12.6 min (cf. n-nonane: 17.9 min). |
| § Application to the synthesis of iloprost methyl ester (20): In a dry Schlenk tube (10 mL), bis(tri-tert-butylphosphine)palladium(0) (0.8 mg, 1.6 µmol) and potassium fluoride (0.2 mg, 3.2 µmol) were placed under Ar. After the addition of DMF (400 µL), the mixture was stirred for 5 min at room temperature followed by successive additions of a methyl iodide solution (32 µL, 50 mM, 1.6 µmol) in DMF and a solution of stannane 19 (5.1 mg, 8.0 µmol) in DMF (400 µL). After stirring at 60 °C for 5 min, the mixture was rapidly cooled in an ice bath, followed by the removal of insoluble materials by filtration through a plug of cotton. The filtrate was azeotropically concentrated with toluene under reduced pressure, and the residual material was filtered through a short column of silica-gel (1.0 g, eluted with n-hexane–ethyl acetate = 1 ∶ 1). Anisole (10 µL, 0.20 M CH3CN solution, 2.0 µmol) was added to the combined eluates as an internal standard, and the mixture was then analyzed by reversed-phase HPLC (Mightysil RP-18 GP column: Kanto Chemical, 150 × 4.6 mm i.d.; eluted by CH3CN/H2O = 45/55; flow rate 1.0 mL min−1; column oven temperature 40 °C; detected by UV at 203 nm); yield of iloprost methyl ester (20): 81% based on starting CH3I; retention time: 21.1 and 22.6 min (16R- and 16S-epimers, respectively) (cf. anisole: 7.4 min). |
| This journal is © The Royal Society of Chemistry 2004 |