Application of Fmoc-amino acid carrying an unmasked carbohydrate to the synthesis of the epidermal growth factor-like domain of bovine blood coagulation factor IX†
Toshio Takemuraa, Hironobu Hojo*a, Yoshiaki Nakaharaa, Takeshi Ishimizub and Sumihiro Haseb
aDepartment of Applied Biochemistry, Institute of Glycotechnology, Tokai University, Hiratsuka, Kanagawa, 259-1292, Japan
bDepartment of Chemistry, Graduate School of Science, Osaka University, Toyonaka, Osaka, 560-0043, Japan
First published on 24th November 2003
Abstract
The epidermal growth factor (EGF)-like domain of the bovine blood coagulation factor IX (45–87) carrying glucose at Ser53 was synthesized by a solid-phase method using 9-fluorenylmethoxycarbonyl (Fmoc)-amino acids. The introduction of Ser53 was carried out using the benzotriazolyl ester of Fmoc-serine carrying an unmasked glucose. The remaining sequence was also introduced using the benzotriazolyl ester. HPLC analysis of the crude peptide shows that acylation of the free hydroxyl group of the glucose was not significant, demonstrating that the amino acid carrying an unmasked carbohydrate is a useful building block for solid-phase synthesis.
Introduction
Natural proteins often retain carbohydrates, which are generally linked to the polypeptide chains via the side chain amide bond of asparagine (N-linked glycans) or the hydroxyl groups of serine and threonine residues (O-glycans). In the latter cases, the reducing end sugar is usually α-linked N-acetylgalactosamine. In 1988 Hase et al. found a new type of O-glycosidic linkage, Xyl-α1, 3-Xyl-α1, 3-Glc-β1-O-Ser, in the EGF-like domains of the blood coagulation factors VII and IX.1 The consensus sequence, which is proposed for the addition of glucose, is C1XSXPC2 (X = any amino acid), where C1 and C2 are the first and second cysteines of the EGF-like domain. A database search for this sequence shows that several secreted and cell surface proteins are supposed to have this carbohydrate modification.2 However, the role of O-glucose on the EGF repeats remains largely unknown, except that the mutation of the carbohydrate-linked serine residue to alanine in factor VII is known to result in a decrease in factor VII clotting activity.3 To understand the function of this unusual O-glycosylation from biological and structural points of view, we synthesized the EGF-like domain carrying glucose as shown in Fig. 1.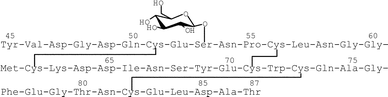 | ||
| Fig. 1 Amino acid sequence of the EGF-like domain of the bovine blood coagulation factor IX (45–87). | ||
The synthesis of the glycopeptide has been mainly carried out by a solid-phase method using the Fmoc protocol. The hydroxyl groups of the carbohydrate portion are usually protected by acetyl or benzyl groups. When acetyl-protected amino acids are used for peptide synthesis, several problems might arise. The acetyl groups have to be removed after the peptide chain assembly by treatment with CH3ONa in methanol, which may cause β-elimination of serine-linked carbohydrates and epimerization of amino acid residues. In addition, the acetyl group tends to migrate to the terminal amino group of the growing peptide chain during solid-phase synthesis, which prevents further elongation of the peptide chain. In the case of the benzyl group, no such problems arise, as its removal can be achieved either by catalytic hydrogenation or mild acid treatment, and also the benzyl group does not migrate under the conditions used for solid-phase synthesis. However, for the synthesis of the EGF-like domain carrying β-linked glucose, the use of benzyl protection would be inconvenient, since the glycosylation of Fmoc-serine by benzyl-protected glucose may not yield the desired β-anomer as a major product.
Recently, several reports appeared on the use of Fmoc-amino acids carrying unprotected carbohydrates for the solid phase synthesis of glycopeptide.4–11 In particular, Yamamoto et al. prepared Fmoc-Asn carrying an oligosaccharide with free hydroxyl groups from eggs and used it for the glycopeptide synthesis.11 This method will open a facile route to N-glycopeptides without laborious chemical synthesis of the carbohydrate moiety. However, when unmasked derivatives are used for the solid-phase synthesis, there is a potential risk of O-acylation on the free hydroxyl groups of the carbohydrate moiety. In previous reports, mild coupling methods, such as pentafluorophenyl or benzotriazolyl esters, are employed to avoid this issue. At present the application of amino acids carrying unmasked carbohydrates for glycopeptide synthesis is limited to relatively short peptides (less than 20 amino acid residues) and thus, its practical use for longer peptides has to be clarified. In this report, we synthesized Fmoc-serine carrying unprotected β-glucose and applied it to the synthesis of the EGF-like domain of the bovine blood coagulation factor IX (45–87). There are many reports on the synthesis of the EGF-like domain of proteins,12–24 but the chemical synthesis of the glycosylated EGF-like domain has not appeared so far.
Results and discussion
Synthesis of the EGF-like domain
Fmoc-Ser(Glc) 7 was prepared as shown in Fig. 2. The Fmoc-serine allyl ester was glycosylated with tetraacetyl glucosyl bromide 3 using silver triflate as an activator to give compound 4. During the glycosylation reaction, a part of the hydroxyl group of the Fmoc-serine allyl ester was acetylated, which decreased the yield of the desired product. This side product might be derived from the orthoester. The purified yield of the desired product 4 was 34%. The allyl group was removed by a Pd(0) catalyst in the presence of N-methylaniline (NMA), and compound 5 was obtained quantitatively. The Fmoc and acetyl groups were simultaneously removed by CH3ONa in methanol. Without purification, the obtained product 6 was reacted with N-(9-fluorenylmethoxycarbonyloxy)succinimide (Fmoc-OSu). The desired building block 7 was obtained after RPHPLC purification in 68% yield from compound 5.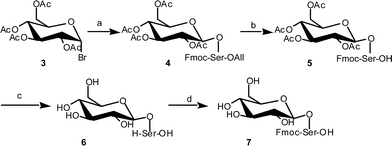 | ||
| Fig. 2 Synthetic procedure for Fmoc-Ser carrying unmasked glucose. a) Fmoc-Ser-OAll, AgOTf; b) Pd(PPh3)4, NMA; c) CH3ONa–MeOH; d) Fmoc-OSu. | ||
The solid-phase synthesis of the EGF-like domain was carried out as shown in Fig. 3. Starting from Fmoc-Thr(But)-OCH2-PEG-PS resin, the peptide chain was elongated using an ABI 433A peptide synthesizer employing the FastMoc protocol. After the introduction of Asn54, a part of the resin was taken and the chain elongation was continued manually. The Fmoc group of the resin was removed by 20% piperidine–NMP and the Fmoc-Ser(Glc) 7 was introduced manually using its benzotriazolyl ester (3 eq.). The reaction mixture was vortexed at room temperature for 30 min and at 50 °C for 15 min. Ninhydrin monitoring indicated the completion of the coupling. The remaining sequence was also introduced manually using the benzotriazolyl ester (20 eq.) in the same manner. After complete assembly of the peptide sequence, the resin was treated with a trifluoroacetic acid (TFA) cocktail (Reagent K25) and the crude peptide was analyzed by RPHPLC. Since the peptide is in reduced form, which is free from the disulfide bond isomer, precise detection of the O-aclyation product would be expected. However, as shown in Fig. 4a, the peptide with six thiol groups was strongly adsorbed to the RPHPLC column at 30 °C. Though the adsorption became weaker as the column temperature increased from 30 °C to 60 °C, complete elution was not possible. Thus, the purity of the crude peptide was not estimated at this stage. The folding and the disulfide bond formation were then carried out using the redox buffer containing reduced and oxidized glutathione at pH 8.0. After 3 days, the same amount of crude peptide was analyzed by RPHPLC. As shown in Figs 4c and d, a sharp peak, which has the desired mass number, was obtained as a major product regardless of the column temperature. Mass analysis of smaller peaks eluted after the major peak in Fig. 4c showed that these peaks mainly consist of the disulfide bond isomers. The main peak was purified by preparative RPHPLC and the desired EGF-like domain carrying glucose was obtained in 3.4% yield. To estimate the efficiency of this synthesis, non-glycosylated EGF-like domain 2 was prepared totally by the synthesizer using the FastMoc protocol, which uses O-benzotriazol-1-yl-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HBTU) as a coupling reagent. As shown in Figs 4e and f, the RPHPLC profile of this peptide is similar to that of glycosylated EGF-like domain 1. The yield of the purified non-glycosylated EGF-like domain 2 was 3.7%, which is comparable to the glycosylated peptide 1. Thus, the syntheses of two EGF-like domains proceeded with comparable efficiency, which means that the acylation of the free hydroxy groups of the carbohydrate portion is not significant. The purified peptides 1 and 2 were homogeneous, which was shown by MALDI-TOF mass spectrometry, RPHPLC (Fig. 5) and by amino acid analyses.
 | ||
| Fig. 3 Synthetic route for the EGF-like domain of bovine factor IX 1. | ||
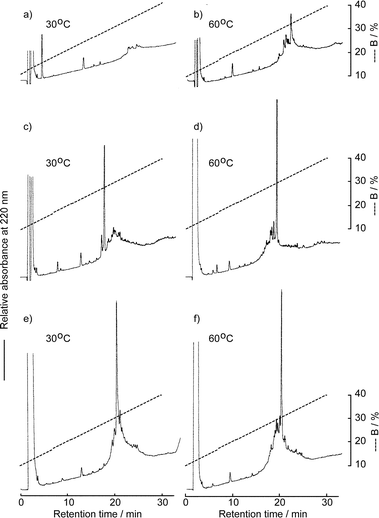 | ||
| Fig. 4 RPHPLC profiles of the crude glycosylated peptide 1 and non-glycosylated peptide 2: a) and b); peptide 1 before oxidation (ca. 90 µg of crude peptide), c) and d); peptide 1 after oxidation (ca. 90 µg of crude peptide), e) and f); peptide 2 after oxidation (ca. 80 µg of crude peptide). Elution conditions: column, Mightysil RP-18GP (4.6 × 150 mm) at a flow rate of 1 ml min−1; eluent A, distilled water containing 0.1% TFA, B, acetonitrile containing 0.1% TFA. | ||
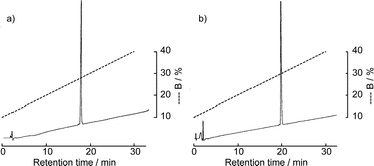 | ||
| Fig. 5 RPHPLC profile of the purified peptide 1 (a) and 2 (b). Elution conditions: column, Mightysil RP-18GP (4.6 × 150 mm) at a flow rate of 1 ml min−1; eluent A, distilled water containing 0.1% TFA, B, acetonitrile containing 0.1% TFA; temperature, 30°C. | ||
Determination of the disulfide pairings
To determine the mode of disulfide linkages, the synthetic peptides were digested with thermolysin at 45 °C for 24 h and the peptide fragments were separated by RPHPLC. Each fraction was analyzed by the mass and amino acid analyses. In the case of the non-glycosylated peptide 2, peptide fragments A and B were obtained (Table 1), which correspond to the correct disulfide bond shown in Fig. 1. In contrast, glycosylated peptide 1 seemed to be somewhat more resistant to the digestion by thermolysin. Even after 24 h digestion, the peptide bond between Cys51-Glu52 was not cleaved. To determine the N-terminal disulfide pairings, peptide 1 was treated with BrCN to cleave Met61-Cys62 and was further digested with Staphylococcus aureus protease V8. The digest was analyzed by RPHPLC and fragment C was obtained, which shows the formation of the disulfide bond between Cys51-Cys62. The C-terminal disulfide bond was determined by the thermolysin digestion for 24 h as described above.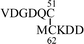
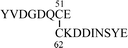
In conclusion, Fmoc-serine carrying an unmasked glucose was prepared by the solution method. This unit was successfully used during the solid-phase synthesis of the EGF-like domain of bovine Factor IX by the benzotriazolyl active ester method, indicating that the glycopeptides can be synthesized without hydroxyl group protection at the carbohydrate moiety, if mild activation methods are employed.
Experimental
Optical rotation values were determined with a DIP-370 polarimeter (Jasco, Tokyo) at 20 °C ± 5 °C. 1H-NMR spectra were recorded with a AL-400 spectrometer (Jeol, Tokyo). MALDI-TOF mass spectra were recorded with a Voyager-DE PRO spectrometer (Applied Biosystems, CA). Amino acid composition was determined with a LaChrom amino acid analyzer (Hitachi, Tokyo) after hydrolysis with 6 M HCl at 150 °C for 2 h in an evacuated sealed tube.N-(9-Fluorenylmethoxycarbonyl)-O-(β-D-glucopyranosyl)-L-serine 7
To the solution of compound 5 (500 mg, 760 µmol) in methanol (2 ml), 1 M CH3ONa solution in methanol (1.0 ml, 1.0 mmol) was added and the reaction mixture was stirred at room temperature for 3 h. The solution was neutralized by 1 M HCl and the solvent was removed in vacuo. The residue was dissolved in distilled water and washed with ether (×3) and lyophilized to give compound 6. The obtained powder was dissolved in 10% Na2CO3 (5 ml) and was dropped into the solution of Fmoc-OSu (385 mg, 1.1 mmol) in dimethoxyethane (5 ml). After the reaction mixture was kept at room temperature overnight, the solution was washed with ether (×3) and acidified by 1 M HCl. The product was extracted by 1-butanol and purified by RPHPLC to give compound 7 (250 mg, 510 µmol, 68%). Rf 0.58 (4 : 1 : 1 1-butanol–AcOH—H2O); [α]D +4.0 (c 0.9 in methanol); Anal. calcd for C24H27NO10·1/2 H2O: C, 57.83; H, 5.66; N, 2.81. Found: C, 57.62; H, 5.47; N, 3.07; 1H NMR (CD3SOCD3): δ 7.90 (d, 2H, J 7.6 Hz, Ar), 7.76–7.73 (m, 2H, Ar), 7.70 (d, 1H, J 9.0, NH) 7.43 (br t, 2H, J 7.4 Hz, Ar), 7.34 (br t, 2H, J 7.4 Hz, Ar), 4.15 (d, 1H, J 7.6 Hz, H-1), 3.68 (br d, 1H, J 10.7 Hz, H-6), 3.62 (dd, 1H, J 3.3 Hz, 9.9 Hz, Ser-βH), 3.44 (dd, 1H, J 5.7 Hz, 11.8 Hz, H-6), 3.16 (br t, 1H, J 8.8 Hz, H-3), 3.05 (t, 1H, J 9.0 Hz, H-4), 2.98 (t, 1H, J 8.3 Hz, H-2); MALDI-TOF MS: calcd for C24H25NO10Na m/z 512.15. Found m/z 512.07.Solid-phase synthesis of the glycosylated EGF-like domain (45–57) 1
Starting from Fmoc-Thr(But)-OCH2-PEG-PS resin (0.25 mmol g−1, 0.4 g), the peptide chain was elongated by an ABI 433A peptide synthesizer (Applied Biosystems, Foster City) employing FastMoc protocol. After Fmoc-Asn(Trt)-Pro-Cys(Trt)-Leu-Asn(Trt)-Gly-Gly-Met-Cys(Trt)-Lys(Boc)-Asp(OBut)-Asp(OBut)-Ile-Asn(Trt)-Ser(But)-Tyr(But)-Glu(OBut)-Cys(Trt)-Trp(Boc)-Cys(Trt)-Gln(Trt)-Ala-Gly-Phe-Glu(OBut)-Gly-Thr(But)-Asn(Trt)-Cys(Trt)-Glu(OBut)-Leu-Asp(OBut)-Ala-Thr(But)-OCH2-PEG-PS-Resin was obtained, a part of this resin (6.3 µmol) was taken and the Fmoc group was removed with 20% piperidine-1-methyl-2-pyrrolidinone (NMP) for 5 and 15 min. Fmoc-Ser(Glc) 7 (9.2 mg, 19 µmol) was preactivated with 1 M 1,3-dicyclohexylcarbodiimide (DCC)–NMP (23 µl) and 1 M 1-hydroxybenzotriazole (HOBt)–NMP (23 µl) for 30 min at room temperature and reacted with the resin at room temperature for 30 min. The reaction mixture was further vortexed at 50 °C for 15 min. A ninhydrin test indicated the complete introduction of the glucosyl serine residue. The remaining sequence was introduced manually using Fmoc-amino acid benzotriazolyl ester (130 µmol). After complete assembly of the peptide sequence, the Fmoc group was removed and the resin was dried in vacuo. A part of the resin (50 mg out of 68 mg obtained) was treated with Reagent K (500 µl) for 2 h at room temperature. TFA was removed by a nitrogen stream and the ether was added to form a precipitate, which was washed twice with ether and dried in vacuo. The peptide was extracted by 50% aqueous acetonitrile containing 0.1% TFA (2 ml) and filtered through a membrane filter. The solution was dropped into 0.1 M ammonium acetate (50 ml, pH 8.0) containing oxidized (3 mg) and reduced forms of glutathione (15 mg) and was stirred for 3 days at room temperature. The solution was acidified by acetic acid and loaded onto a RPHPLC column. The main fraction was collected and lyophilized to give the desired peptide 1. The yield was 160 nmol (3.4% based on the Thr content on the starting resin) MADLI-TOF MS found: m/z 4853.7 (M + H)+. Calcd: m/z 4853.8 (M + H)+; amino acid analysis; Asp9.13Thr1.85Ser1.66Glu5.88Pro0.86Gly5Ala2.04Cys0.99 Val0.97Met0.94 Ile1.00Leu2.08Tyr2.00Phe1.01Lys1.00.The determination of the disulfide bridge of the EGF-like domain
EGF-like domain 1 or 2 (100 µg) was dissolved in 0.1 M pyridine acetate (pH 6.5, 100 µl) and 20 µg of thermolysin was added. The solution was kept at 45 °C for 24 h. The peptide fragment was separated by RPHPLC and analyzed by MALDI-TOF mass and amino acid analyzer. In the case of peptide 1, the peptide (100 µg) was dissolved in 70% formic acid and treated with BrCN (100 µg, 1.0 µmol) for 24 h at room temperature in the dark. The solvent was removed in vacuo and the residual mass was dissolved in distilled water and lyophilized. The obtained powder was dissolved in 0.1 M NH4HCO3 (40 µl) and digested with Staphylococcus aureus protease V8 (20 µg) at 40 °C for 3 h and purified by RPHPLC. Each fraction was analyzed by MADLI-TOF MS.Acknowledgements
The authors acknowledge Tokai University for a grant-aid for high-technology research.References
- S. Hase, S. Kawabata, H. Nishimura, H. Takeya, T. Sueyoshi, T. Miyata, S. Iwanaga, T. Takao, Y. Shimonishi and T. Ikenaka, J. Biochem., 1988, 104, 867–868 CAS.
- L. Shao, Y. Luo, D. J. Moloney and R. S. Haltiwanger, Glycobiology, 2002, 12, 763–770 CrossRef CAS.
- S. Bjoern, D. C. Foster, L. Thim, F. C. Wiberg, M. Christensen, Y. Komiyama, A. H. Pedersen and W. Kisiel, J. Biol. Chem., 1991, 266, 11051–11057 CAS.
- L. J. Otvos, L. Urge, M. Hollosi, K. Wroblewski, G. Graczyk, G. D. Fasman and J. Thurin, Tetrahedron Lett., 1990, 31, 5889–5892 CrossRef CAS.
- K. B. Reimer, M. Meldal, S. Kusumoto, K. Fukase and K. Bock, J. Chem. Soc., Perkin Trans. 1, 1993, 925–932 RSC.
- O. Seitz and C.- H. Wong, J. Am. Chem. Soc., 1997, 119, 8766–8776 CrossRef CAS.
- M. Mizuno, K. Haneda, R. Iguchi, I. Muramoto, T. Kawakami, S. Aimoto, K. Yamamoto and T. Inazu, J. Am. Chem. Soc., 1999, 121, 284–290 CrossRef CAS.
- T. Ichiyanagi, M. Takatani, K. Sakamoto, Y. Nakahara, Y. Ito, H. Hojo and Y. Nakahara, Tetrahedron Lett., 2002, 43, 3297–3300 CrossRef CAS.
- M. Gobbo, L. Biondi, F. Filira, R. Gennaro, M. Benincasa, B. Scolaro and R. Rocchi, J. Med. Chem., 2002, 45, 4494–4504 CrossRef CAS.
- J. Xue and Z. Guo, J. Org. Chem., 2003, 68, 2713–2719 CrossRef CAS.
- N. Yamamoto, Y. Ohmori, T. Sakakibara, K. Sasaki, L. R. Juneja and Y. Kajihara, Angew. Chem., Int. Ed., 2003, 42, 2537–2540 CrossRef CAS.
- L. H. Huang, X.-H. Ke, W. Sweeney and J. P. Tam, Biochem. Biophys. Res. Commun., 1989, 160, 133–139 CAS.
- Y. Yang, W. V. Sweeney, K. Schneider, B. T. Chait and J. P. Tam, Protein Sci., 1994, 3, 1267–1275 CAS.
- S. Y. Shin, T. Takenouchi, T. Yokoyama, T. Ohtaki and E. Munekata, Int. J. Peptide Protein Res., 1994, 44, 485–490 CAS.
- J. C. Spetzler and R. Chang, Int. J. Peptide Protein Res., 1994, 43, 351–358 CAS.
- D. P. Meininger, M. J. Hunter and E. A. Komives, Protein Sci., 1995, 4, 1683–1695 CAS.
- Y. Stenberg, K. Julenius, I. Dahlqvist and J. Stenflo, Int. J. Peptide Protein Res., 1997, 248, 163–170 CAS.
- M. Lohmeyer, P. M. Harrison, S. Kannan, M. DeSantis, N. J. O`Reilly, M. J. E. Sternberg, D. S. Salomon and W. J. Gullick, Biochemistry, 1997, 36, 3837–3845 CrossRef CAS.
- T. M. Hackeng, P. E. Dawson, S. B. H. Kent and J. H. Griffin, Biopolymers, 1998, 46, 53–63 CrossRef CAS.
- H. Chen, A. L. Pyluck, M. Janik and N. S. Sampson, Biopolymers, 1998, 47, 299–307 CrossRef CAS.
- A. Muranyi, B. E. Finn, G. P. Gippert, S. Forsen, J. Stenflo and T. Drakenberg, Biochemistry, 1998, 37, 10605–10615 CrossRef CAS.
- R. P. Diaugustine, R. Henry, C. H. Sewall, C. A. Suarez-Quian and M. P. Walker, Growth Factors, 1999, 17, 37–48 Search PubMed.
- T. J. Zamborelli, W. S. Dodson, B. J. Harding, J. Zhang, B. D. Bennet, D. M. Lenz, Y. Young, M. Haniu, C.-F. Liu, T. Jones and M. A. Jarosinski, J. Peptide Res., 2000, 55, 359–371 CrossRef CAS.
- M. Husbyn and A. Cuthbertson, J. Peptide Res., 2002, 60, 121–127 CrossRef.
- D. S. King, C. G. Fields and G. B. Fields, Int. J. Peptide Protein Res., 1990, 36, 255–266 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: experimental details. See http://www.rsc.org/suppdata/ob/b3/b312413d/ |
| This journal is © The Royal Society of Chemistry 2004 |