Conformation–activity relationships in polyketide natural products. Towards the biologically active conformation of epothilone
Richard E.
Taylor
*,
Yue
Chen†
,
Gabriel M.
Galvin‡
and
Praveen K.
Pabba§
Department of Chemistry & Biochemistry and the Walther Cancer Research Center, 251 Nieuwland Science Hall, University of Notre Dame, Notre Dame, IN, 46556-5670, USA. E-mail: taylor.61@nd.edu
First published on 20th November 2003
Abstract
The conformation–activity relationships for the biologically active polyketide, epothilone, have been determined. Computer-based molecular modeling and high field NMR techniques have provided the solution preferences for epothilones 1 and 2. For the C1–C8 polypropionate region, two conformational families, conformers 1 and 2, have been identified as having significant populations in polar and non-polar solvents. In the C11–C15 region, additional flexibility was observed and two local conformations have been identified as important, conformers 3 and 4. Epothilone analogues with altered conformational profiles have been designed and synthesized. Conformational analysis and the results of biological assays have been correlated to provide increased understanding of the biologically active conformation for the epothilone class of natural product. Conformation–activity relationships have been shown to be an important complement to structure–activity data.
Introduction
The epothilones represent an important lead in the search for improved chemotherapeutic agents, which has led to intense interest from the synthetic, medicinal, and biological communities.1 Biological experiments have shown that epothilones interact with β-tubulin at the paclitaxel-binding site.2 However, despite enormous efforts over the past several decades, only a low resolution (4 Å) electron diffraction structure of the tubulin dimer has been obtained.3 The lack of detailed structural information with regards to the interaction of paclitaxel (and other microtubule-stabilizing natural products)4 with tubulin at the molecular level has made rational design of analogues more difficult.5 Epothilone's relatively simple structure and accessibility from biosynthetic expression has allowed the preparation of hundreds of synthetic or semi-synthetic analogues. In fact, several compounds in the epothilone class are in the early stages of Phase I and Phase II clinical trials.6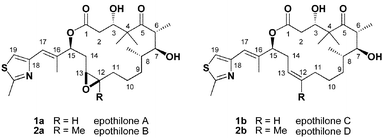
Many epothilone analogues that have been prepared were accessible due to their isolation as intermediates in total synthesis or as undesired by-products from unselective reactions. Most significantly, modifications to the C3–C8 polypropionate region, particularly stereochemical changes, drastically reduce their cytotoxicity and tubulin polymerization ability. Therefore, we proposed that the conformation of the macrolide ring along with its spatial relationship to the thiazole side chain are key elements, essential for the biological activity of epothilones. Moreover, these conformational requirements are controlled by the C3–C8 polypropionate region and the stereochemistry at C15. The overall goal of this project was the identification of the biologically active conformation of epothilone. Critical to the success of this novel approach would be the rational incorporation of functionality capable of affecting conformational preferences with minimal structural alteration. In this report, we demonstrate several examples of analogue design. One set of analogues probes the biologically active conformation in the C1–C8 polypropionate region. An additional four analogues were prepared to investigate the conformational flexibility of the C11–C15 region. From a general perspective this approach demonstrates that conformation–activity relationships are an important complement to classic SAR.
Results and discussion
Conformational preferences in the C1–C8 region
Several years ago, we reported a study on the conformational properties of the epothilones, which utilized a combination of computational methods and high field NMR experiments.7 Our report concentrated on the critical polypropionate region and concluded that, in solution, the C1–C11 of epothilones preferred to exist in at least two conformational families controlled by syn-pentane interactions and intramolecular hydrogen bonds.8 Since the epothilone conformation while bound to microtubules may not be similar to the solution structure, we felt it important to use computational modeling methods to gain insight into the available conformational space. This was accomplished by using internal coordinate Monte Carlo searching available in MacroModel (ver. 5.0 and 7.0).9 We determined that 10000 structures are sufficient to reproduce search convergence from different starting geometries. The conformations were minimized using the MM2* force field with both the generalized Born/surface area (GB/SA) water and chloroform models. However, we found that the chloroform model over-estimated the importance of internal hydrogen bonds and chose to use the water model on a routine basis. Analysis of the output file showed that the conformation of the C1–C11 region could be clustered into two low energy conformers within 2 kcal mol−1 of the global minimum (conformers 1 and 2, Fig. 1).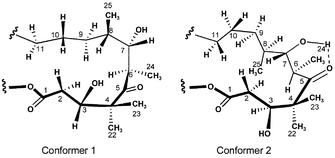 | ||
| Fig. 1 Calculated conformational preferences of the C1–C8 region. | ||
Conformer 1 is characterized by the C3–OH residing above the plane of the macrolide, a C5–C8 gauche− torsion, and a series of anti torsions across the C8–C12 region. In contrast, conformer 2 has the C3-hydoxyl roughly in the plane of the macrolide ring. An anti C5–C8 torsion is compensated by a C8–C11 gauche− torsion.
The computationally predicted conformer 1 is quite similar to the conformation of epothilone B in the solid state.10 However, proton NMR coupling constants as well as distinctive NOE data supported the existence of both conformers 1 and 2 in both polar (DMSO/D2O) and non-polar solvents. A strong NOE was observed between H3 and H6, which supports a significant population of conformations where the C3-hydroxyl resides in the plane of the macrolide. Thus, a putative hydrogen bond between the C3-hydroxyl and the C5 carbonyl (or C1 carbonyl) is not a controlling feature. In fact, it was recently reported that C3-cyano epothilones, which lack the ability to hydrogen bond, retain significant biological activity.11 A second important marker of conformational preference in solution is the H6–H7 coupling constant. Computational models suggested a H–C6–C7–H dihedral angle of −174° (Jcalc = 10.4 Hz) in conformer 1 and 66° (Jcalc = 1.0 Hz) in conformer 2. The Boltzman-weighted coupling constant for H6–H7 was 7.5 Hz. In excellent agreement with calculated predictions, the H6–H7 coupling constants of epothilones 1 and 2 have values of J6–7 = 2–5 Hz in chlorinated solvents and J6–7 = 8–10 Hz in more polar solvent such as DMSO-d6. These intermediate values support significant population of both conformers 1 and 2 with an increased population of conformations related to conformer 2 in non-polar solvents. This is to be expected due to the presence of an intramolecular hydrogen bond between the C7-hydroxyl and the C5 carbonyl, which is more likely in non-polar solvents.
Destabilization of conformer 1 through a designed syn-pentane interaction
Our approach to the identification of the conformational constraints to epothilone's biological activity would be based on the preparation of analogues with altered conformational preferences. Consideration of an appropriately positioned methyl group had a number of positive attributes and (S)-10-methylepothilone C 3 was selected as an initial target, Fig. 2.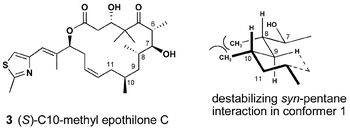 | ||
| Fig. 2 Conformer 2—stabilized analogue. | ||
This designed molecule 3 is “natural product-like” as it retains a polyketide pattern. In fact, methyl substitution at C10 represents an acetate → propionate switch in the biogenetic pattern. This has two interesting implications. During the evolution of the organism that produces the epothilones it is possible that this substitution pattern was previously biosynthesized and abandoned for lack of beneficial activity. Alternatively, if found to be active, the recent advances in genetic engineering of polyketide synthases (PKS) may provide an alternative synthetic route to this compound through manipulation of the epothilone PKS gene cluster.12 Finally, the S-stereochemistry was chosen to induce a destabilizing syn-pentane interaction with the methyl group at C8. Through this indirect manner we hoped to influence the conformational preferences of the polyketide region by minimal and distal structural modifications. In fact, computer-based conformational analysis of 3 showed a reversal of preference and conformer 2 became the global minimum. Thus, it was planned that solution conformational analysis and the biological activity of analogue 3, accessible via total synthesis, would provide critical information with regards to the tubulin-bound conformation of epothilone's C1–C8 region.
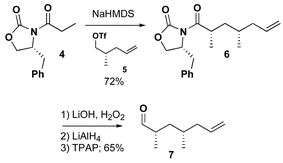
The strategy we chose for the synthesis of analogue 3 was related to our early efforts in the total synthesis of epothilone A and C, and highlighted by a ring-closing metathesis to form a C12–C13 olefin. While several groups have used a similar disconnection strategy for the total synthesis of epothilone A, we reported original and independent approaches to each of three fragments.13 For the synthesis of analogue 3, aldehyde 7 was prepared as shown in Scheme 1. The methyl-substituted stereogenic centers were created using Evans' oxazolidinone chemistry.14 Each alkylation proceeded in good yield with excellent stereocontrol. The chiral auxiliary was removed by hydrolysis and the resulting carboxylic acid reduced to the primary alcohol, which proved to be a convenient point for storage. When appropriate, oxidation with TPAP provided the aldehyde 7 in good yield.
 | ||
| Scheme 1 | ||
The coupling of the three fragments is summarized in Scheme 2. Aldol reaction between the lithium enolate of ketone13a8 and aldehyde 7 provided the desired 6,7-syn-7,8-anti stereochemistry of the aldol product 9 in 70% yield with 5 : 1 diastereoselectivity. A comparison of proton NMR data between 9 and related compounds prepared in our laboratory as well as compounds prepared by others in the field supported the expected anti-Felkin selectivity. The rationalization of the facial selectivity is based upon the detailed analysis of the aldol reaction presented by Roush15a and explored in detail by Danishefsky15b as applied to the epothilone system. After the aldol and protecting group manipulation, the primary alcohol was oxidized to carboxylic acid 10, which was necessary for coupling to thiazole13b11. The sequence to (S)-10-methylepothilone C was then completed in 3 steps. Thiazole 11 was coupled to acid 10 under Yamaguchi conditions.16 Ring-closing olefin metathesis of the diene 12 provided the 16-membered macrolide as a 1.5 : 1 mixture of Z- and E-isomers. The designed analogue 3 was then generated by deprotection of the silyl ethers with TFA. Compound 3 and its E-12,13-olefin isomer were easily separated by chromatography.
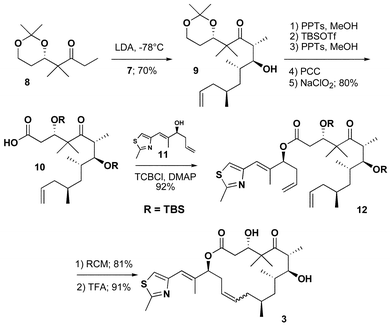 | ||
| Scheme 2 | ||
With analogue 3 in hand, solution NMR experiments were used to determine its conformational preferences. As highlighted in Fig. 3 a number of key transannular NOEs were observed to support the presence of conformations related to conformer 2. The observed H6–H7 coupling constant of 0 Hz matched calculated predictions for conformer 2 (1.0 Hz). In contrast, conformer 1 has a calculated H6–H7 coupling constant of 10.0 Hz. NMR experiments at varied temperatures (10–60 °C) showed key differences between the C3 and C7 hydroxyl protons. The change in chemical shift was much greater for C3–OH (Δδ = 0.7 ppm) than for C7–OH (Δδ = 0.3 ppm). This supports a strong hydrogen bond between the C7–OH and the C5 carbonyl. Attempts to obtain crystals suitable for X-ray crystallographic analysis were unsuccessful. Despite the lack of information with regard to the solid-state conformation, the solution data do support the population of conformations related to conformer 2.
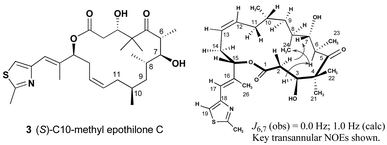 | ||
| Fig. 3 Solution conformation of (S)-10-methyl epothilone C. | ||
Purified samples of (S)-10-methylepothilone C 3, as well as the 12,13-E isomer, were submitted to Bristol-Myers Squibb for biological evaluation. As shown in Table 1, neither compound showed any cytotoxicity in the explored range of tumor cell lines. As with a classic SAR profile much can be learned from inactive compounds. The significant decrease in activity in the C10-methyl series may not be attributed to the minimal structural alteration provided by the C10-methyl. A significant lowering of the population of conformations related to conformer 1 is more likely responsible for the attenuation of cytotoxicity. In fact, cyclopropane-containing analogue 13, Fig. 4, has been recently shown to possess significant activity despite having C10-subsitution.17 In contrast to 3 the C10-alkyl substituent of 13 is tied back relieving the syn-pentane interaction with the methyl group at C8 and repopulating conformer 1. Moreover, the trans-cyclopropane ring rigidifies the C9–C10–C11–C12 torsion to ∼180° similar to active analogue epo 490 (14).17
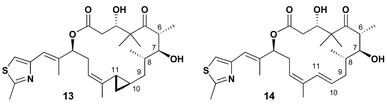 | ||
| Fig. 4 Biologically active epothilone analogues. | ||
| A2780 | SKBR3 | HCT116 | LX-1 | K562 | |
|---|---|---|---|---|---|
| 1a | 7.36 | 6.57 | 6.88 | 5.30 | 5.92 |
| 1b | 0.57 | 1.65 | 1.33 | 0.25 | 0.38 |
| 2a | 71.0 | 46.4 | 47.9 | 47.8 | 56.9 |
| 2b | 17.3 | 22.5 | 10.8 | 8.4 | 12.3 |
| 3 | >1012 | >1012 | >1012 | >1012 | >1012 |
| E-3 | >1012 | >1012 | >1012 | >1012 | >1012 |
Stabilization of conformer 1 through torsional rigidity (C8–C11)
There is additional evidence that conformations related to conformer 1 are important for biological activity. The Danishefsky group reported the synthesis of 9,10-dehydroepothilone D 15, which was prepared via a ring-closing metathesis as an alternative route to the natural series through selective hydrogenation of the 9,10-olefin, Fig. 5.18 Biological assays of the intermediate unsaturated analogue 15 showed increased cytotoxicity relative to epothilone D. More recently, epoxide 16 was shown to be one of the most active epothilone derivatives prepared to date with >3× the activity of epothilone B itself.19 Our contributed analysis of NMR and computational studies concluded that while the presence of the 9,10-trans-olefin stabilized conformer 1 in the polypropionate region (C1–C8), it also increased the flexibility of the C11–C15 olefin region.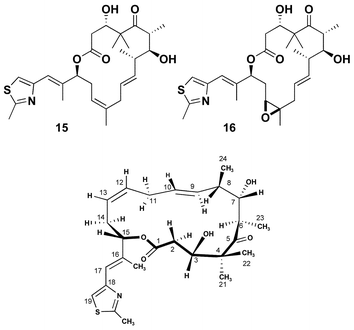 | ||
| Fig. 5 Structure and preferred conformation of 9,10-dehydro-epothilones. | ||
Conformational flexibility of the C11–C15 region
Exploration of the conformational properties of the C11–C15 region of this molecule was a critical next phase in the analysis of the epothilone pharmacophore.20 Coupled with our understanding of the polypropionate region, this would complete our studies and, hopefully, allow the design of structurally simplified analogues, which could mimic the biologically active conformation of epothilone and still retain significant activity. Conformational analysis of the C11–C15 region of epothilones B and D suggested conformational flexibility. The two extreme conformations, conformer 3 and conformer 4, are represented in Fig. 6. Conformer 3 is characterized by an anti relationship between C13 and C16 while, on the other hand, conformer 4 contains a gauche relationship. Computational and NMR studies suggested the presence of each, in solution, but that conformer 3 is more highly populated than conformer 4. This is also supported by experimental findings where epoxidation of epothilone D gave good β-selectivity providing direct access to epothilone B.21 Several groups have reported models of the epothilone pharmacophore and its relationship to other classes of microtubule-stabilizing natural products based on computational methods and reported structure–activity relationships.22 In fact, two of these studies have proposed that the bioactive conformation of the epothilone epoxide region is similar to conformer 4.22a,b,23,24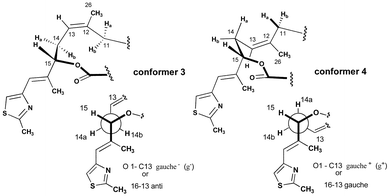 | ||
| Fig. 6 Conformation of the C11–C14 region of epothilones. | ||
In an effort to distinguish conformers 3 and 4 and determine which is more closely related to the biologically active conformation of the epothilones, we designed and prepared simple C14-methyl substituted compounds 17–20, Fig. 7. We rationalized that 17/18 and 19/20 would preferentially exist in conformers 3 and 4, respectively. In addition, the presence of the C14-methyl substituent would destabilize the alternative conformer. These preferences were supported by computer modeling techniques similar to our previous analysis of the natural products. It is important to again point out that the methyl analogues 17–20 represent an acetate → propionate switch in the biogenetic pathway to these compounds. Thus, they could represent compounds accessible to the microorganism in its evolutionary history or by genetic manipulation of its polyketide synthase machinery in the future.
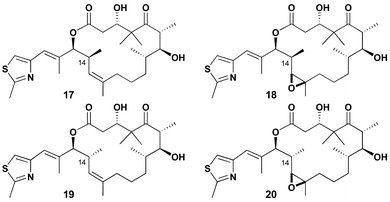 | ||
| Fig. 7 Conformer 3 and 4 stabilized analogues. | ||
The four analogues were prepared using the identical methodology we developed and successfully applied to the total synthesis of epothilones B and D.25 Thiazole aldehyde 21 is the point of divergence for the synthesis of 22 and 23, Scheme 3. Brown asymmetric crotylboration26 efficiently controlled the enantio- and diastereoselectivity of the C14, C15 stereogenic centers. Model systems suggested that the steric hindrance provided by the additional methyl substituent at C14 would cause selectivity issues with the late stage deprotection of a C15 TBS ether. Therefore, we protected the secondary hydroxyl as a tert-butoxydiphenylsilyl ether which is more easily removed under basic conditions. Oxidative cleavage provided 22 and 23 in good yield.
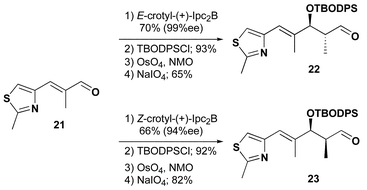 | ||
| Scheme 3 | ||
As detailed in Scheme 4, intermolecular Ni/Cr coupling27 of vinyl iodide 24 with the thiazole aldehyde 22 (2 equiv.) provided the desired allylic alcohol 25 in 75% yield as a single diastereomer. The relative stereochemistry at this position was inconsequential since exposure to thionyl chloride in ether–pentane provided the desired primary chloride 26 in 85% yield with excellent stereochemical control. LiEt3BH not only reductively cleaved the chiral auxiliary but also efficiently reduced the allylic chloride in 72% yield providing the primary alcohol. Oxidation with Dess–Martin periodinane28 then provided aldehyde 27. The tandem Ni/Cr coupling followed by thionyl chloride rearrangement has become a powerful method for natural product fragment coupling and the control of trisubstituted olefin geometry.
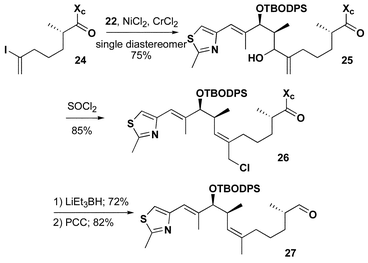 | ||
| Scheme 4 | ||
We had previously reported an efficient route to the chiral ketone necessary for the preparation of epothilone congeners.13a However, the synthesis of the chiral starting material for that route was dependent on an enzymatic resolution of a secondary alcohol which was found to make large-scale production tedious. Scheme 5 describes an alternative route, which exploits the elegant chemistry of Kiyooka.29 The reaction of 2-methyl-1-phenoxy-1-trimethylsilyloxy-1-propene with readily available aldehyde 28 under the influence of Kiyooka's chiral boron reagent provided aldol adduct 29 in 71% yield (96% ee by chiral GC analysis). The use of the phenoxy-ketene acetal not only provides higher enantioselectivity than the commercially available methoxy-ketene acetal but also allows for a one-step ester-to-ketone conversion. Protection of the secondary alcohol was followed by exposure to ethylmagnesium bromide in the presence of triethylamine which provided the desired ethyl ketone 30.
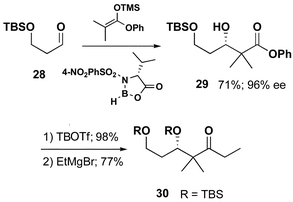 | ||
| Scheme 5 | ||
Completion of the route to (S)-14-methylepothilone D 17 was accomplished by aldol condensation, functional group manipulation, and lactonization as detailed in Scheme 6.
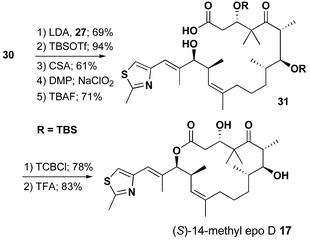 | ||
| Scheme 6 | ||
The conversion of aldehyde 23 to (R)-14-methyl epo D 19 preceeded with similar efficiency through an identical synthetic sequence as outlined in Schemes 7 and 8.
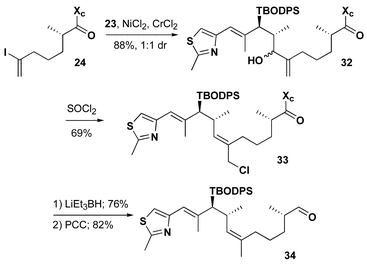 | ||
| Scheme 7 | ||
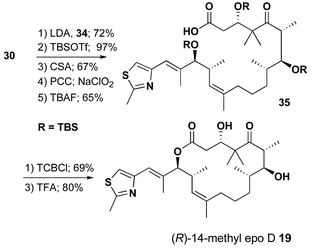 | ||
| Scheme 8 | ||
(S)-14-Methylepothilone D 17 underwent a highly selective epoxidation with mCPBA to provide (R)-14-methylepothilone B 18 in 57% yield, Scheme 9. Epoxidation of (R)-14-methylepothilone D 19 also proceeded in a highly selective fashion. However, the C11–C15 region of 19 exists primarily in conformer 4 due to the conformational constraints imposed by the C14-methyl substituent. Therefore, the stereochemistry of the epoxide 36 is epimeric to epothilone B and analogue 18.
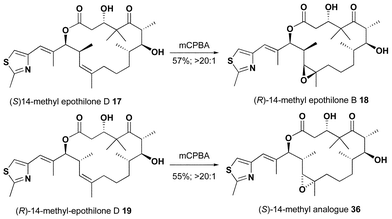 | ||
| Scheme 9 | ||
X-Ray diffraction studies of single crystals of 17 and 18·H2O showed that the conformation of the C11–C15 region of each in the solid-state was quite similar to that reported for epothilone B and thus conformer 3.20 Proton NMR coupling constants (J14–15 = 9.9, 10.5 Hz, respectively) also supported this preference in solution. In contrast, proton NMR coupling constants of analogues 19 and 36 had the expected values for conformer 4 (J14–15 = 3.3, < 2.0 Hz, respectively). Computational models, Fig. 6, suggested a H–C14–C15–H dihedral angle of 175° (Jcalc = 10.9 Hz) in 17 (conformer 3) and 62° (Jcalc = 3.1 Hz) in 19 (conformer 4). A strong NOE enhancement was observed between the H14 and one of the C11 protons in both 17 and 19. In addition a strong NOE enhancement was observed between H15 and H13 in epoxide 18 but not in epoxide 36. Attempts to obtain single crystals of 19 and 36 were unsuccessful. However, additional NMR evidence for these conformational differences was observed in ROESY experiments on all four analogues. An NOE enhancement between H13 and the C16–CH3 was only observed in analogues 19 and 36 but not in 17 and 18. These data not only support the preference for conformer 4 but also the configuration of epoxide 36.
As we previously reported, the biological activity of epothilone analogues 17, 18, 19, and 36 were evaluated against a panel of human tumor cell lines by collaborators at Kosan Biosciences.20 Compounds 17 and 18 (conformer 3 preference) maintain significant cytotoxicity similar to epothilone D. In marked contrast, analogues 19 and 36 (conformer 4 preference) showed no measurable cytotoxicity. This study is thus another clear example that conformation has a significant impact on the biological activity.
The conformation–activity relationship study carried out on the C11–C15 region of epothilones strongly supports the importance of conformer 3 as the bioactive conformation of the 12,13-epoxide (olefin) region of the epothilones. Moreover, investigation of the polypropionate region found strong evidence for the activity of conformers closely related to conformer 1 despite alternative conformations, with significant population, found in solution. It is clear from this study that the conformation originally observed in the solid state is closely related to the bound structure. Recently, the Novartis group reported the conformation of epothilone A while bound to tubulin through elegant use of transfer NOE and cross correlation relaxation data.30 In fact, their reported structure is consistent with conformers 1 and 3 although their proposed orientation of the C3-hydroxyl is in the plane of the macrolide as in conformer 2. Of additional significance is their proposed orientation of the thiazole side chain, a region previously shown to be flexible in solution.7
Overall, the approach presented here offers a new perspective on rational design of modified biologically active polyketide macrolides which complements transfer NOE studies and can directly provide new leads for further investigation. In addition, the conformational analogues such as those presented here are designed such that the recent advances in genetic engineering of polyketide synthases (PKS) may provide an alternative synthetic route through manipulation of the PKS gene cluster. This work has demonstrated that the successful design of polyketide analogues is reliant on an understanding of the structural and conformational constraints of the pharmacophore.
Acknowledgements
Dr. Jaroslav Zajicek is thanked for his assistance with NMR experiments. Dr. Craig Fairchild and Dr. Barry Long (Bristol-Myers Squibb) are acknowledged for the biological activity data on compound 3. Support provided by the National Cancer Institute/National Institutes of Health (CA85499). YC thanks the University of Notre Dame for support through a Reilly Fellowship. RET acknowledges support from Bristol-Myers Squibb, Kosan Biosciences, and Eli Lilly through a Lilly Grantee award.References
- (a) S. Borman, Chem. Eng. News, 2002, 80, 35; (b) K. C. Nicolaou, A. Ritzen and K. Namoto, Chem. Commun., 2001, 1523 RSC; (c) K. C. Nicolaou, F. Roschanger and D. Vourloumis, Angew. Chem., Int. Ed., 1998, 37, 2014 CrossRef CAS.
- D. M. Bollag, P. A. McQueney, J. Zhu, O. Hensens, L. Koupal, J. Leiesch, M. Goetz, E. Lazarides and C. W. Woods, Cancer Res., 1995, 55, 2325 CAS.
- (a) E. Nogales, S. G. Wolf and K. H. Downing, Nature, 1998, 391, 199 CrossRef CAS; (b) P. Muerer-Grob, J. Kasparian and R. H. Wade, Biochemistry, 2001, 40, 8000 CrossRef.
- For a recent review of microtubule-stabilizing compounds see: D. C. Myles, Annu. Rep. Med. Chem., 2002, 37, 125 Search PubMed.
- J. P. Snyder, J. H. Nettles, B. Cornett, K. H. Downing and E. Nogales, Proc. Natl. Acad. Sci. USA, 1999, 96, 5312 CrossRef.
- R. M. Borzilleri and G. D. Vite, Drugs Future, 2002, 27, 1149 Search PubMed.
- R. E. Taylor and J. Zajicek, J. Org. Chem., 1999, 64, 7224 CrossRef CAS.
- (a) R. W. Hoffman, Angew. Chem., Int. Ed. Engl., 1992, 31, 1124 CrossRef; (b) R. W. Hoffman, Angew. Chem., Int. Ed., 2000, 39, 2054 CrossRef.
- F. Mohamadi, N. G. J. Richards, W. C. Guida, R. Liskamp, M. Lipton, C. Caufield, G. Chang, T. Hendrickson and W. C. Still, J. Comput. Chem., 1990, 11, 440 CrossRef CAS.
- G. Hoefle, N. Bedorf, H. Steinmetz, D. Schomburg, K. Gerth and H. Reichenbach, Angew. Chem., Int. Ed. Engl., 1996, 35, 1567 CrossRef.
- A. Regueiro-Ren, K. Leavitt, S. Kim, G. Höfle, M. Kiffe, J. Z. Gougoutas, J. D. DiMarco, F. Y. F. Lee, C. R. Fairchild, B. H. Long and G. D. Vite, Org. Lett., 2002, 4, 3815 CrossRef CAS.
- (a) C. Khosla, J. Org. Chem., 2000, 65, 8127 CrossRef CAS; (b) B. Julien, S. Shah, R. Ziermann, R. Goldman, L. Katz and C. Khosla, Gene, 2000, 249, 153 CrossRef CAS; (c) L. Tang, S. Shah, L. Shung, J. Carney, L. Katz, C. Khosla and B. Julien, Science, 2000, 287, 640 CrossRef CAS.
- (a) R. E. Taylor, G. M. Galvin, K. A. Hilfiker and Y. Chen, J. Org. Chem., 1998, 63, 9580 CrossRef CAS; (b) R. E. Taylor and J. D. Haley, Tetrahedron Lett., 1997, 38, 2061 CrossRef CAS.
- D. A. Evans, M. D. Ennis and D. J. Mathre, J. Am. Chem. Soc., 1982, 104, 1737 CrossRef CAS.
- (a) W. R. Roush, J. Org. Chem., 1991, 56, 4151 CrossRef CAS; (b) C. B. Lee, Z. Wu, F. Zhang, M. D. Chapell, S. J. Stachel, T.-C. Chou, Y. Guan and S. J. Danishefsky, J. Am. Chem. Soc., 2003, 123, 5249 CrossRef CAS.
- J. Inanaga, K. Hirata, H. Saeki, T. Katsuki and M. Yamaguchi, Bull. Chem. Soc. Jpn., 1979, 52, 1989 CAS.
- K. Biswas, H. Lin, J. T. Njardarson, M. D. Chappell, T.-C. Chou, Y. Guan, W. P. Tong, L. He, S. B. Horwitz and S. J. Danishefsky, J. Am. Chem. Soc., 2002, 124, 9825 CrossRef CAS.
- A. Rivkin, F. Yoshimura, A. E. Gabarda, T.-C. Chou, H. Dong, W. P. Tong and S. J. Danishefsky, J. Am. Chem. Soc., 2003, 125, 2899 CrossRef CAS.
- F. Yoshimura, A. Rivkin, A. E. Gabarda, T.-C. Chou, H. Dong, G. Sukenick, F. F. Morel, R. E. Taylor and S. J. Danishefsky, Angew. Chem., Int. Ed., 2003, 42, 2518 CrossRef CAS.
- R. E. Taylor, Y. Chen, A. Beatty, D. C. Myles and Y. Zhou, J. Am. Chem. Soc., 2003, 125, 26 CrossRef CAS.
- S. J. Stachel and S. J. Danishefsky, Tetrahedron Lett., 2001, 42, 2895 CrossRef CAS.
- (a) P. Giannakakou, R. Gussio, E. Nogales, K. H. Downing, D. Zaharevitz, B. Bollbuck, G. Poy, D. Sackett, K. C. Nicolaou and T. Fojo, Proc. Natl. Acad. Sci. USA, 2000, 97, 2904 CrossRef CAS; (b) M. Wang, X. Xia, D. Hwang, J. M. Jansen, M. Botta, D. C. Liotta and J. P. Snyder, Org. Lett., 1999, 1, 43 CrossRef CAS; (c) L. Ojima, S. Chakravarty, T. Inoue, S. N. Lin, L. F. He, S. B. Horwitz, S. D. Kuduk and S. J. Danishefsky, Proc. Natl. Acad. Sci. USA, 1999, 96, 4256 CrossRef CAS; (d) P. Ballone and M. Marchi, J. Phys. Chem. A, 1999, 103, 3097 CrossRef CAS.
- The biological activity of 12,13-cyclopropyl and 12,13-aziridinyl epothilones strongly support the importance of conformer 3 in the C11–C15 region. (a) J. Johnson, S.-H. Kim, M. Bifano, J. DiMarco, C. Fairchild, J. Z. Gougoutas, F. Lee, B. Long, J. Tokarski and G. D. Vite, Org. Lett., 2000, 2, 1537 CrossRef CAS; (b) A. Regueiro-Ren, R. M. Borzilleri, X. Zheng, S.-H. Kim, J. A. Johnson, C. R. Fairchild, F. Y. F. Lee, B. H. Long and G. D. Vite, Org. Lett., 2001, 3, 2693 CrossRef CAS; (c) K. C. Nicolaou, K. Namoto, A. Ritzen, T. Ulven, M. Shoji, J. Li, G. D'Amico, D. C. Liotta, C. T. French, M. Wartmann, K.-H. Altmann and P. Giannakakou, J. Am. Chem. Soc., 2001, 123, 9313 CrossRef CAS.
- Conformer 4 has recently been observed in the solid-state structure of an epothilone B analogue; see reference 11.
- (a) R. E. Taylor and Y. Chen, Org. Lett., 2001, 3, 2221–2224 CrossRef CAS; (b) the complete experimental details and characterization data for the synthesis of 17, 18, 19, and 36 are included in supporting information of reference 20.
- H. C. Brown and S. B. Krishna, J. Am. Chem. Soc., 1986, 108, 5919 CrossRef CAS.
- (a) K. Takai, M. Tagashira, T. Kuroda, K. Oshima, K. Utimoto and H. Nozaki, J. Am. Chem. Soc., 1986, 108, 6048 CrossRef CAS; (b) H. Jin, J. Uenishi, W. J. Christ and Y. Kishi, J. Am. Chem. Soc., 1986, 108, 5644 CrossRef CAS.
- D. B. Dess and J. C. Martin, J. Org. Chem., 1983, 48, 4155 CrossRef CAS.
- S.-i. Kiyooka, H. Kira and M. A. Hena, Tetrahedron Lett., 1996, 37, 2597 CrossRef CAS.
- T. Carlomagno, M. J. J. Blommers, J. Meiler, W. Jahnke, T. Schupp, F. Peterson, D. Schinzer, K.-H. Altmann and C. Griesinger, Angew. Chem., Int. Ed., 2003, 42, 2511 CrossRef CAS.
Footnotes |
| † Current address: Kosan Biosciences, Hayward, CA. |
| ‡ Current address: Bristol-Myers Squibb, Syracuse NY. |
| § Current address: Lexicon Pharmaceuticals, Princeton, NJ. |
| This journal is © The Royal Society of Chemistry 2004 |