Synthesis and cytotoxicity of 9-(2-deoxy-2-alkyldithio-β-D-arabinofuranosyl)purine nucleosides which are stable precursors to potential mechanistic probes of ribonucleotide reductases
Stanislaw F. Wnuk*a, Elzbieta Lewandowska†a, Dania R. Companionia, Pedro I. Garcia Jra and John A. Secrist IIIb
aDepartment of Chemistry and Biochemistry, Florida International University, Miami, FL 33199, USA. E-mail: wnuk@fiu.edu; Fax: (305) 348-3772
bSouthern Research Institute, Birmingham, AL 35205, USA. E-mail: secrist@sri.org
First published on 20th November 2003
Abstract
A series of 2′-thionucleosides, as potential inhibitors of ribonucleotide reductases, has been synthesized. Treatment of the 3′,5′-O-TPDS-2′-O-(trifluoromethanesulfonyl)adenosine with potassium thioacetate gave the arabino epimer of 2′-S-acetyl-2′-thioadenosine which was deacetylated to give 9-(3,5-O-TPDS-2-thio-β-D-arabinofuranosyl)adenine in high yield. Treatment of the latter with diethyl azodicarboxylate–C3H7SH–THF gave 2′-propyl disulfide which was desilylated to give 9-(2-deoxy-2-propyldithio-β-D-arabinofuranosyl)adenine. Subsequent tosylation (O5′) and displacement of the tosylate with pyrophosphate afforded the 5′-O-diphosphate in a stable form as propyl mixed-disulfide, which upon treatment with dithiothreitol releases 9-(2-thio-β-D-arabinofuranosyl)adenine 5′-diphosphate. The arabino 2′-mercapto group might interact with the crucial thiyl radical at cysteine 439 leading to the inhibition of ribonucleotide reductases via formation of a Cys439–2′-mercapto disulfide bridge. The 2,6-diamino-, 2-amino-6-chloro- and 2-amino-6-methoxypurine ribosides were also converted to the corresponding 2′-deoxy-2′-propyldithio-β-D-arabinofuranosyl nucleosides, which might serve as convenient precursors to the arabino epimer of 2′-thioguanosine. Analogously, 2′-deoxy-2′-propyldithioadenosine was prepared from 9-(β-D-arabinofuranosyl)adenine. The nucleoside disulfides show modest cytotoxicity in a panel of human tumor cell lines.
Introduction
Ribonucleotide reductases (RNRs) are ubiquitous enzymes that execute conversion of 5′-(di- or tri)phosphate esters of ribonucleosides into the requisite 2′-deoxy building blocks.1 Inhibition of RNRs disrupts the primary source of DNA components and is an appealing target for the rational drug design.2 The ribonucleoside diphosphate reductase (RDPR) from Escherichia coli consists of two subunits (R1 and R2) whose X-ray structure have been determined.3 The R1 subunit contains allosteric control sites and cysteine residues that participate in catalytic turnover and/or as redox dithiol/disulfide pairs. The R2 subunit contains an essential tyrosine (Tyr122) free radical that is responsible for generation of a proximate thiyl radical (Cys439) on R1. A thiyl radical initiates nucleotide reduction by abstraction of H3′ from the substrate ribonucleotide and water (O2′) is then lost from C2′ of the resulting hydrogen-bonded C3′ radical.1a,4In spite of numerous attempts using modified substrates as radical traps for both enzyme and substrate radicals, direct observation of nucleotide radical intermediates has remained elusive until very recently.5–7 Elegant EPR studies with (E)-2′-deoxy-2′-(fluoromethylene)cytidine 5′-diphosphates and its [2H] and [13C] isotopomers has allowed detection and characterization of a substrate-derived allyl radical during inactivation of RDPR.2,5 Spectroscopic evidence has been provided for new radical intermediates in the reduction of cytidine 5′-diphosphate (CDP) by Glu441Gln R1 mutant of RDPR.6,7 Furthermore, radicals at C3′ of adenosine,8 and 2′-substituted homonucleosides9a and homoribofuranose9b have been selectively generated to simulate the initiation–elimination cascades that occur during enzymatic 2′-deoxygenation of ribonucleotides.
Fontecave and Decout and their coworkers demonstrated that 2′-thiouridine 5′-diphosphate A is a potent inactivator of RDPR (Fig. 1).10 They postulated that the mode of inactivation involved substrate-protein disulfide B which after generation of a radical at C3′ C underwent fragmentation to the protein-based perthiyl radical R1–SS·D.10a The perthiyl radical D can then add to the proximal 2′-deoxy-3′-ketouridine intermediate generated at the active site. Such an addition may produce new radicals which would resemble structures that were proposed to be formed during inactivation of RDPR by 2′-azido-2′-deoxyuridine 5′-diphosphate.11
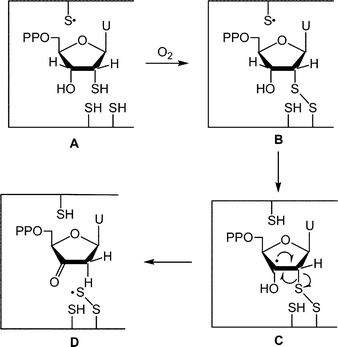 | ||
| Fig. 1 The proposed mechanism10a for the inhibition of RDPR by 2′-deoxy-2′-thiouridine 5′-diphosphate A. | ||
We now report the syntheses of stable mixed-disulfide precursors to 9-(2-thio-β-D-arabinofuranosyl)adenine 5′-diphosphate (E, Fig. 2). The arabino 2′-mercapto group in E may interact with Cys439 leading to the inhibition of RDPR via formation of R1 Cys439–2′-SH disulfide bridge F. Alternatively, competitive abstraction of hydrogen from the 2′-thiol group (path a) rather than the H3′ (path b) by Cys439 radical G may also be considered as a possible interaction pathway. The homolytic bond dissociation energies of 381 kJ mol−1 for S–H and 393 kJ mol−1 for H–C–O make hydrogen atom abstraction from the 2′-mercapto group (path a) thermodynamically more feasible.4 It is noteworthy that arabinonucleosides have some preferences12 for C2′ endo conformation which is conducive for the formation of disulfide bridge F or abstraction of a proton from the 2′-thiol (G, path a).
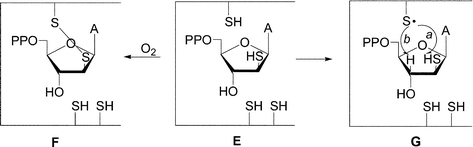 | ||
| Fig. 2 Possible interaction of RDPR with 9-(2-thio-β-D-arabinofuranosyl)adenine 5′-diphosphate. | ||
Results and discussion
From the available arabino thionucleosides,13 we chose an adenosine 2′-mercapto analog (e.g., 414) because attempts to synthesize 1-(2-thio-β-D-arabinofuranosyl)uracil failed due to the rapid Michael addition of the arabino 2′-mercapto function across the double bond of the uracil moiety to give 2′-deoxy-2′,6-epithio-5,6-dihydro derivative.15 The synthesis of arabino 2′-thio analogs was attempted using two different approaches. In our first approach, treatment of the 3′,5′-O-[1,1,3,3-tetraisopropyldisiloxane(TPDS)-1,3-diyl]-2′-O-(trifluoromethanesulfonyl)adenosine16 (1) with potassium thioacetate gave the arabino epimer of the protected 2′-S-acetyl-2′-thioadenosine (2, Scheme 1). Sequential removal of the 3′,5′-O-TPDS [tetrabutylammonium fluoride (TBAF)] and 2′-S-acetyl (NH3–MeOH) groups afforded 9-(2-thio-β-D-arabinofuranosyl)adenine14 (4; 41% from 1) after purification on Dowex 1 × 2 (OH−) column. Compound 4 had spectroscopic data as reported14b but slow formation of the corresponding disulfide was apparent since treatment of the mixture with dithiothreitol (DTT) led to a sharper spot on TLC.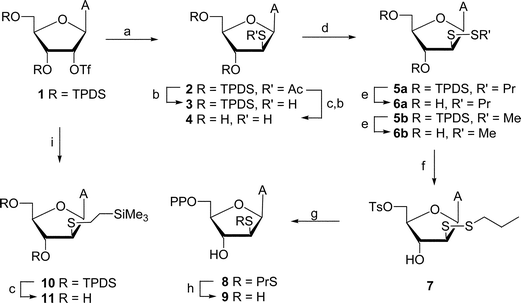 | ||
| Scheme 1 Reagents and conditions: (a) CH3COSK–DMF; (b) NH3–MeOH; (c) TBAF–THF; (d) DEAD–PrSH or MeSH–THF; (e) NH4F–MeOH; (f) TsCl–pyridine; (g) (Bu4N)3·HP2O7–CH3CN; (h) DTT–H2O; (i) Me3SiCH2CH2SH–K2CO3–DMF. | ||
The thiols protected as mixed-disulfides were prepared using diethyl azodicarboxylate (DEAD) as an oxidizing agent.10b,17 Thus, treatment of 4 with DEAD and 1-propanethiol in THF gave disulfide 6a (18%) plus unchanged 4 (25%). In order to overcome the limited solubility of deprotected thiol 4 in THF, 9-(3,5-O-TBDS-2-thio-β-D-arabinofuranosyl)adenine 3 was prepared by deacetylation of 2 with NH3–MeOH (97%). Treatment of 3 with DEAD–C3H7SH–THF gave protected disulfide 5a (45%) but separation from the diethyl hydrazinedicarboxylate byproduct was tedious. Desilylation (NH4F–MeOH18) gave 6a (60%) which was readily purified. Diagnostic peaks for the propyl group were observed in the 1H and 13C NMR spectra of 5a and 6a.
In the second approach, chemistry utilizing 2-(trimethylsilyl)ethyl sulfides19 was employed for the attempted preparation of mixed-methyl disulfide 6b. Thus, displacement of triflate from 1 with 2-(trimethylsilyl)ethanethiolate in DMF proceeded smoothly at 60 °C to give 2′-S-[(2-trimethylsilyl)ethyl]-2′-thionucleoside 10 in good yield. Deprotection of 10 with TBAF gave 11, which also demonstrated stability of the 2-(trimethylsilyl)ethyl group towards fluoride.20 However, reaction of 10 or 11 with dimethyl(methylthio)sulfonium tetrafluoroborate19,20 in the presence of large excess of dimethyl sulfide failed to produce methyl mixed-disulfides 5b or 6b. Instead unchanged 10 or 11 and fluorescent-type byproduct(s) were isolated from the reaction mixtures indicating instability of the purine ring (as opposed to pyrimidine19) under the reaction conditions. However, treatment of thiol 3 with MeSH–DEAD–THF gave 5b that was deprotected to yield less sterically hindered 6b.
From the methods available for the 5′-phosphorylation of nucleosides,21 we chose the methodology of Poulter and coworkers22 that is based on the displacement of the 5′-O-tosylate ester with the corresponding pyrophosphate ion. Thus, disulfide 6a was converted to the 5′-O-tosylate 7 using standard chemistry with the bulky propyl group allowing selective 5′-O-tosylation. Treatment of 7 with tris(tetrabutylammonium) hydrogen pyrophosphate22 effected displacement to give 5′-diphosphate 8. The nucleotide 8 was purified by ion-exchange chromatography on a DEAE Sephadex A-25 (HCO3−) column using triethyl ammonium bicarbonate (TEAB, 0.05 → 0.50 M) as an elutant. The 31P NMR spectrum showed two doublets (J = 23.5 Hz) at δ −9.75 and −5.26 and the 1H NMR spectrum confirmed the structure. This material was converted to its sodium salt by passage through a Dowex 50 (Na+) column. Reduction of diphosphate 8 with a slight excess of DTT immediately generated10a the mercaptonucleotide 9. Dithiothreitol was chosen as a reducing agent since it is one of the electron sources used in the assays for ribonucleotide reductase activity in vitro.
In addition to the propyl 6a and methyl 6b mixed-disulfides, the ribo analogue 16 was synthesized in order to test differences in the biological activities between ribo and arabino epimers. Subjection of 9-(3,5-O-TPDS-2-O-triflate-β-D-arabinofuranosyl)adenine1612 to the similar dithiolation sequence (12 → 16, Scheme 2) gave ribo disulfide 16 in 11% overall yield.
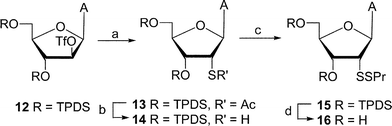 | ||
| Scheme 2 Reagents and conditions: (a) CH3COSK–DMF; (b) NH3–MeOH; (c) DEAD–PrSH–THF; (d) NH4–MeOH. | ||
The 2-amino-6-chloro-, 2-amino-6-methoxy-, and 2,6-diaminopurine ribosides are known to be substrates of adenosine deaminase.23 The deaminase-mediated hydrolysis at C6 of 2-amino-6-(substituted) mixed-disulfides 23a–c (Scheme 3) would generate the arabino epimer of 2′-thioguanosine. Subjection of 2-amino-6-chloro- (17a), 2-amino-6-methoxy- (17b) and 2,6-diaminopurine riboside (17c) to our triflation–dithiolation sequence (17 → 23), produced arabino mixed-disufides 23a (14%), 23b (10%), and 23c (24%; overall yield).
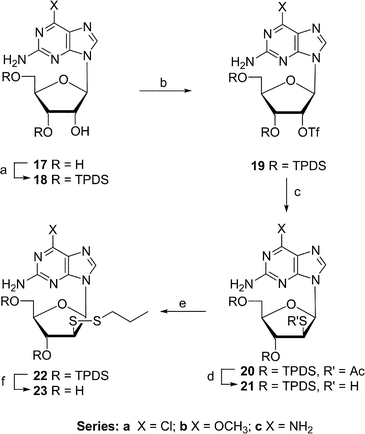 | ||
| Scheme 3 Reagents and conditions: (a) TPDS-Cl2–pyridine; (b) Tf2NPh–DMAP–CHCl2; (c) CH3COSK–DMF; (d) NH3–MeOH; (e) DEAD–PrSH–THF; (f) NH4F–MeOH. | ||
Cytotoxic activities
The disulfide nucleosides 6a, 6b, 16, and 23a–c were evaluated for their cytotoxicities in a panel of human tumor cell lines. This panel was comprised of SNB-7 (CNS), DLD-1 (colon), CCRF-CEM (leukemia), NCI-H23 (lung), ZR-75-1 (mammary), LOX IMVI (melanoma), PC-3 (prostate), and CAKI-1 (renal) cells. The two mixed arabino disulfides with adenine as the base, 6a and 6b, were not cytotoxic up to the highest doses evaluated (>150 µM) in any of the eight cell lines. The mixed ribo disulfide 16 had a weak IC50 of 110 µM in CCRF-CEM cells, and exhibited no cytotoxicity up to the highest doses in any other cell lines. The three mixed arabino disulfides containing 2,6-disubstituted purines were similarly non-toxic to most of the cell lines. In CCRF-CEM cells, which are known to be sensitive to many nucleosides, 23a had an IC50 of 14 µM and 23b had an IC50 of 19 µM. The only other cytotoxicities seen were for 23a (100 µM, NCI-H23), 23b (160 µM, DLD-1, and 140 µM, NCI-H23), and 23c (130 µM, CCRF-CEM). The lack of or very modest cytotoxicity seen with these analogs may be the result of their inability to be activated by the appropriate enzymes to the monophosphate level and then up to the di- and/or triphosphate levels.We have synthesized 9-(2-deoxy-2-propyldithio-β-D-arabinofuranosyl)adenine and 2-amino-6-substituted(amino/chloro/methoxy)purine analogues which are stable precursors of arabino 2′-deoxy-2′-mercaptonucleotides. The mixed propyl-disulfides were chosen as in general they could be deprotected under mild reductive conditions compatible with biological assays to form the corresponding mercaptonucleosides. These mercaptonucleosides can serve as valuable probes for understanding the role of the Cys439 radical during enzymatic deoxygenation reaction catalyzed by ribonucleotide reductases. Studies on inhibition of RDPR with 9 are in progress.24
Experimental
1H (Me4Si) NMR spectra were determined with solution in CDCl3 at 400 MHz, 13C (Me4Si) at 100.6 MHz and 31P (H3PO4) at 161.9 MHz unless otherwise noted. Mass spectra (MS) were obtained by atmospheric pressure chemical ionization (APCI) technique. Reagent grade chemicals were used and solvents were dried by reflux over and distillation from CaH2 under an argon atmosphere except THF (K–benzophenone). TLC was performed on Merck kieselgel 60-F254 with MeOH–CHCl3 (1 : 19) and EtOAc–hexane (4 : 1) as developing systems, and products were detected with 254 nm light. Merck kieselgel 60 (230–400 mesh) was used for column chromatography. Elemental analyses were determined by Galbraith Laboratories, Knoxville, TN. The 2,6-(disubstituted)purine ribosides 17a–c were purchased from Berry & Associates, Inc, Ann Arbor, MI.9-[2-S-Acetyl-3,5-O-(1,1,3,3-tetraisopropyl-1,3-disiloxanyl)-2-thio-β-D-arabinofuranosyl]adenine (2)
9-[3,5-O-(1,1,3,3-Tetraisopropyl-1,3-disiloxanyl)-2-thio-β-D-arabinofuranosyl]adenine (3)
9-(2-Thio-β-D-arabinofuranosyl)adenine (4)
Treatment of 2 (283 mg, 0.50 mmol) with TBAF (1 M solution in THF; 1 mL) in THF (15 mL) at 0 °C for 30 min followed by NH3–MeOH (10 mL; 2 h, ambient temperature) gave crude 4. This material was partitioned (EtOAc–H2O) and the water layer was chromatographed [Dowex 1 × 2 (OH−); H2O → 25% MeOH–H2O) to give 414b [89 mg, 55%; contaminated (1H NMR) by the corresponding dimer-disulfide].9-[3,5-O-(1,1,3,3-Tetraisopropyl-1,3-disiloxanyl)-2-deoxy-2-propyldithio-β-D-arabinofuranosyl]adenine (5a)
9-[3,5-O-(1,1,3,3-Tetraisopropyl-1,3-disiloxanyl)-2-deoxy-2-methyldithio-β-D-arabinofuranosyl]adenine (5b)
A solution of 3 (100 mg, 0.19 mmol) and DEAD (29 µL, 0.19 mmol) in THF (5 mL) was stirred overnight at ambient temperature and then methanethiol (183 mg, 3.8 mmol) in THF (3 mL) was added. Stirring was continued at ambient temperature for 96 h, and then volatiles were evaporated and the residue was column chromatographed (EtOAc–hexane; 1 : 1) to give 5b (41 mg, 38%): 1H NMR δ 1.04–1.14 (m, 28, 4 × i-Pr), 2.32 (s, 3, SCH3), 3.92–3.99 (m, 2, H2′,4′), 4.05–4.21 (m, 2, H5′,5″), 4.76 (t, J = 7.8 Hz, 1, H3′), 6.07 (br s, 2, NH2), 6.53 (d, J = 7.1 Hz, 1, H1′), 8.01 (s, 1, H2), 8.33 (s, 1, H8); MS m/z 572 (100, MH+).9-(2-Deoxy-2-propyldithio-β-D-arabinofuranosyl)adenine (6a)
Treatment of 4 (159 mg, 0.56 mmol) with DEAD (98 mg, 0.57 mmol) and propane-1-thiol (2.9 mL, 2.4 g, 32 mmol) in THF (15 mL), as described for 5a, also gave 6a (36 mg, 18%).
9-(2-Deoxy-2-methyldithio-β-D-arabinofuranosyl)adenine (6b)
Treatment of 5b (19 mg, 0.033 mmol) by procedure D (chromatography: EtOAc → 4% MeOH–EtOAc) gave 6b (7.3 mg, 67%) as a white powder: UV max 261 nm (ε 12 700), min 230 nm (ε 2800); 1H NMR (MeOH-d4) δ 2.33 (s, 3, CH3S), 3.87–4.00 (m, 4, H2′,4′,5′,5″), 4.57 (t, J = 7.7 Hz, 1, H3′), 6.57 (d, J = 7.0 Hz, 1, H1′), 8.20 (s, 1, H2), 8.39 (s, 1, H8); MS m/z 330 (100, MH+). Anal. calcd. for C11H15N5O3S2·0.5 C4H8O2 (373.45; 1H NMR integration of residual EtOAc): C, 41.81; H, 5.13; N, 18.75. Found: C, 41.47; H, 5.38; N, 18.39%.9-[2-Deoxy-2-propyldithio-5-O-(p-tolylsulfonyl)-β-D-arabinofuranosyl]adenine (7)
p-Tolylsulfonyl chloride (106 mg, 0.55 mmol) was added to a solution of 6a (40 mg, 0.11 mmol) in anhydrous pyridine (2 mL) and the reaction mixture was stirred for 3 h at 0 °C. Volatiles were evaporated, and the residue was partitioned (NaHCO3–H2O–EtOAc). The organic layer was washed (AcOH–H2O, NaHCO3–H2O), dried (MgSO4), evaporated and column chromatographed (EtOAc → 2.5% MeOH–EtOAc) to give 7 (28 mg, 52%): 1H NMR δ 0.90 (t, J = 7.2 Hz, 3, CH3), 1.56 (sextet, J = 7.2 Hz, 2, CH2), 2.42 (s, 3, PhCH3), 2.59 (m, 2, CH2S), 3.90 (t, J = 8.2 Hz, 1, H2′), 4.16 (m, 1, H4′), 4.42 (dd, J = 3.1, 11.2 Hz, 1, H5′), 4.45 (dd, J = 4.5, 11.3 Hz, 1, H5″), 4.81 (t, J = 8.2 Hz, 1, H3′), 6.48 (d, J = 7.2 Hz, 1, H1′), 7.27 (d, J = 8.0 Hz, 2, HAr), 7.87 (d, J = 8.1 Hz, 2, HAr), 7.99 (s, 1, H2), 8.29 (s, 1, H8); MS m/z 512 (100, MH+).9-(2-Deoxy-2-propyldithio-β-D-arabinofuranosyl)adenine 5′-diphosphate (8)
Tris(tetra-n-butylammonium) hydrogenpyrophosphate22 (31 mg, 0.035 mmol) was added to a stirred solution of 7 (12 mg, 0.023 mmol) in CH3CN (0.5 mL) and stirring was continued at ambient temperature for 72 h. Volatiles were removed in vacuo and the residue was dissolved in water and was purified on an ion exchange resin [Sephadex-DEAE A-25, 40–125 µm; gradient elution with triethylammonium bicarbonate (0.05 → 0.5 M)]. Appropriated fractions were evaporated, and the residue was dissolved in H2O and was converted to a sodium salt by passing the solution through a Dowex (Na+) column to yield 8 (4 mg, 29%) as a trisodium salt: 1H NMR (MeOH-d4) δ 0.64 (t, J = 6.3 Hz, 3, CH3), 1.27 (sextet, J = 6.5 Hz, 2, CH2), 2.32 (t, J = 6.7 Hz, 2, CH2S), 3.88 (dd, J = 7.4, 8.8 Hz, 1, H2′), 4.03–4.07 (m, 1, H4′), 4.19–4.23 (m, 2, H5′,5″), 4.49 (t, J = 8.3 Hz, 1, H3′), 6.84 (d, J = 7.1 Hz, 1, H1′), 8.11 (s, 1, H2), 8.33 (s, 1, H8); 31P NMR δ −9.75 (d, J = 23.5 Hz, 1, Pα), −5.26 (d, J = 23.5 Hz, 1, Pβ).Treatment of 8 (1 mg) with dithiothreitol (1 mg) in H2O (1 mL) produced 9 as judged by the appearance of a new spot on TLC and immediate releasing of the PrSH odor.
9-[2-S-(2-Trimethylsilylethyl)-3,5-O-(1,1,3,3-tetraisopropyl-1,3-disiloxanyl)-2-thio-β-D-arabinofuranosyl]adenine (10)
2-(Trimethylsilyl)ethanethiol (0.1 mL, 90 mg, 0.7 mmol) was added to a stirred suspension of 1 (371 mg, 0.60 mmol) and K2CO3 (276 mg, 2.0 mmol) in DMF (3 mL) at ambient temperature, and the reaction mixture was heated at 60 °C overnight. Volatiles were evaporated and the residue was partitioned [CHCl3–NaHCO3–H2O (ice-cold)]. The organic layer was washed (NaCl–H2O), dried (MgSO4), evaporated and column chromatographed (EtOAc–hexanes; 1 : 1) to give 2 (108 mg, 30%): 1H NMR δ −0.07 (s, 9, SiMe3), 0.57 (dt, J = 5.2, 14.0 Hz, 1, CH2Si), 0.61 (dt, J = 5.2, 14.0 Hz, 1, CH2Si), 0.80–1.31 (m, 28, 4 × i-Pr), 2.49 (dt, J = 5.6, 12.7 Hz, 1, SCH2), 2.53 (dt, J = 5.6, 12.7 Hz, 1, SCH2), 3.72 (dd, J = 7.0, 9.7 Hz, 1, H2′), 3.89 (dt, J = 3.0, 7.9 Hz, 1, H4′), 4.09 (dd, J = 2.8, 12.9 Hz, 1, H5′), 4.16 (dd, J = 3.2, 12.9 Hz, 1 H5″), 4.55 (dd, J = 8.3, 9.3 Hz, 1, H3′), 6.50 (d, J = 7.0 Hz, 1, H1′), 8.08 (s, 1, H2), 8.32 (s, 1, H8); MS m/z 626 (100, MH+). Anal. calcd for C27H51N5O4SSi3·H2O (644.07): C, 50.35; H, 8.29; N, 10.87. Found: C, 50.39; H, 8.11; N, 10.49%.Treatment (ambient temperature to ∼45 °C, 2 h to overnight) of 10 (97 mg, 0.16 mmol) [or 11 (27 mg, 0.07 mmol)] with dimethyl(methylthio)sulfonium tetrafluoroborate (219 mg, 1.12 mmol) and dimethyl sulfide (0.72 mL, 496 mg, 8 mmol) in anhydrous THF (5 mL) gave recovered 10 or 11 (∼20% to 80% depends on reaction conditions) plus other byproduct(s).
9-[2-S-(2-Trimethylsilylethyl)-2-thio-β-D-arabinofuranosyl]adenine (11)
TBAF–THF (1M; 0.3 mL, 0.3 mmol) was added to a solution of 10 (50 mg, 0.08 mmol) in THF (2 mL) and was stirred for 2h at ambient temperature. Volatiles were evaporated and the residue was column chromatographed (EtOAc → 8% MeOH–EtOAc) to give 11 (27 mg, 90%): 1H NMR δ −0.08 (s, 9, SiMe3), 0.59–0.63 (m, 2, SiCH2), 2.40–2.45 (m, 2, SCH2), 3.74 (dd, J = 6.9, 8.8 Hz 1, H2′), 3.80–4.01 (m, 3, H4′,5′,5″), 4.32–4.38 (m, 1, H3′), 6.57 (d, J = 6.9 Hz, 1, H1′), 8.20 (s, 1, H2), 8.41 (s, 1, H8); MS m/z 384 (100, MH+).2′-S-Acetyl-3′,5′-O-(1,1,3,3-tetraisopropyl-1,3-disiloxanyl)-2′-thioadenosine (13)
Treatment of 1216 (290 mg, 0.45 mmol) by procedure A gave 1325 (200 mg, 78%): 1H NMR δ 1.05–1.12 (m, 28, 4 × i-Pr), 2.31 (s, 3, Ac), 4.02–4.09 (m, 3, H4′,5′,5″), 4.70 (dd, J = 4.8, 7.4 Hz, 1, H2′), 5.26 (dd, J = 5.4, 7.2 Hz, 1, H3′), 6.07 (d, J = 4.8 Hz, 1, H1′), 7.97 (s, 1, H2), 8.29 (s, 1, H8); MS m/z 568 (100, MH+).3′,5′-O-(1,1,3,3-Tetraisopropyl-1,3-disiloxanyl)-2′-thioadenosine (14)
Treatment of 13 (180 mg, 0.31 mmol) by procedure B gave 14 (160 mg, 96%): 1H NMR δ 1.03–1.15 (m, 28, 4 × i-Pr ), 2.19 (d, J = 6.4 Hz, 1, SH), 4.08 (d, J = 4.7 Hz, 1, H2′), 4.17–4.25 (m, 3, H4′,5′,5″), 4.88 (t, J = 6.4 Hz, 1, H3′), 5.98 (d, J = 7.1 Hz, 1, H1′), 6.22 (br s, 2, NH2), 8.01 (s, 1, H2), 8.34 (s, 1, H8); MS m/z 526 (100, MH+).3′,5′-O-(1,1,3,3-Tetraisopropyl-1,3-disiloxanyl)-2′-deoxy-2′-propyldithioadenosine (15)
Treatment of 14 (140 mg, 0.27 mmol) by procedure C gave 15 (67 mg, 42%): 1H NMR δ 0.89 (t, J = 7.3 Hz, 3, CH3), 1.07–1.12 (m, 28, 4 × i-Pr), 1.59 (sextet, J = 7.2 Hz, 2, CH2), 2.56 (t, J = 7.4 Hz, 2, SCH2), 4.05–4.07 (m, 2, H2′,4′), 4.19–4.26 (m, 2, H5′,5″), 5.20 (t, J = 7.3 Hz, 1, H3′), 6.07 (br s, 2, NH2), 6.34 (d, J = 3.5 Hz, 1, H1′), 8.03 (s, 1, H2), 8.29 (s, 1, H8); MS m/z 600 (100, MH+).2′-Deoxy-2′-propyldithioadenosine (16)
Treatment of 15 (60 mg, 0.1 mmol) by procedure D gave 16 (12.4 mg, 35%): mp 145–147 °C (dec; EtOAc–MeOH); UV max 260 nm (ε 15 400), min 229 nm (ε 4100); 1H NMR (MeOH-d4) δ 0.74 (t, J = 7.3 Hz, 3, CH3), 1.36 (sextet, J = 7.1 Hz, 2, CH2), 2.18–2.24 (m, 2, CH2S), 3.78 (dd, J = 2.4, 12.6 Hz, 1, H5′), 3.92 (dd, J = 2.6, 12.6 Hz, 1, H5″), 4.19–4.25 (m, 1, H4′), 4.30 (dd, J = 5.0, 9.2 Hz, 1, H2′), 4.57 (d, J = 5.0 Hz, 1, H3′), 6.20 (d, J = 9.2 Hz, 1, H1′), 8.22 (s, 1, H2), 8.35 (s, 1, H8); MS m/z 358 (100, MH+). Anal. calcd. for C13H19N5O3S2·0.5 C4H8O2 (401.50; 1H NMR integration of residual EtOAc): C, 44.87; H, 5.77; N, 17.44. Found: C, 44.48; H, 5.71; N, 17.21%.2-Amino-6-methoxy-9-[3,5-O-(1,1,3,3-tetraisopropyl-1,3-disiloxanyl)-β-D-ribofuranosyl]purine (18b)
TPDS-Cl2 (317 mg, 0.32 mL, 1.01 mmol) was added to a stirred solution of 2-amino-6-methoxy-(β-D-ribofuranosyl)purine (17b; 300 mg, 1.01 mmol) in dry pyridine (8 mL), and the mixture was stirred overnight at ambient temperature. Volatiles were evaporated and the residue was partitioned (EtOAc–H2O). The organic phase was washed [cold 0.1 M HCl–H2O (2 × 30 mL), H2O, saturated NaHCO3–H2O, and brine], dried (Na2SO4), and evaporated. Column chromatography (CHCl3 → 4% MeOH–CHCl3) gave 18b (375 mg, 69%): 1H NMR δ 0.93–1.12 (m, 28, 4 × i-Pr), 4.03–4.15 (m, 6, H4′,5′,5″, OCH3), 4.49 (d, J = 4.7 Hz, 1, H2′), 4.82 (m, 1, H3′), 5.32 (br s, 2, NH2), 6.04 (s, 1, H1′), 7.82 (s, 1, H8); MS m/z 540 (100, MH+). Anal. calcd for C23H41N5O6Si2 (539.77): C, 51.18; H, 7.66; N, 12.97. Found: C, 51.38; H, 7.88; N, 12.88%.2-Amino-6-chloro-9-[2-O-trifluoromethanesulfonyl-3,5-O-(1,1,3,3-tetraisopropyl-1,3-disiloxanyl)-β-D-ribofuranosyl]purine (19a)
2-Amino-6-methoxy-9-[2-O-trifluoromethanesulfonyl-3,5-O-(1,1,3,3-tetraisopropyl-1,3-disiloxanyl)-β-D-ribofuranosyl]purine (19b)
Treatment of 18b (200 mg, 0.37 mmol) by procedure E gave 19b (156 mg, 64%): 1H NMR δ 1.04–1.12 (m, 28, 4 × i-Pr), 4.03–4.23 (m, 6, H4′,5′,5″,OCH3), 4.85 (br s, 2, NH2), 4.94 (dd, J = 4.7, 9.0 Hz, 1, H3′), 5.70 (d, J = 4.7 Hz, 1, H2′), 6.04 (s, 1, H1′), 7.82 (s, 1, H8); MS m/z 672 (100, MH+).2,6-Diamino-9-[2-O-trifluoromethanesulfonyl-3,5-O-(1,1,3,3-tetraisopropyl-1,3-disiloxanyl)-β-D-ribofuranosyl]purine (19c)
Treatment of 18c27 (370 mg, 0.706 mmol) by procedure E (column chromatography: EtOAc–hexane, 75 : 25) gave amorphous 19c (360 mg, 78%), which failed to crystallize (MeOH): 1H NMR δ 1.01–1.12 (m, 28, 4 × i-Pr), 4.04–4.20 (m, 3, H4′,5′,5″), 4.66 (br s, 2, NH2), 4.99 (dd, J = 4.5, 8.9 Hz, 1, H3′), 5.41 (br s, 2, NH2), 5.81 (d, J = 4.5 Hz, 1, H2′), 6.05 (s, 1, H1′), 7.71 (s, 1, H8); 13C NMR δ 13.2–17.4 (12, 4 × i-Pr ), 60.1 (C2′), 68.6 (C5′), 81.6 (C3′), 86.9 (C4′), 88.4 (C1′), 115.0 (C5), 136.7 (C8), 151.7 (C4), 155.8 (C2), 159.7 (C6); MS m/z 657 (100, MH+). Anal. calcd for C23H39F3N6O7SSi2·CH4O (688.86, 1H NMR integration of MeOH): C, 41.84; H, 6.29; N, 12.20. Found: C, 41.56; H, 5.99; N, 12.25%.2-Amino-6-chloro-9-[2-S-acetyl-3,5-O-(1,1,3,3-tetraisopropyl-1,3-disiloxanyl)-2-thio-β-D-arabinofuranosyl]purine (20a)
Treatment of 19a (250 mg, 0.37 mmol) by procedure A (column chromatography: EtOAc–hexanes, 20 : 80) gave 20a (150 mg, 67%): mp 78–86 °C; 1H NMR δ 0.8–1.3 (m, 28, 4 × i-Pr), 2.20 (s, 3, Ac), 3.94–3.97 (m, 1, H4′), 4.09 (dd, J = 3.2, 12.0 Hz, 1, H5′), 4.16 (dd, J = 3.2, 12.0 Hz, 1, H5″), 4.56 (dd, J = 7.0, 10.5 Hz, 1, H2′), 4.68 (t, J = 10.5 Hz, 1, H3′), 5.24 (br s, 2, NH2), 6.39 (d, J = 7.0 Hz, 1, H1′), 7.96 (s, 1, H8); 13C NMR δ 13.3–17.6 (12, 4 × i-Pr), 30.7 (CH3) 52.2 (C2′), 61.4 (C5′), 71.66 (C3′), 83.7 (C4′), 83.9 (C1′), 125.7 (C5), 141.3 (C8), 151.5 (C4), 153.8 (C2), 159.1 (C6), 194.0 (CO); MS m/z 602 (100, MH+[35Cl]), 604 (40, MH+[37Cl]). Anal. calcd for C24H40ClN5O5SSi2 (602.30): C, 47.86; H, 6.69; N, 11.63. Found: C, 47.87; H, 6.67; N, 11.24.2-Amino-6-methoxy-9-[2-S-acetyl-3,5-O-(1,1,3,3-tetraisopropyl-1,3-disiloxanyl)-2-thio-β-D-arabinofuranosyl]purine (20b)
Treatment of 19b (100 mg, 0.15 mmol) by procedure A (column chromatography: EtOAc–hexanes, 20 : 80) gave 20b (65 mg, 73%): 1H NMR δ 1.04–1.20 (m, 28, 4 × i-Pr), 2.19 (s, 3, Ac), 3.93–3.96 (m, 1, H4′), 4.05–4.09 (m, 4, H5′, OCH3), 4.16 (dd, J = 3.8, 12.8 Hz, 1, H5″), 4.55 (dd, J = 7.2, 10.4 Hz, 1, H2′), 4.77 (dd, J = 8.2, 10.4 Hz, 1, H3′), 4.87 (br s, 2, NH2), 6.35 (d, J = 7.2 Hz, 1, H1′), 7.79 (s, 1, H8); MS m/z 598 (100, MH+).2,6-Diamino-9-[2-S-acetyl-3,5-O-(1,1,3,3-tetraisopropyl-1,3-disiloxanyl)-2-thio-β-D-arabinofuranosyl]purine (20c)
Treatment of 19c (340 mg, 0.52 mmol) by procedure A (column chromatography: EtOAc–hexanes, 20 : 80) gave 20c (220 mg, 73%): 1H NMR δ 1.04–1.20 (m, 28, 4 × i-Pr), 2.22 (s, 3, Ac), 3.95 (dt, J = 3.2, 7.9 Hz, 1, H4′), 4.06 (dd, J = 3.1, 12.8 Hz, 1, H5′), 4.09 (dd, J = 4.2, 12.8 Hz, 1, H5″), 4.57 (dd, J = 7.2, 10.4 Hz, 1, H2′), 4.65 (br s, 2, NH2), 4.84 (t, J = 10.2 Hz, 1, H3′), 5.35 (br s, 2, NH2), 6.30 (d, J = 7.2 Hz, 1, H1′), 7.72 (s, 1, H8); 13C NMR δ 12.8–17.8 (12, 4 × i-Pr ), 30.6 (CH3), 52.4 (C2′), 61.9 (C5′), 72.6 (C3′), 83.4 (C4′), 83.8 (C1′), 114.9 (C5), 137.3 (C8), 152.2 (C4), 156.0 (C2), 159.9 (C6), 194.0 (CO); MS m/z 583 (100, MH+).2-Amino-6-chloro-9-[3,5-O-(1,1,3,3-tetraisopropyl-1,3-disiloxanyl)-2-thio-β-D-arabinofuranosyl]purine (21a)
Treatment of 20a (110 mg, 0.18 mmol) by procedure B (column chromatography: EtOAc–hexanes, 35 : 65) gave 21a (95 mg, 93%): mp 270 °C (dec.); 1H NMR δ 1.02–1.19 (m, 28, 4 × i-Pr), 1.42 (d, J = 8.0 Hz, 1, SH), 3.83–3.89 (m, 2, H2′,4′), 4.12 (dd, J = 2.8, 13.2 Hz, 1, H5′), 4.15 (dd, J = 2.8, 13.2 Hz, 1, H5″), 4.44 (t, J = 8.5 Hz, 1, H3′), 5.25 (br s, 2, NH2), 6.32 (d, J = 7.0 Hz, 1, H1′), 8.10 (s, 1, H8); MS m/z 560 (100, MH+[35Cl]), 562 (40, MH+[37Cl]).2-Amino-6-methoxy-9-[3,5-O-(1,1,3,3-tetraisopropyl-1,3-disiloxanyl)-2-thio-β-D-arabinofuranosyl]purine (21b)
Treatment of 20b (120 mg, 0.2 mmol) by procedure B (column chromatography: EtOAc–hexane, 35 : 65) gave 21b (102 mg, 91%): 1H NMR δ 1.03–1.18 (m, 28, 4 × i-Pr), 1.39 (d, J = 8.2 Hz, 1, SH), 3.80–3.86 (m, 2, H2′,4′), 4.04–4.07 (m, 4, H5′,OCH3), 4.15 (dd, J = 2.6, 13.0 Hz, 1, H5″), 4.46 (t, J = 8.5 Hz, 1, H3′), 5.02 (br s, 2, NH2), 6.31 (d, J = 7.1 Hz, 1, H1′), 7.95 (s, 1, H8); MS m/z 556 (100, MH+).2,6-Diamino-9-[3,5-O-(1,1,3,3-tetraisopropyl-1,3-disiloxanyl)-2-thio-β-D-arabinofuranosyl]purine (21c)
Treatment of 20c (210 mg, 0.36 mmol) by procedure B (column chromatography: EtOAc–hexane, 75 : 15) gave amorphous 21c (160 mg, 82%), which failed to crystallize (MeOH): 1H NMR δ 1.03–1.19 (m, 28, 4 × i-Pr), 1.51 (d, J = 8.0 Hz, 1, SH), 3.81 (dd, J = 7.5, 13.2 Hz, 1, H2′), 3.87–3.89 (m, 1, H,4′), 4.07 (dd, J = 2.6, 13.0 Hz, 1, H5′), 4.19 (dd, J = 2.6, 13.0 Hz, 1, H5″), 4.51 (t, J = 8.7, Hz, 1, H3′), 4.75 (br s, 2, NH2), 5.46 (br s, 2, NH2), 6.27 (d, J = 7.1 Hz, 1, H1′), 7.84 (s, 1, H8); 13C NMR δ 12.8–17.9 (12, 4 × i-Pr ), 48.5 (C2′), 61.2 (C5′), 75.7 (C3′), 83.9 (C4′), 84.4 (C1′), 114.5 (C5), 137.0 (C8), 152.3 (C4), 155.8 (C2), 159.6 (C6); MS m/z 541 (100, MH+). Anal. calcd for C22H40N6O4SSi2·CH4O (572.87; 1H NMR integration of MeOH): C, 48.22; H, 7.74; N, 14.67. Found: C, 48.65; H, 7.56; N, 14.92%.2-Amino-6-chloro-9-[3,5-O-(1,1,3,3-tetraisopropyl-1,3-disiloxanyl)-2-deoxy-2-propyldithio-β-D-arabinofuranosyl]purine (22a)
Treatment of 21a (122 mg, 0.212 mmol) by procedure C (after thiol was added, the reaction mixture was stirred overnight at ambient temperature) and column chromatography (EtOAc–hexane, 30 : 70) gave 22a (66 mg, 48%): 1H NMR δ 0.92 (t, J = 7.2 Hz, 3, CH3), 1.05–1.27 (m, 28, 4 × i-Pr ), 1.60 (sextet, J = 7.2 Hz, 2, CH2), 2.56 (t, J = 7.2 Hz, 2, SCH2), 3.86–3.89 (m, 2, H2′,4′), 4.08 (dd, J = 3.0, 12.7 Hz, 1, H5′), 4.10 (dd, J = 3.0, 12.7 Hz, 1, H5″), 4.51 (dd, J = 8.0, 9.6 Hz, 1, H3′), 5.20 (br s, 2, NH2), 6.42 ( d, J = 7.0 Hz, 1, H1′), 7.98 (s, 1, H8); MS m/z 634 (100, MH+[35Cl]), 636 (55, MH+[37Cl]).2-Amino-6-methoxy-9-[3,5-O-(1,1,3,3-tetraisopropyl-1,3-disiloxanyl)-2-deoxy-2-propyldithio-β-D-arabinofuranosyl]purine (22b)
Treatment of 21b (110 mg, 0.2 mmol) by procedure C (after thiol was added, the reaction mixture was stirred overnight at ambient temperature) and column chromatography (EtOAc–hexane, 30 : 70) gave 22b (52 mg, 42%): 1H NMR δ 0.90 (t, J = 7.2 Hz, 3, CH3), 1.05–1.27 (m, 28, 4 × i-Pr), 1.58 (sextet, J = 7.2 Hz, 2, CH2), 2.56 (t, J = 7.2 Hz, 2, SCH2), 3.85–3.90 (m, 2, H2′,4′), 4.07–4.09 (m, 5, H5′,5″,OCH3), 4.59 (dd, J = 8.4, 9.5 Hz, 1, H3′), 4.82 (br s, 2, NH2), 6.41 (d, J = 7.0 Hz, 1, H1′), 7.80 (s, 1, H8); MS m/z 630 (100, MH+).2,6-Diamino-9-[3,5-O-(1,1,3,3-tetraisopropyl-1,3-disiloxanyl)-2-deoxy-2-propyldithio-β-D-arabinofuranosyl]purine (22c)
Treatment of 21c (145 mg, 0.268 mmol) by procedure C (after thiol was added, the reaction mixture was stirred overnight at ambient temperature) and column chromatography (EtOAc–hexane, 90 : 10) gave 22c (110 mg, 67%): 1H NMR δ 0.87 (t, J = 7.2 Hz, 3, CH3), 1.04–1.27 (m, 28, 4 × i-Pr), 1.55 (sextet, J = 7.2 Hz, 2, CH2), 2.53 (t, J = 7.2 Hz, 2, SCH2), 3.84–3.87 (m, 2, H2′,4′), 4.06–4.12 (m, 2, H5′,5″), 4.56 (t, J = 8.2 Hz, 1, H3′), 4.92 (br s, 2, NH2), 5.86 (br s, 2, NH2), 6.39 (d, J = 7.0 Hz, 1, H1′), 7.70 (s, 1, H8); MS m/z 615 (100, MH+).2-Amino-6-chloro-9-(2-deoxy-2-propyldithio-β-D-arabinofuranosyl)purine (23a)
Treatment of 22a (30 mg, 0.047 mmol) by procedure D (column chromatography: CHCl3–MeOH, 96 : 4) gave 23a (17 mg, 91%): mp 190–192 °C (dec.); UV max 309 nm (ε 8800), max 248 nm (ε 8200), min 272 nm (ε 1900), min 236 nm (ε 6800); 1H NMR (MeOH-d4) δ 0.89 (t, J = 7.2 Hz, 3, CH3), 1.53 (sextet, J = 7.2 Hz, 2, CH2), 2.58 (t, J = 7.2 Hz, 2, CH2S), 3.84–3.92 (m, 4, H2′,4′,5′,5″), 4.49 (t, J = 8.0 Hz, 1, H3′), 6.52 (d, J = 7.0 Hz, 1, H1′), 8.35 (s, 1, H8); 13C NMR (MeOH-d4) δ 12.1 (CH3), 22.0 (CH2), 41.0 (SCH2), 60.3 (C2′), 62.7 (C5′), 72.7 (C3′), 84.8 & 84.9 (C4′ & C1′), 123.7 (C5), 142.4 (C8), 150.6 (C4), 154.0 (C2), 160.6 (C6); MS m/z 392 (100, MH+[35Cl]), 394 (55, MH+[37Cl]). Anal. calcd. for C13H18ClN5O3S2 (391.90): C, 39.84; H, 4.63; N, 17.87. Found: C, 39.63; H, 4.79; N, 17.49%.2-Amino-6-methoxy-9-(2-deoxy-2-propyldithio-β-D-arabinofuranosyl)purine (23b)
Treatment of 22b (40 mg, 0.064 mmol) by procedure D (column chromatography: CHCl3–MeOH, 96 : 4) gave 23b (20 mg, 81%): mp 81–85 °C; UV max 282 nm (ε 9700), max 249 nm (ε 9600), min 262 nm (ε 5300), min 227 nm (ε 4600); 1H NMR (MeOH-d4) δ 0.89 (t, J = 7.3 Hz, 3, CH3), 1.53 (sextet, J = 7.3 Hz, 2, CH2), 2.58 (t, J = 7.3 Hz, 2, CH2S), 3.82–3.96 (m, 4, H2′,4′,5′,5″), 4.07 (s, 3, OCH3), 4.52 (t, J = 8.2 Hz, 1, H3′), 6.47 (d, J = 7.1 Hz, 1, H1′), 8.08 (s, 1, H8); 13C NMR (MeOH-d4) δ 12.1 (CH3), 22.0 (CH2), 41.0 (SCH2), 53.3 (OCH3), 60.4 (C2′), 63.1 (C5′), 72.4 (C3′), 84.7 & 84.9 (C4′ & C1′), 114.1 (C5), 139.1 (C8), 153.7 (C4), 160.7 (C2), 161.7 (C6); MS m/z 388 (100, MH+). Anal. calcd. for C14H21N5O4S2·CH4O (419.52; 1H NMR integration of MeOH): C, 42.94; H, 6.01; N, 16.69. Found: C, 43.01; H, 5.76; N, 16.66%.2,6-Diamino-9-(2-deoxy-2-propyldithio-β-D-arabinofuranosyl)purine (23c)
Treatment of 22c (110 mg, 0.18 mmol) by procedure D (column chromatography: CHCl3–MeOH, 96 : 4) gave 23c (57 mg, 85%): mp 82–90 °C (soften), 110 °C; UV max 283 nm (ε 11 200), max 257 nm (ε 9600), min 267 nm (ε 7900), min 238 nm (ε 6300); 1H NMR (MeOH-d4) δ 0.89 (t, J = 7.2 Hz, 3, CH3), 1.53 (sextet, J = 7.2 Hz, 2, CH2), 2.58 (t, J = 7.2 Hz, 2, CH2S), 3.81–3.96 (m, 4, H2′,4′,5′,5″), 4.51 (t, J = 8.8 Hz, 1, H3′), 6.41 (d, J = 7.2 Hz, 1, H1′), 7.98 (s, 1, H8); 13C NMR (MeOH-d4) δ 12.1 (CH3), 22.0 (CH2), 41.0 (SCH2), 60.4 (C2′), 63.2 (C5′), 72.4 (C3′), 84.7 & 84.9 (C4′ & C1′), 113.1 (C5), 137.8 (C8), 151.6 (C4), 156.6 (C2), 160.7 (C6); MS m/z 373 (100, MH+). Anal. calcd. for C13H20N6O3S2·H2O (390.49): C, 39.99; H, 5.68; N, 21.52. Found: C, 40.35; H, 5.78; N, 21.33%.Cell culture
The eight human cell lines used were obtained from the Developmental Therapeutics Program Tumor Repository, National Cancer Institute (Frederick, MD). The cell lines were grown in RPMI 1640 medium containing 9% fetal bovine serum, 1% iron-supplemented calf serum, and 2 mmol L-glutamine. For the in vitro evaluation of the sensitivity of the human cell lines to the compounds, cells were plated in 96-well microtiter plates (5.0 × 103 cells per well) and then were exposed continuously to various concentrations of the compounds for 72 h at 37 °C. Cell viability was based on the reduction of MTS (in conjunction with phenazine ethosulfate) to a water soluble formazan product, which was measured with a microplate reader at 490 nm. The background absorbance mean was subtracted from the data followed by conversion to percent of control. The drug concentrations producing survival just above and below the 50% level were used in a linear regression analysis to calculate the IC50.Acknowledgements
This work was partially supported by an award from the American Heart Association, Florida/Puerto Rico Affiliate. D.R.C and P.I.G were sponsored by MBRS RISE program (NIH/NIGMS; R25 GM61347). We thank Professor J. Stubbe for helpful suggestions on the manuscript. We thank Dr William R. Waud and his staff at the Southern Research Institute for supplying the cytotoxicity data.References
- (a) J. Stubbe and W. A. van der Donk, Chem. Biol., 1995, 2, 793–801 CrossRef CAS; (b) A. Jordan and P. Reichard, Annu. Rev. Biochem., 1998, 67, 71–98 CrossRef CAS; (c) M. D. Sintchak, G. Arjara, B. A. Kellogg, J. Stubbe and C. Drennan, Nature Struct. Biol., 2002, 9, 293–3000 CrossRef CAS.
- J. R. McCarthy and P. S. Sunkara, in Design Synthesis, and Antitumor Activity of an Inhibitor of Ribonucleotide Reductas, eds. D. B. Weiner and W. B. Williams, CRC Press, Boca Raton, Fl, 1995, pp 3–32 Search PubMed.
- (a) P. Nordlund, B.-M. Sjöberg and H. Eklund, Nature, 1990, 345, 593–598 CrossRef CAS; (b) U. Uhlin and H. Eklund, Nature, 1994, 370, 533–539 CAS; (c) M. Eriksson, U. Uhlin, S. Ramaswamy, M. Ekberg, K. Regnstrom, B. M. Sjöberg and H. Eklund, Structure, 1997, 5, 1077–1092 CrossRef CAS.
- S. S. Mao, T. P. Holler, G. X. Yu, J. M. Bollinger Jr., S. Booker, M. I. Johnston and J. Stubbe, Biochemistry, 1992, 31, 9733–9743 CrossRef CAS.
- (a) G. J. Gerfen, W. A. van der Donk, G. Yu, J. R. McCarthy, E. T. Jarvi, D. P. Matthews, C. Farrar, R. G. Griffin and J. Stubbe, J. Am. Chem. Soc., 1998, 120, 3823–3835 CrossRef CAS; (b) W. A. van der Donk, G. Gerfen and J. Stubbe, J. Am. Chem. Soc., 1998, 120, 4252–4253 CrossRef CAS.
- (a) A. L. Persson, M. Eriksson, B. Katterle, S. Pötsch, M. Sahlin and B. M. Sjöberg, J. Biol. Chem., 1997, 272, 31533–31541 CrossRef CAS; (b) A. L. Persson, M. Sahlin and B. M. Sjöberg, J. Biol. Chem., 1998, 273, 31016–31020 CrossRef CAS.
- C. C. Lawrence, M. Bennati, H. V. Obias, G. Bar, R. G. Griffin and J. Stubbe, Proc. Natl. Acad. Sci. USA, 1999, 96, 8879–8984 CrossRef.
- R. Lenz and B. Giese, J. Am. Chem. Soc., 1997, 119, 2784–2794 CrossRef CAS.
- (a) M. J. Robins, Z. Guo, M. C. Samano and S. F. Wnuk, J. Am. Chem. Soc., 1999, 121, 1425–1433 CrossRef CAS; (b) M. J. Robins and G. J. Ewing, J. Am. Chem. Soc., 1999, 121, 5823–5824 CrossRef CAS.
- (a) J. Coves, L. Le Hir de Fallois, L. Le Pape, J.-L. Decout and M. Fontecave, Biochemistry, 1996, 35, 8595–8602 CrossRef CAS; (b) L. Le Hir de Fallois, J.-L. Decout and M. Fontecave, J. Chem. Soc., Perkin Trans. 1., 1997, 2587–2595 RSC; (c) B. Roy, S. Chambert, M. Lepoivre, A.-M. Aubertin, J. Balzarini and J.-L. Décout, J. Med. Chem., 2003, 46, 2565–2568 CrossRef CAS.
- (a) S. Salowe, J. M. Jr. Bollinger, M. Ator, J. Stubbe, J. McCracken, J. Peisach, M. C. Samano and M. J. Robins, Biochemistry, 1993, 32, 12749–12760 CrossRef CAS; (b) W. A. van der Donk, J. Stubbe, G. J. Gerfen, B. F. Bellew and R. G. Griffin, J. Am. Chem. Soc., 1995, 117, 8908–8916 CrossRef CAS; (c) S. F. Wnuk, S. M. Chowdhury, P. I. Garcia Jr. and M. J. Robins, J. Org. Chem., 2002, 67, 1816–1819 CrossRef CAS.
- (a) A. Jaworski, I. Ekiel and D. Shugar, J. Am. Chem. Soc., 1978, 100, 4357–4361 CrossRef CAS; (b) I. Ekiel, M. Remin, E. Darzynkiewicz and D. Shugar, Biochim. Biophys. Acta, 1979, 562, 177–191 CrossRef CAS; (c) N. Yathindra and M. Sundaralingam, Biochim. Biophys. Acta, 1979, 564, 301–310 CrossRef CAS.
- (a) S. F. Wnuk, Tetrahedron, 1993, 49, 9877–9936 CrossRef CAS; (b) S. Chambert and J.-L. Decout, Org. Prep. Proced. Int., 2002, 34, 27–85 CAS.
- (a) R. Ranganathan and D. Larwood, Tetrahedron Lett., 1978, 4341–4344 CrossRef CAS; (b) J. H. Marriott, M. Mottahedeh and C. B. Reese, Carbohydr. Res., 1991, 216, 257–269 CrossRef CAS.
- (a) M. Imazawa, T. Ueda and T. Ukita, Chem. Pharm. Bull., 1975, 23, 604–610 CAS; (b) S. Shibuya and T. Ueda, J. Carbohydr. Nucleosides Nucleotides, 1980, 7, 49–56 Search PubMed.
- M. J. Robins, S. D. Hawrelak, A. E. Hernandez and S. F. Wnuk, Nucleosides Nucleotides, 1992, 11, 821–834 CAS.
- T. Mukaiyama and K. Takahashi, Tetrahedron Lett., 1968, 5907–5908 CrossRef CAS.
- W. Zhang and M. J. Robins, Tetrahedron Lett., 1992, 33, 1177–1180 CrossRef CAS.
- S. Chambert, I. Gautier-Luneau, M. Fontecave and J.-L. Decout, J. Org. Chem., 2000, 65, 249–253 CrossRef CAS.
- M. B. Anderson, M. G. Ranasinghe, J. T. Palmer and P. L. Fuchs, J. Org. Chem., 1988, 53, 3127–3129 CrossRef.
- D. W. Hutchinson, in Chemistry of Nucleosides, and Nucleotides, ed. L. B. Townsend, Plenum Press, New York, 1991, vol. 2 Search PubMed.
- J. J. Davisson, D. R. Davis, V. M. Dixit and C. D. Poulter, J. Org. Chem., 1987, 52, 1794–1801 CrossRef CAS.
- M. J. Robins, J. S. Wilson, D. Madej, D. L. J. Tyrrel, W. P. Gati, R. J. Lindmark and S. F. Wnuk, J. Heterocycl. Chem., 2001, 38, 1297–1306 CAS.
- In collaboration with Professor J. Stubbe at Massachusetts Institute of Technology.
- K. Fukukawa, T. Ueda and T. Hirano, Chem. Pharm. Bull., 1883, 31, 1582–1592 Search PubMed.
- V. Nair and D. F. Purdy, Tetrahedron, 1991, 47, 365–382 CrossRef CAS.
- L. Beigelman, P. Haeberli, D. Sweedler and A. Karpeisky, Tetrahedron, 2000, 56, 1047–1056 CrossRef CAS.
Footnote |
| † On faculty leave from the Department of Chemistry, University of Agriculture, Poznan, Poland. |
| This journal is © The Royal Society of Chemistry 2004 |