NMR and UV studies of 3′-S-phosphorothiolate modified DNA in a DNA : RNA hybrid dodecamer duplex; implications for antisense drug design†
Andrew P. G.
Beevers
a,
Kevin J.
Fettes
b,
Ghalia
Sabbagh
b,
Fatima K.
Murad
a,
John R. P.
Arnold
c,
Richard
Cosstick
b and
Julie
Fisher
*a
aDepartment of Chemistry, University of Leeds, Leeds, UK LS2 9JT. E-mail: julief@chem.leeds.ac.uk; Fax: 44 (0) 113 343 6565; Tel: 44 (0) 113 343 6577
bDepartment of Chemistry, University of Liverpool, Liverpool, UK L69 7ZD
cYorkshire Cancer Research Laboratory of Drug Design, Tom Connors Cancer Research Centre, University of Bradford, Bradford, UK BD7 1DP
First published on 20th November 2003
Abstract
High-resolution NMR spectroscopy has been used to establish the conformational consequences of the introduction of a single 3′-S-phosphorothiolate link in the DNA strand of a DNA : RNA hybrid. These systems are of interest as potential antisense therapeutic agents. Previous studies on similarly modified dinucleotides have shown that the conformation of the sugar to which the sulfur is attached shifts to the north (C3′-endo/C2′-exo). Comparisons made between NOESY cross-peak intensities, and coupling constants from PE-COSY spectra, for both non-modified and modified duplexes confirm that this conformational shift is also present in the double helical oligonucleotide system. In addition it is noted that in both the dinucleotides and the modified duplex, the conformation of the sugar ring 3′ to the site of modification is also shifted to the north. That this pattern is observed in the small monomeric system as well as the larger double helix is suggestive of some pre-ordering of the sequences. The conclusion is supported by consideration of the 1H chemical shifts of the heterocyclic bases near the site of the modification. The enhanced stability that these conformational changes should bring was confirmed by UV thermal melting studies. Subsequently a series of singly and doubly 3′-S-phosphorothiolate-modified duplexes were investigated by UV. The results are indicative of an additive effect of the modification with thermodynamic benefit being derived from alternate spacing of two modified linkers.
Introduction
Chemical modification of nucleic acids is a subject which has seen huge developments over the past 10–15 years,1 spurred on by our increased knowledge of the fundamental importance of nucleic acid–nucleic acid complexes in biology, and the potential for manipulating these in the pursuit of better therapeutic agents and/or bio-analytical tools.2The possible therapeutic utilization of chemically modified nucleic acids is an area that continues to attract interest.2 Although perhaps still awaiting a major breakthrough many oligonucleotide drugs have now progressed through Phase I clinical trials—one drug, Vitravene™, a 21-mer phosphorothioate-DNA,3 has been approved for clinical use in the USA. The mode of action of each of these drugs is proposed to involve the formation of a hybrid structure between the chemically modified oligonucleotide and a specific site on a mRNA or pre-mRNA. The drug recognises its target via Watson–Crick base pairing and is therefore complementary (and anti-parallel) to the mRNA (sense) sequence. Consequently such drugs are referred to as antisense oligonucleotides (asONs).
The formation of mRNA : asON complexes may inhibit or prevent RNA transport, splicing or translation4 and may promote degradation of the mRNA by, e.g., RNaseH.5 Whatever process is to be induced many hurdles have to be overcome to form the required complex—and hence the need for chemical variants on natural nucleic acids. The majority of the modifications reported have been based on deoxynucleic acid systems as these are synthetically more convenient.
In designing a DNA-based drug for use in an antisense therapeutic strategy a number of factors have to be considered.6 These include the selectivity and specificity of mRNA recognition. It is of course necessary to know the sequence of the mRNA and the location within this of the target. To find a ‘particular’ codon an asODN of ca. 15 bases is required. The drug has to be sufficiently chemically stable to withstand attack from endogenous agents such as 3′-exo- and endo- nucleases and also able to be transported across the hydrophobic cell membrane.7 Assuming that these issues can be addressed it is then necessary to enhance the lifetime of the asODN : mRNA complex; the target DNA : RNA system, when of mixed sequence, is thermodynamically less stable than its DNA : DNA and RNA : RNA counterparts.8 If RNaseH recruitment is important5c,9 then this has to be considered in the design process also. RnaseH recognises and cleaves the RNA in DNA : RNA systems thus removing the mRNA and completely inactivating the gene.5c
Amongst the huge range of chemical variants reported in the literature, those involving an N3′-P5′ phosphoramidate10 or a locked nucleic acid (LNA)11,9 have shown considerable potential. In addition modified nucleotides that involve the replacement of one of the non-bridging oxygens by a sulfur atom12 have shown very promising antisense activity (e.g. as in Vitravene™).3 Although such a modification decreases the hydrophilicity and increases the nuclease stability of the asODN, it does not enhance the stability of the asODN : RNA hybrid. A modification which retains these properties but which has the potential for greater RNA binding affinity is the 3′-S-phosphorothiolate analogue.13 3′-S-Phosphorothiolate modified oligonucleotides have been used as tools to study the cleavage of phosphodiester bonds by nucleic acid processing enzymes, but are stable under physiological conditions.13–15 The enhanced RNA binding was proposed on the basis that the 3′-sulfur atom should induce a conformational shift in its attached sugar; making the dexoyribose (south-pucker) look like a ribose (north pucker) (Fig. 1).16 In studies of dinucleotides containing this modification it has been found that the sugar pucker of the ring to which the sulfur is attached is indeed predominantly north (C3′-endo/C2′-exo).17 Moreover this shift in the north–south conformational equilibrium was found to be transmitted to the subsequent (n + 1) sugar. Clearly if both of these features were to be retained in a longer sequence, and in a duplex, we would expect such sequences to have enhanced RNA binding affinity and therefore considerable potential as antisense agents. To investigate this, initially a single 3′-S-phosphorothiolate link has been included in a DNA dodecamer and its complex with its complementary RNA dodecamer (1) (see Scheme 1) has been analysed using 2D NMR methods. The non-modified DNA : RNA hybrid (2) has been similarly analysed for comparison. UV thermal melting studies18 have been conducted to further probe the thermodynamic behaviour of these sequences and those with the 3′-S-modification singly and doubly incorporated at different locations (see Scheme 1) for which further NMR studies are planned. We now describe the detailed results of these studies, some parts of which have already been published in a preliminary form.19
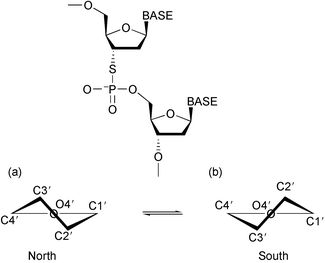 | ||
| Fig. 1 A schematic representation of the 3′-S-phosphorothiolate linkage, and the (a) north and (b) south sugar pucker. | ||
 | ||
| Scheme 1 Sequences of the DNA strands under investigation. In each case the RNA strand has the sequence 5′ r(G13G14C15 A16A17A18 U19U20U21 A22G23G24). The (S) indicates the position of the 3′-S-phosphorothiolate linkage. | ||
Results
Spectral analysis
An analysis of the conformational features of the dinucleotides, thymidylyl(3′-5′) thymidine (dTpT), its phosphorothiolate analogue, 3′-deoxy-3′-thiothymidylyl(3′-5′) thymidine (dTSpT) and their ribonucleotide analogues, rIpU and rISpU (where I represents inosine), based on 1H–1H coupling constants has been reported elsewhere.17 Variable temperature 1H and 31P spectra have been recorded for dTpT and dTSpT. The H6 protons of dTSpT are both deshielded relative to those of dTpT. This is consistent with an increase in the population of the type of stacking arrangement that occurs in nucleic acid double helices, rather than the tilted stacking arrangement that is often seen for other aromatic systems.20 The differences in chemical shift between the H6 protons in dTSpT and the same protons in dTpT are 0.042 and 0.026 ppm respectively. This difference is essentially temperature independent over the temperature range 27 °C to 57 °C (0.039 and 0.021 ppm respectively at 57 °C). The H6 protons in each dinucleotide experience increased shielding as the temperature is increased, however the effect is more significant for dTSpT (Δδ −0.15 ppm) than for dTpT (Δδ −0.08 ppm). 31P spectra recorded over the same temperature range displayed a significant upfield shift for the phosphorus of dTSpT (Δδ −0.313 ppm) and a slight downfield shift for the phosphorus of dTpT (Δδ 0.020 ppm). A similar observation has been made for the 5′-S-phosphorothiolate system (dTpST).2131P–1H couplings were not analysed, however it was noted that the width of the 31P multiplet in dTSpT was greater by 8 Hz than that for dTpT.Proton NMR chemical shift assignment for (1) and (2) was achieved using standard procedures and required the recording of DQF-COSY, TOCSY and NOESY spectra.22 The recording of NOESY spectra with a range of mixing times enabled the deoxy sugar 2′ and 2″ protons to be distinguished. Only limited assignment of the 5′/5″ protons was achieved and these are not reported.23 For both the modified and non-modified systems numerous inter-strand NOEs were observed between non-exchangable protons. These together with similar connections amongst imino, amino, thymine methyl and adenine H2 protons enabled confirmation of Watson–Crick base-pairing patterns. Chemical shifts are tabulated in the Electronic Supplementary Information† (Tables S1 and S2).
The proton chemical shifts are essentially identical for (1) and (2) apart from at or near the modification site.19 The most dramatic change is a highfield shift for T8 H3′ (in (1)) of 1.29 ppm. This is primarily due to the introduction of the less electronegative sulfur atom and is identical to the shift change noted for the same proton in both the deoxy- and ribo-dinucleotides.17 The H2 proton of A17 (the base-paired partner of T8) is shielded by 0.11 ppm. The H6 proton of T9 is deshielded by 0.33 ppm and that of T8 by 0.08 ppm. These observations are consistent with an increase in the intrastrand stacking of T9 with that of T8.20a,21
The COSY-type spectra recorded for (1) and (2) are essentially identical (with the chemical shift exceptions referred to) with one notable difference. The cross-peak connecting H1′ to H2′ of T8 in (1) is missing. A very low intensity peak is observed in the TOCSY spectrum and the through-space connection is clearly observed in the NOESY spectrum.† These observations are consistent with the pucker of the T8 sugar adopting a predominantly north conformation.24
NOESY spectra recorded for (1) and (2) were also essentially identical in cross-peak connections and relative intensities except at and around the site of modification. Particular attention was paid to correlations between H2′ and H6/H8, H1′ to intra-nucleoside H2′/H2″ and adenine H2 to H1′, in residues adjacent to, or opposite, the site of modification.19 In comparison with observations made for (2) T8 H6 is closer to T7 H2′ than to its own H2′. For both sequences comparison of H1′(n)–H2′(n) and H1′(n)–H2″(n) NOE intensities for residues remote from the sulfur substitution indicated that the H1′ is always closer to its own H2″ than its H2′. Due to resonance overlap this comparison could not be extended to T8 in (1). The relative intensities of NOEs connecting adenine H2 protons to H1′ protons of its intrastrand (n + 1) sugar or cross strand sugar were compared. For each of the deoxy-adenines the H2 proton was found to be closer in space to (the H1′ proton of) its cross strand partner than to its same strand neighbour. In contrast, for the ribo-adenines the converse was true. These observations were made for both sequences, the only difference in analysis occurring at A18 in (1) where the cross peak intensities were essentially identical.† These observations are consistent with a conformational change at T8 in (1) and a predominantly north sugar pucker for this residue.25,26
PE-COSY spectra were recorded for both (1) and (2) to enable measurement of scalar proton–proton couplings. Vicinal coupling constants arrived at through spectral simulation have previously been used in a Karplus-type relationship to provide a full pseudorotational analysis of the dinucleotides.17 The resonance overlap inherent in the 1D spectra of the oligonucleotides precluded such an approach here. However the simplified cross-peak patterns of PE-COSY spectra did provide coupling data upon which important conformational conclusions could be made. Particular attention was paid to H1′–H2′ and H1′–H2″ cross-peaks as these couplings readily report changes in sugar conformation.†27 For (2) it was found that the H1′–H2′ and H1′–H2″ couplings were very similar to each other for each deoxy residue, with the H1′–H2′ generally being the larger of the two. In a true B-type helix, with pure C2′-endo (or south) sugar conformation, typical of double helical DNA, a great disparity would be expected, with the H1′–H2′ coupling being much larger (and of the order 9–10 Hz; in (2) all the couplings fall in the range 5–8.7 Hz).27 For (1) similar observations were made with two key exceptions. No cross-peak was observed for T8 H1′–H2′ (see earlier comments) and the T9 H1′–H2′ coupling was significantly reduced (and was less than its H1′–H2″ coupling). In a true A-type helix, with C3′-endo sugar (or north) puckers, typical of RNA molecules, the H1′–H2′ coupling would be very small as the dihedral angle between these protons approaches 90°. These observations are therefore consistent with the T8 sugar adopting a pure north conformation, and a shift in the conformational equilibrium for T9, also moving to the north. It is possible to go even further than this. If a two state equilibrium between north and south is assumed, it is possible to determine the actual population of the two states by considering the sums of the H1′–H2′ and H1′–H2″ couplings (ΣH1′).28 When this analysis is performed for (2), with the exception of residues C1 and C2, the conformational equilibrium lies to the south, as expected for natural deoxy-sugars. For (1) similar conclusions may be drawn, except at positions T8 and T9. These two residues display a significant increase in north character, with populations of 100% and 70% respectively. These observations mirror those made for the dinucleotide systems previously studied, in which the north population of similarly related deoxy-sugars was 85% and 54% (cf. 28% and 37% in dTpT).17
UV thermal melting studies
Duplexes (1) and (2), and subsequently duplexes (3)–(7), were subjected to UV thermal melting analyses with the aim of establishing the thermodynamic consequences of the presence of one or two 3′-S-phosphorothiolate links. The melting temperature, Tm (the mid-point in the sigmoidal curve produced when UV absorption is plotted as a function of temperature), is considered a good indicator of the thermodynamic stability of the system under investigation.18 Many repetitions of the thermal melting of (1) and (2) were performed, with the Tm for (1) being 1.6–2.5 °C higher than that of (2) (Table 1).† For (3) and (4) Tm increases relative to (2) were also observed although slightly smaller (1–1.3 °C). A further increase in Tm was noted for (5) and (6) in which the modified link is incorporated in adjacent residues. This increase in Tm when compared to (2) is additive; i.e. is the sum of the effect observed for the modification at T8 and T7 or T9 (3 °C in each case). A further increase is noted when the modified residues are separated by a single non-modified residue (ΔTm 4 °C). Studies on differently modified DNA : RNA hybrids have also shown that there is a thermodynamic advantage to this ‘spaced’ arrangement and indeed that there is a saturation level in terms of the extra stability additional modified units can bring.2b,29| Sequence number | T m/°C |
|---|---|
| a T m values are measured as the maximum of the first derivative of the melting curve (A260versus temperature). Samples were dissolved in 10 mM phosphate buffer. b Single strand concentrations were 3 µM. c Single strand concentrations were 4 µM. | |
| 1 | 26.8,b 28.7c |
| 2 | 25.2,b 26.2c |
| 3 | 26.5b |
| 4 | 26.5b |
| 5 | 28.8b |
| 6 | 28.8b |
| 7 | 29.2b |
Thus our UV thermal melting data for (1) and (2) are as we would predict from the NMR analysis. NMR studies of (3)–(7) are planned.
Discussion
The chemistry involved in placing a sulfur atom at the 3′ position of a sugar ring was originally developed to explore problems unrelated to antisense drug design.13–15 Nevertheless this was a logical progression, as the established biochemical stability the sulfur substitution endowed on nucleic acids was just that sought for antisense drugs. In addition the sulfur substitution offered the possibility of alleviating another problem faced with DNA-based asONs; that is the relatively poor thermodynamic stability of DNA : RNA hybrids.8 A significant effort has been made by researchers to make the asON look more RNA-like, as the RNA : RNA complex is more stable; the LNA systems have perhaps been the most notable in this respect.2b,9,11,29 In principal a 3′-S substituent should also facilitate this. It is well known that sugar rings in nucleic acids generally exist in equilibrium between two extreme conformations; labelled north and south (Fig. 1).16 The position of the equilibrium depends on several factors which include the anomeric effect between the sugar's ring oxygen and the nitrogen of the nucleobase; this is optimised in a purely north conformation.30 Several gauche effects also operate and in particular that between the sugar's 3′ oxygen and ring oxygen is optimised when a south conformation is adopted.16 The contribution of the gauche effect to the equilibrium has been found to increase as the electronegativity of the 3′ substituent increases.31 Consequently the less electronegative and larger sulfur atom should reduce the contribution of the gauche effect and push the equilibrium further to the (desired) north. It has been shown previously that a 3′-phosphoramidate link follows this trend.32 Studies of dinucleotide systems containing a 3′-S link have indeed revealed this conformational preference.17 The 5′ sugars of both dTSpT and rISpU have been shown to adopt predominantly north puckers; the population being 100% in the case of the modified ribose sugar. Thus this very conservative modification was considered to be worth testing in a longer sequence.Studies on the dinucleotides have revealed another interesting feature of the 3′-S modification. The conformational shift is also transmitted, though reduced, to the 3′ sugar. The question thus arises as to the cause of this. UV thermal melting studies of dTpT, dTSpT, rIpU and rISpU display a less than 5% increase in absorption with temperature; distinct Tm's are not evident. This rules out the presence of dimerization or higher association and the possibility of intermolecular interactions playing a significant part in this 3′ sugar change. The 31P chemical shift change with temperature increase for dTSpT appears anomalous, suggesting that the phosphate backbone is distorted and perhaps this is the reason for the sugar pucker. It is generally observed that 31P chemical shifts for deoxynucleotides in DNA double helices move downfield as temperature is increased.33 The converse has been observed when, for example, a G : A mismatch is introduced to the duplex. In this case an upfield shift is observed initially and then a lowfield shift at higher temperatures. This trend is attributed to the presence of some ‘high energy, more trans P–O ester’ conformation at low temperature.33 It is difficult to see how this could come about in such a small and unimolecular system as dTSpT. We have no evidence to suggest this is the case, but if it was a factor (and the cause of the upfield shift) is it the reason for the conformational change in the 3′ sugar? The answer to this is provided by the results of the conformation analysis of dTpST; the 5′-S system.21 The 31P chemical shift change with temperature for this molecule is almost identical in magnitude and direction to that for dTSpT and yet this 5′-S link does not change the conformational preferences of the two deoxy sugars. Thus a change in the phosphate backbone is not the source of the 3′ transmission of the north puckering.
A ‘transmitted’ conformational change has been observed for other chemical variants, most notably the LNA system.11 When complexed with complementary RNA the sugar ring 3′ to the modification is found to have an essentially north pucker. Various reasons for this have been proposed and rejected.11c Amongst these a water-mediated hydrogen bond between the O2′ and O4′ atoms has been proposed. This would not be expected to have a significant lifetime in our unimolecular dinucleotides, and in any case dTSpT does not have an O2′. The possibility of enhanced intramolecular base stacking has been rejected for the LNA : RNA hybrid where cross-strand stacking is apparent. However this is the most likely source of the feature that was reported for dTSpT and rISpU. The temperature dependence of the chemical shift of the H6 protons in dTpT has been shown to be more significant than observed for dTpST and this has been attributed to preferential base stacking, previously predicted for pyrimidine–pyrimidine systems.34 The shift change observed for the H6 protons of dTSpT is even more dramatic. This is consistent with a higher population of the stacked structure at room temperature than is observed for dTpT. Recently a study of 1-propynylated oligodeoxypyrimidine : RNA complexes resulted in the conclusion that the modified single strand oligomers had ‘order’ which could explain a smaller unfavourable entropy change on duplex formation.35 Similarly for LNA systems the lower entropy of the ssLNA compared to ssDNA has been proposed to be one of the reasons for the enhanced thermostability of the LNA : RNA complex.11c The 3′-S modification is very subtle, any enhanced stability this substitution brings to a double helical system must come from the enhanced intramolecular base stacking also apparent in the dinucleotide systems. Evidence in support of this was obtained from the NMR and UV studies performed for (1) and (2).19,23
The success of the resonance assignment procedure for the hybrids is proof of the DNA strand adopting an essentially B-type helix, and the RNA strand A-type.36 The COSY, TOCSY and NOESY spectra for (1) and (2) are identical except at or near the 3′-S substitution site. This in itself is an important result. Although the intention is to make the DNA strand adopt a more A-type helical structure it may not be beneficial to push the conformational equilibrium too far north. Studies of hybrids in which the DNA strand has complete phosphoramidate incorporation,37 and of those which have a high LNA content38 in the DNA strand, have shown that these are unable to elicit RNase H activity, although they do still display ‘antisense’ activity; this is a characteristic of many ‘second-generation’ drugs.39 The sugar protons of the modified T8 residue experience a shielding and deshielding pattern, relative to that observed for (2) that is reminiscent of what was observed for the dinucleotides;17 the largest shift change being for H3′. Of all the other protons only T9 H6 experiences a shift change greater than 0.3 ppm. This shift difference is almost three times that observed for TSpT, presumably this is due to an even higher population of the (intrastrand) stacked arrangement in (1)20a,21 than in dTSpT. A17 H2 is shielded slightly. This may be due to a change in interstrand base-stacking but this is more likely to be with T8; A17 H2 has a strong NOE connection to the H1′ of T8 which is consistent with the conformation of T8 changing to C3′-endo and a closer proximity of these residues. Resonance assignment for (1) was therefore straight forward once (2) had been analysed.
A comparison of assigned COSY spectra immediately revealed the conformational consequences of the inclusion of the 3′-S link; the correlation between T8 H1′ and T8 H2′ had reduced to zero. The H1′–H2′ correlation of ribose sugars is at best weak, as these protons bear an approximately 90° dihedral angle relationship to each other.36 This strongly suggested that the T8 sugar pucker was also predominantly north. Further evidence of this was forthcoming from the analysis of the intensities of NOE cross peaks connecting protons near T8 in (1) and (2); more specifically the pattern of NOE's observed amongst H2′ and H6/H8 protons, H1′ and H2′/H2″ protons, and adenine H2 and H1′ protons. The same NOE patterns were seen for all mixing times used (50–300 ms). For nucleotides in which the sugar adopts a north conformation, the distance between an H6/H8 and the H2′ proton (intra-residue) is longer than that to the H2′ of the preceding residue. Also, in a north-puckered sugar the difference between H1′–H2′ and H1′–H2″ distances is less than observed for a south puckered sugar, with the H1′–H2″ always shorter. Additionally an adenine with a south-type sugar pucker has an H2 which is closer in space to its cross-strand H1′ than to the H1′ of its n + 1 (intrastrand) sugar unit.25,26 When these comparisons were made for (1) and (2) it was clear that the RNA strand in both molecules had north-type sugar puckers, and the DNA strand had south-type sugar puckers except at T8 where the NOE pattern was as expected for a significantly north population. The situation was less clear for T9 as resonance overlap precluded analysis of the H2′ to H6/H8 NOE; the H1′–H2′ and H1′–H2″ NOE observations suggested T9 adopted a south sugar pucker. The difference in the NOE intensities was greater than for the RNA strands but less than was observed for the DNA residues, perhaps suggesting some intermediate conformation, it is unsafe to draw strong conclusions from this single piece of data. However chemical shift changes observed between (1) and (2) suggested that T9 had indeed undergone some structural change and this was established through measurement of H1′–H2′ and H1′–H2″ coupling constants in PE-COSY spectra. Summation of these coupling constants enabled the conformational preferences for each of the DNA sugars in (1) and (2) to be determined. With the exception of end residues where the effects of duplex fraying would be expected to impact on the measurements, in (2) sugars displayed an essentially south pucker (varying between 60–90%). In (1) the trend was the same except at T8 and T9. For T8 the percentage south population had reduced to zero and at T9 it had reduced to 30% (from 70% in (2)). These observations follow the trends reported for the dinucleotides17 although the conformational shift is even more dramatic. It is tempting to speculate that this enhancement is simply due to preferential base stacking within the DNA 12-mer as compared to the 2-mer (dinucleotide). Alternatively the enhanced change could be due to differing interactions with the RNA strand. That is, an RNA : RNA-type (A17 : T8(S)) base-pair may induce the formation of a similar pairing (A16 : T9) with the associated required structural change; we have no evidence for this for T9. One way to test this would be to perform studies of the all DNA equivalent of (2) and indeed such studies are planned. However there is some literature that suggests that this is not the case. NMR-based structural studies of [d(CG)r(C)d(TAGCG)]2 have revealed that “even the insertion of a single RNA residue in DNA persists in the north pucker and causes some perturbation of the conformation of neighbouring residues (T4 intra-strand and C7 cross-strand)”.40 In this40 example the base pairing is ribose : deoxyribose. Thus ‘special’ base-pairing interactions seem unlikely to be the cause of the observations we have made; rather our data point towards intra-strand base-stacking as being the most important factor.
The hoped-for increased thermal stability that the 3′-S- link should bring was established via the UV thermal melting studies. The single 3′-S substitution at T8 resulted in an increase in Tm of up to 2.5 °C. This increase was less when the substitution was made at T7 or T9. This may reflect a ‘context’ dependence of the substitution. The sequence used here is heterogeneous and has an ApT step; such steps are known to induce bends and indeed this has been observed for the all DNA duplex.41 We are unaware of such mixed sequences being investigated in this manner previously hence we cannot report a literature precedent for this. However it would seem reasonable that if there is structural variation to start with the effect of a chemical modification would have differing structural consequences and hence differing thermodynamic consequences. Thus NMR studies of sequences (3)–(7) are planned and work is in progress in synthesising a more ‘repetitive’ sequence for Tm studies. Although there may be a context or sequence dependence nevertheless the contributions of the different substitutions to Tm are additive; Tm for the T7T8 double 3′-S substitution is the same as for the T8T9 double substitution and this is encouraging. Additionally it was noted that when the 3′-S substitutions were separated (in this sequence only one intervening residue was possible) a further increase in Tm was observed. This is in line with trends observed by other workers in this area. With the synthesis of a different sequence as mentioned it will be possible to explore the optimal separation of modified linkers and indeed the optimal number of substitutions for a given chain length.
In conclusion, the aim of these studies was to establish the structural and thermodynamic consequences of the introduction of a 3′-S-phosphorothiolate to a DNA molecule on its complex with its complementary RNA strand. It was anticipated that the link would result in a conformational shift in its attached sugar with the effect that the DNA : RNA hybrid would be more stable. The results reported here do indeed confirm this local structural change and its beneficial thermodynamic consequences. In addition as has been found with a number of other modifications, the conformational affect of the 3′-S linker is transmitted to the sugar 3′ to it. However, in contrast to studies of other modifications, we have been able to rule out a number of the hypotheses on the cause of this transmission, due to the very conservative nature of the sulfur substitution. It has thus proved possible to advance the discussion on factors important in the thermodynamic behaviour of nucleic acids in general.
Experimental
Sample preparation
The natural dinucleotides were purchased from Sigma Chemicals (Dorset, UK) and used without further purification. The modified dinucleotides were prepared as previously described. The samples were dissolved in D2O, lyophilised twice from D2O and subsequently dissolved in 20 mM sodium phosphate buffer (pH 6.8 in H2O). Final sample concentrations were 5 mM.The natural DNA dodecamer and the RNA dodecamer were prepared on an Applied Biosystems ABI 391 PCRmate solid phase oligonucleotide synthesiser. The oligomers were prepared on a 1 µM scale, using the standard protocol. All reagents for the synthesis, including phosphoramidites, were purchased from Glen Research (Virginia, USA). Subsequently deprotection, purification and desalting steps were performed as described previously.42 Modified oligonucleotides were synthesised on an Expedite™ 8909 DNA synthesiser on a 1 µM scale. Introduction of the phosphorothiolate linkage was accomplished using thymidine 3′-S-phosphorothioamidite activated with 4,5-dicyanoimidazole (1 M in acetonitrile) in a 15 min coupling step as previously outlined.43 In all other respects the procedure used the standard capping, oxidation and deblocking reagents and protocols. Deprotection and purification were performed as described previously.42 Purified and desalted oligonucleotides were concentrated and sodium phosphate buffer (pH 6.8) added to give a concentration of 10 mM or 20 mM buffer for NMR and UV studies respectively. The duplexes were obtained by combining equimolar amounts of the complementary strands. For NMR studies samples also contained 100 mM sodium chloride. For experiments carried out in D2O the duplex solutions were lyophilised twice and redissolved in 0.5 ml of 99.99% D2O (Sigma, Dorset, UK) giving final hybrid concentrations of 0.4 and 0.6 mM for (1) and (2) respectively. The samples were annealed by heating to 90 °C for 2 minutes and cooling slowly before each experiment. For experiments carried out in water, the D2O samples were lyophilised and redissolved in 90% HPLC grade H2O (Sigma, Dorset, UK) and 10% D2O (Sigma, Dorset, UK). To check the 1 : 1 ratio of the DNA and RNA strands aliquots of the NMR samples were checked on either analytical ion-exchange or reverse-phase HPLC as described previously. Sample concentrations for UV thermal melting studies were approximately 30 µM.
NMR experiments
NMR experiments were performed on a GE OMEGA 500, a Bruker DRX 500, and a Varian INOVA 500, at 25 °C. 31P spectra for the dinucleotides dissolved in phosphate buffer/D2O were recorded at 202 MHz, over the temperature range 27–57 °C. A spectral width of 4050 Hz was employed together with a relaxation delay of 1 s and 64 transients were acquired. For the duplexes NOESY spectra were recorded with mixing times ranging from 50 to 300 ms. Typically 512 t1 increments of 128–200 transients over 5000 Hz, in 1024 data points were acquired, with hypercomplex phase cycling. Solvent suppression was achieved by presaturation during the preacquisition delay of 1.5 s. For experiments in H2O NOE data were acquired at 5–15 °C using the WATERGATE sequence, a spectral width of 10 000 Hz, up to 256 transients, and 2048 data points. TPPI phase cycling was used. For TOCSY spectra spin lock times of 80–100 ms were utilised with hypercomplex phase cycling. DQFCOSY spectra were collected in TPPI mode. PE-COSY experiments were performed with 1024 t1 increments in 4096 data points. A small flip angle of 35° was employed.The acquired data were processed using FELIX95 (Biosym Technologies, San Diego, CA, USA) mounted on a Sun Sparc IPC station. Gaussian window functions were generally used with line-broadening and Gaussian factors of −10 and 0.4 respectively. Zero filling prior to Fourier transformation (giving a 2k by 2k matrix) resulted in a digital resolution of 2.4 Hz. The PE-COSY data were processed using the Xwinnmr package (Bruker, UK) mounted on a Silicon Graphics O2. Strip processing of the H1′–H2′/H2″ region of the spectrum gave a apparent matrix of 16k × 8k, and a digital resolution of 0.3 Hz in F1.
UV thermal melting
UV thermal melting studies were performed using a Hewlett Packard 8452A diode array spectrophotometer equipped with a Peltier device. Both spectrophotometer and Peltier unit were controlled by a PC using software provided by HP. Absorbance was followed at 260 nm. The sample temperature was increased in half degree increments from 12 to 90 °C, with a five minute equilibration period before each reading.Acknowledgements
This is a contribution from the Leeds Centre for Chemical Dynamics. We thank Dr. Ji-Chun Yang (MRC Laboratory for Molecular Biology, Cambridge, UK) for assistance with the PE-COSY sequence. JRPA acknowledges Yorkshire Cancer Research for support. FKM is supported by an EPSRC studentship. The work at Liverpool was supported by the EPSRC (GRM46181).References
- C. Hélène and J-J. Toulmè, Biochim. Biophys. Acta, 1990, 1049, 99 CrossRef CAS; E. Uhlmann and A. Peyman, Chem. Rev., 1990, 90, 543 CrossRef CAS; S. Agrawal, Trends Biotechnol., 1996, 14, 376 CrossRef CAS; Antisense Research and Application, ed. S. T. Crooke, Springer-Verlag, New York, 1998, vol. 113 Search PubMed; S. Agrawal and E. R. Kandimalla, Mol. Med. Today, 2000, 6, 72 Search PubMed; E. Uhlmann, Curr. Opin. Drug Discovery Dev., 2000, 3, 203 CrossRef CAS.
- (a) K. F. Pirollo, A. Rait, L. S. Sleer and E. H. Chang, Pharmacol. Therapeut., 2003, 99, 55 Search PubMed; (b) M. Petersen and J. Wengel, Trends Biotechnol., 2003, 21, 74 CrossRef CAS.
- S. T. Crooke, Antisense Nucleic Acid Drug Dev., 1998, 8, 7.
- E. T. Kool, Chem. Rev., 1997, 97, 1473 CrossRef CAS.
- (a) S. T. Crooke, Biochim. Biophys. Acta, 1999, 1489, 31 CrossRef CAS; (b) S. Agrawal, Biochim. Biophys. Acta, 1999, 1489, 53 CrossRef CAS; (c) E. Zamaratski, P. I. Pradeepkumar and J. Chattopadhyaya, J. Biochem. Biophys. Methods, 2001, 48, 189 CrossRef CAS; (d) J-J. Toulmè and D. Tidd, Ribonuclease H, ed. R. J. Crouch, ISERM, Paris, 1998, p. 225 Search PubMed.
- M. Manoharan, Antisense Nucleic Acid Drug Dev., 2002, 12, 128 CrossRef CAS.
- A. De Mesmaeker, P. M. Haner and H. E. Moser, Acc. Chem. Res., 1995, 28, 366 CrossRef CAS.
- K. B. Hall and L. W. McLaughlin, Biochemistry, 1991, 30, 10606 CrossRef CAS.
- L. Kværnø and J. Wengel, Chem. Commun., 2001, 1419 RSC.
- S. Gryaznov and J. K. Chen, J. Am. Chem. Soc., 1994, 116, 3143 CrossRef CAS; T. Skorski, D. Perrotti, M. Neiborowska, S. Gryaznov and B. Calabretta, Proc. Natl. Acad. Sci. USA, 1997, 94, 3966 CrossRef CAS.
- (a) K. Bondensgaard, M. Petersen, S. K. Singh, R. K. Rajwanshi, R. Kumar, J. Wengel and J. P. Jacobsen, Chem. Eur. J., 2000, 6, 2687 CrossRef CAS; (b) J. Wengel, Acc. Chem. Res., 1999, 32, 301 CrossRef CAS; (c) M. Petersen, K. Bondensgaard, J. Wengel and J. P. Jacobsen, J. Am. Chem. Soc., 2002, 124, 5974 CrossRef CAS.
- G. Ehrlich, D. Patinkin, D. Ginsberg, H. Zakut, F. Eckstein and H. Soreq, Antisense Res. Dev., 1994, 4, 173 Search PubMed; B. Hatmann, H-O. Betrand and S. Fermandjian, Nucleic Acids Res., 1999, 27, 3342 CrossRef; L. Kværnø, R. Kumar, B. M. Dahl, C. E. Olsen and J. Wengel, J. Org. Chem., 2000, 65, 5167 CrossRef CAS.
- R. Cosstick and J. S. Vyle, Nucleic Acids Res., 1990, 18, 829 CAS.
- C. A. Brautigam, S. Sun, J. A. Piccirilli and T. A. Steitz, Biochemistry, 1999, 38, 696 CrossRef CAS; R. Shah, R. Cosstick and S. C. West, EMBO J., 1997, 16, 1464 CrossRef CAS; J. S. Vyle, D. Kemp, R. Cosstick and B. A. Connolly, Biochemistry, 1992, 31, 3012 CrossRef CAS.
- J. M. Warnecke, E. J. Sontheimer, J. A. Piccirilli and R. K. Hartmann, Nucleic Acids Res., 2000, 28, 720–727 CrossRef CAS; L. B. Weinstein, B. C. N. M. Jones, R. Cosstick and T. R. Cech, Nature, 1997, 388, 805–808 CrossRef CAS; E. J. Sontheimer, S. G. Sun and J. A. Piccirilli, Nature, 1997, 388, 801–805 CrossRef CAS.
- W. Saenger, Principles of Nucleic Acid Structure, Springer-Verlag, New York, 1984 Search PubMed.
- A. P. G. Beevers, E. M. Witch, B. C. N. M. Jones, R. Cosstick, J. R. P. Arnold and J. Fisher, Magn. Reson. Chem., 1999, 37, 814 CrossRef CAS.
- J. D. Puglisi and I. Tinoco, Methods Enzymol., 1989, 80, 304 CrossRef.
- A. P. G. Beevers, K. J. Fettes, I. A. O'Neil, S. M. Roberts, J. R. P. Arnold, R. Cosstick and J. Fisher, Chem. Commun., 2002, 1458 RSC.
- (a) Y-P. Yang, J. L. Miller and P. A. Kollman, J. Am. Chem. Soc., 1999, 121, 1717 CrossRef CAS; (b) C. A. Hunter, K. R. Lawson, J. Perkins and C. J. Urch, J. Chem. Soc., Perkin Trans. 2, 2001, 651 RSC.
- C. Glemarec, Ă. Nyilas, C. Sund and J. Chattopadhyaya, J. Biochem. Biophys. Methods, 1990, 21, 311 CrossRef CAS.
- Methods in Enzymology, ed. T. L. James, Academic Press, New York, 1995, vol. 261 Search PubMed.
- A. P. G. Beevers, PhD Thesis, University of Leeds, 2000.
- S. G. Kim, L. J. Lin and B. R. Reid, Biochemistry, 1992, 31, 3564 CrossRef CAS.
- K. Wuthrich, NMR of Proteins and Nucleic Acids, Wiley, New York, 1986 Search PubMed.
- M. R. Conte, T. C. Jenkins and A. N. Lane, Eur. J. Biochem., 1995, 229, 433 CAS.
- M. Salazar, O. Y. Fedoroff, J. M. Miller, N. S. Ribeiro and B. R. Reid, Biochemistry, 1993, 32, 4207 CrossRef CAS.
- L. J. Rinkel and C. Altona, J. Biomol. Struct. Dyn., 1987, 4, 621 Search PubMed.
- S. K. Singh, P. Nielsen, A. A. Koshkin and J. Wengel, Chem. Commun., 1998, 455 RSC.
- C. Altona and M. Sundaralingam, J. Am. Chem. Soc., 1972, 74, 8205 CrossRef; J. Vanwijk, B. D. Huckriede, J. H. Ippel and C. Altona, Methods Enzymol., 1992, 211 Search PubMed.
- P. A. Frey and R. D. Sammons, Science, 1985, 228, 541 CAS.
- D. Ding, S. M. Gryaznov, D. H. Lloyd, S. Chandrasekaran, S. Yao, L. Ratmeyer, Y-Q. Pan and J. D. Wilson, Nucleic Acids Res., 1996, 24, 354 CrossRef CAS.
- V. A. Roongta, C. R. Jones and D. G. Gorenstien, Biochemistry, 1990, 29, 5245 CrossRef CAS.
- C. Gressner-Prettre, B. Pullman, P. N. Borer, L. S. Lan and P. O. P. Ts'o, Biopolymers, 1976, 15, 2277 CrossRef CAS.
- B. M. Znosko, T. W. Barnes III, T. R. Krugh and D. H. Turner, J. Am. Chem. Soc., 2003, 125, 6090 CrossRef CAS.
- G. Varani and I. Tinoco, Quart. Rev. Biophys., 1991, 24, 479 CAS.
- L. Dedionisio and S. M. Gryaznov, J. Chromatogr. B, 1995, 669, 125 CrossRef CAS.
- C. Wahlestedt, P. Slami, L. Good, J. Kela, T. Johnsson, T. Hokfeldt, C. Broberger, F. Porreca, J. Lai, K. Ren, M. Ossipov, A. Koshkin, N. Jakobsen, J. Skour, H. Oerum, M. H. Jacobsen and J. Wengel, Proc. Natl,. Acad. Sci. USA., 2000, 97, 5633 CrossRef CAS.
- M. Manoharan, Antisense Nucleic Acids Drug Dev., 2002, 12, 128 Search PubMed.
- T. N. Jaishree, G. A. Van der Marel, J. H. Van Boom and A. H. J. Wang, Biochemistry, 1993, 32, 4903 CrossRef CAS.
- S. A. Fawthrop, J-C. Yang and J. Fisher, Nucleic Acids Res., 1993, 21, 4860 CAS; S. A. Fawthrop, PhD Thesis, University of Leeds, 1993.
- J. B. Murray, A. K. Collier and J. R. P. Arnold, Anal. Biochem., 1994, 218, 177 CrossRef CAS.
- K. J. Fettes, S. M. Roberts, I. A. O'Neil and R. Cosstick, Nucleosides Nucleotides, 2001, 20, 1351 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: chemical shift assignments for (1) and (2), expansions of NOESY/TOCSY spectra showing the H1′–H2′/H2″ connections for (1) and (2), sections through NOESY spectra illustrating the differing H2 to H1′ cross-peak intensities for (1) and (2), sections of PE-COSY spectra, sample UV thermal melting curve for (1) and (2). See http://www.rsc.org/suppdata/ob/b3/b311923h/ |
| This journal is © The Royal Society of Chemistry 2004 |