Direct synthesis of unprotected phenols using palladium-catalysed cross coupling reactions of functionalised organozinc reagents
Richard F. W.
Jackson
*a,
Ian
Rilatt
a and
P. John
Murray
b
aDepartment of Chemistry, Dainton Building, University of Sheffield, Brook Hill, Sheffield, UK S3 7HF. E-mail: r.f.w.jackson@shef.ac.uk
bOSI Pharmaceuticals, Watlington Road, Oxford, UK OX4 6LT
First published on 18th November 2003
Abstract
Palladium-catalysed reaction of unprotected 2-, 3-, and 4-iodophenols with a range of amino acid derived organozinc reagents (not used in excess) gives the expected products in good to excellent yield, demonstrating that carbon–zinc bonds are not protonated by acidic phenols under the conditions of palladium-catalysed coupling reactions.
Introduction
A key goal in improving the efficiency of synthetic routes is to reduce, as far as is possible, the need for protecting groups. In the context of reactions of electrophiles with organometallic nucleophiles, protection of alcohols in the electrophile is almost always considered necessary, and was one of the key driving forces for the development of a wide range of hydroxyl protecting groups,1,2 notably silyl ethers. The enhanced acidity of phenols (the pKa of phenol measured in water is 10) means that the need for protection of the phenolic hydroxyl group in reactions involving organometallic reagents goes largely unquestioned.It has been known for some time that carbon–zinc bonds can tolerate the presence of acidic protons, for example in the organozinc reagents 2–5.3–5 Knochel et al. have shown that organozinc halides can be formed in the presence of a range of additives containing acidic protons, although the presence of these additives does reduce the yield of the organozinc reagent.6 Most interestingly, it was demonstrated that reactions that proceed rapidly at low temperature, for example reaction of organocopper reagents with allylic electrophiles, can be carried out in the presence of additives containing free hydroxyl groups, including phenol (although the yield of product was slighly reduced).6 These results imply that, under the right circumstances, protection of free hydroxyl groups may not be required. Since this is important for the application of organozinc chemistry to the chemical modification of peptides, which may incorporate a range of acidic protons, we have decided to test it experimentally.
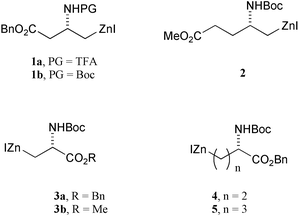
Our recent discovery that it is possible to prepare the methyl ester analogue of the trifluoroacetyl-protected organozinc reagent 1a,7 in which the pKa of the NH proton is estimated to be around 17 (in DMSO),8 prompted us to consider whether other protons of comparable acidity might also be tolerated in the electrophile, and the most obvious and useful functional group that fulfils this criterion appeared to be phenol (the pKa of phenol measured in DMSO is 18).8 We have therefore explored the cross-coupling of free iodophenols with a range of functionalised zinc reagents, since this would give direct access to tyrosine analogues, without the need for hydroxyl protection.†
Results and discussion
Our first experiment involved the attempted coupling of N-TFA protected zinc reagent 1a with 4-iodophenol (1.3 eq.) under standard Pd-catalysed conditions (Scheme 1). We were delighted to isolate the desired product 6 in 66% yield, comparable with the yields obtained from the reaction of the same zinc reagent with electrophiles not containing acidic protons.7 The fact that the yield was greater than 50% established that protonation of the organozinc reagent by the phenol was occurring at a slower rate than the cross coupling reaction, and did not compromise the viability of the reaction.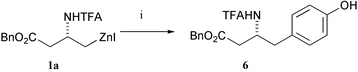 | ||
| Scheme 1 Reagents and conditions: 4-IC6H4OH, DMF, Pd2(dba)3 (2.5 mol%), P(o-tol)3 (10 mol%), r.t. | ||
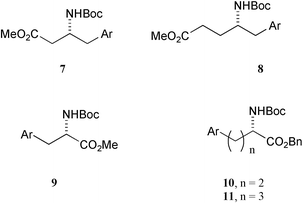
Having established this principle, we have explored the coupling of a range of other amino acid-derived organozinc reagents with unprotected 2-, 3- and 4-iodophenol, which allowed us to isolate the free phenols 7–11, and our results are given in Table 1. The very high yields obtained in the cross coupling with 3- and 4-iodophenol using zinc reagent 1b are a striking demonstration of the tolerance exhibited by this reagent towards acidic protons. The lower yield obtained with 2-iodophenol reflects the behaviour of other 2-substituted iodobenzene derivatives in similar coupling reactions.4 Finally, attempted reaction of 3b with 3,4-dihydroxyiodobenzene unfortunately gave none of the desired product, L-DOPA, indicating the enhanced acidity of catechol is sufficient to ensure competitive protonation of the zinc reagent.
| Organozinc | Electrophile | Product | Ar | Yield (%)a |
|---|---|---|---|---|
| a Isolated yields refer to homogeneous material purified by flash chromatography. | ||||
| 1a | 4-IC6H4OH | 6 | 4-HOC6H4 | 66 |
| 1b | 2-IC6H4OH | 7a | 2-HOC6H4 | 42 |
| 1b | 3-IC6H4OH | 7b | 3-HOC6H4 | 88 |
| 1b | 4-IC6H4OH | 7c | 4-HOC6H4 | 85 |
| 2 | 4-IC6H4OH | 8 | 4-HOC6H4 | 64 |
| 3b | 4-IC6H4OH | 9a | 4-HOC6H4 | 59 |
| 3b | 3-IC6H4OH | 9b | 3-HOC6H4 | 54 |
| 4 | 4-IC6H4OH | 10 | 4-HOC6H4 | 48 |
| 5 | 4-IC6H4OH | 11 | 4-HOC6H4 | 49 |
In a single experiment, we have also established that an unprotected benzylic alcohol functionality can be tolerated; the cross-coupling reaction of zinc reagent 1b with 3-iodobenzyl alcohol gave the product 12 (Scheme 2), although the yield was modest (42%).
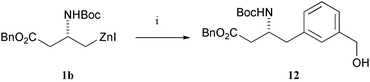 | ||
| Scheme 2 Reagents and conditions: 3-IC6H4CH2OH, DMF, Pd2(dba)3 (2.5 mol%), P(o-tol)3 (10 mol%), r.t. | ||
Conclusions
In summary we have demonstrated that the protection of iodophenols is unnecessary in the Pd-catalysed cross coupling of organozinc iodides, and thereby broadened the potential scope of these reagents in synthesis. These results reinforce the notion that pKa values measured in water need to be used with care, especially when reactions are conducted in aprotic solvents, and that pKa values measured in DMSO8 are a more reliable guide to potential incompatibility.Experimental
The iodide precursors to the zinc reagents 2,53b,444 and 54 were prepared by literature methods. The precursor to 1b was prepared from Boc-L-aspartic acid β-benzyl ester by an analogous method to that used for the preparation of the corresponding methyl ester.5 An alternative method has been described.9Synthesis of iodide precursor to zinc reagent 1a
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1708 (CO); 1180 (C–O). Found C, 48.85; H, 3.70; N, 4.32. C13H12NO5F3 requires C, 48.91; H, 3.79; N, 4.39%. δH
(250 MHz, CDCl3) 2.98 (1H, dd, J 17.5 and 4.0), 3.22 (1H, dd, J 17.5 and 4.5), 4.82–4.98 (1H, m), 5.27 (2 H, s), 7.28–7.41 (5H, m) and 7.45 (1H, d, J 8.5); δC
(62.5 MHz, CDCl3) 35.3, 48.7, 67.6, 115.5 (q, J 283), 128.4, 128.7, 129.7, 134.7, 157.3 (q, J 34), 170.8 and 173.9; m/z
(EI) 319.0668 (8%, M+, C13H12NO5F3 requires 319.0667), 184(6), 166(3), 140(5), 108(100), 107(56) and 99(10); [α]22.5D
+59.1 (c 1.02 in CHCl3).
O); 1710 (CO); 1209 (C–O). Found C, 49.19; H, 3.53; N, 6.69. C17H15O7N2F3 requires C, 49.05; H, 3.63; N, 6.73%. δH
(250 MHz, CDCl3) 2.85 (4H, s), 3.02 (1H, dd, J 17.5 and 4.5), 3.27 (1H, dd, J 17.5 and 4.5), 5.11–5.28 (2H, m), 5.28–5.38 (1H, m), 7.36 (5H, s) and 7.72 (1H, d, J 8.5); δC
(62.5 MHz, CDCl3) 25.5, 35.5, 47.4, 67.7, 115. 5 (q, J 285), 128.6, 128.7, 129.6, 134.8, 157.0 (q, J 39), 165.3, 168.5 and 169.7; m/z
(EI) 416.0835 (12%, M+, C17H15O7N2F3 requires 416.0835), 273(4), 224(6), 184(24), 166(4), 139(18), 115(11), 108(25) and 107(43); [α]22.5D
+ 7.0 (c 1.01 in CHCl3).
O). Found C, 51.25; H, 4.66; N, 4.60. C13H14O4NF3 requires C, 51.15; H, 4.62; N, 4.59%. δH
(250 MHz, CDCl3) 2.22 (1H, br t), 2.77 (2H, d, J 6), 3.69–3.87 (2H, m), 4.28–4.43 (1H, m), 5.15 (2H, s) and 7.36 (5H, s); δC
(62.5 MHz, CDCl3) 34.8, 48.7, 62.9, 67.1, 115.8 (q, J 287), 128.4, 128.6, 128.7, 135.1, 157.4 (q, J 38), and 171.3. m/z
(EI) 306.0955 (1 %, MH+, C13H15O4NF3 requires 306.0953), 305(3), 198(7), 139(24), 108(91), 107(43), 91(100) and 79(44); [α]22.5D
+ 9.1 (c 0.99 in CHCl3).
O); 1702 (CO); 1186 (C–O). Found C, 37.88; H, 3.06; N, 3.21; I, 30.57. C13H13O3NF3I requires C, 37.61; H, 3.16; N, 3.37; I, 30.57%. δH
(250 MHz, CDCl3) 2.75 (1H, dd, J 17.0 and 5.5), 2.95 (1H, dd, J 17.0 and 5.0), 3.30–3.50 (2H, m), 4.23–4.40 (1H, m), 5.17 (2H, s), 7.14 (1H, d, J 17.0) and 7.30–7.50 (5H, m); δC
(62.5 MHz, CDCl3) 7.1, 37.7, 47.7, 67.2, 115.7 (q, J 288), 128.4, 128.6, 128.7, 135.1, 156.8 (q, J 37), and 170.2; m/z
(EI) 414.9906 (13%, M+, C13H13O3NF3I requires 414.9892), 308(5), 266(7), 195(9), 154(3), 139(7), 108(100) and 107(32); [α]22.5D
+ 7.0 (c 1.00 in CHCl3).
O); 1708 (CO); 1180 (C–O). Found C, 48.85; H, 3.70; N, 4.32. C13H12NO5F3 requires C, 48.91; H, 3.79; N, 4.39%. δH
(250 MHz, CDCl3) 2.98 (1H, dd, J 17.5 and 4.0), 3.22 (1H, dd, J 17.5 and 4.5), 4.82–4.98 (1H, m), 5.27 (2 H, s), 7.28–7.41 (5H, m) and 7.45 (1H, d, J 8.5); δC
(62.5 MHz, CDCl3) 35.3, 48.7, 67.6, 115.5 (q, J 283), 128.4, 128.7, 129.7, 134.7, 157.3 (q, J 34), 170.8 and 173.9; m/z
(EI) 319.0668 (8%, M+, C13H12NO5F3 requires 319.0667), 184(6), 166(3), 140(5), 108(100), 107(56) and 99(10); [α]22.5D
+59.1 (c 1.02 in CHCl3).
O); 1710 (CO); 1209 (C–O). Found C, 49.19; H, 3.53; N, 6.69. C17H15O7N2F3 requires C, 49.05; H, 3.63; N, 6.73%. δH
(250 MHz, CDCl3) 2.85 (4H, s), 3.02 (1H, dd, J 17.5 and 4.5), 3.27 (1H, dd, J 17.5 and 4.5), 5.11–5.28 (2H, m), 5.28–5.38 (1H, m), 7.36 (5H, s) and 7.72 (1H, d, J 8.5); δC
(62.5 MHz, CDCl3) 25.5, 35.5, 47.4, 67.7, 115. 5 (q, J 285), 128.6, 128.7, 129.6, 134.8, 157.0 (q, J 39), 165.3, 168.5 and 169.7; m/z
(EI) 416.0835 (12%, M+, C17H15O7N2F3 requires 416.0835), 273(4), 224(6), 184(24), 166(4), 139(18), 115(11), 108(25) and 107(43); [α]22.5D
+ 7.0 (c 1.01 in CHCl3).
O). Found C, 51.25; H, 4.66; N, 4.60. C13H14O4NF3 requires C, 51.15; H, 4.62; N, 4.59%. δH
(250 MHz, CDCl3) 2.22 (1H, br t), 2.77 (2H, d, J 6), 3.69–3.87 (2H, m), 4.28–4.43 (1H, m), 5.15 (2H, s) and 7.36 (5H, s); δC
(62.5 MHz, CDCl3) 34.8, 48.7, 62.9, 67.1, 115.8 (q, J 287), 128.4, 128.6, 128.7, 135.1, 157.4 (q, J 38), and 171.3. m/z
(EI) 306.0955 (1 %, MH+, C13H15O4NF3 requires 306.0953), 305(3), 198(7), 139(24), 108(91), 107(43), 91(100) and 79(44); [α]22.5D
+ 9.1 (c 0.99 in CHCl3).
O); 1702 (CO); 1186 (C–O). Found C, 37.88; H, 3.06; N, 3.21; I, 30.57. C13H13O3NF3I requires C, 37.61; H, 3.16; N, 3.37; I, 30.57%. δH
(250 MHz, CDCl3) 2.75 (1H, dd, J 17.0 and 5.5), 2.95 (1H, dd, J 17.0 and 5.0), 3.30–3.50 (2H, m), 4.23–4.40 (1H, m), 5.17 (2H, s), 7.14 (1H, d, J 17.0) and 7.30–7.50 (5H, m); δC
(62.5 MHz, CDCl3) 7.1, 37.7, 47.7, 67.2, 115.7 (q, J 288), 128.4, 128.6, 128.7, 135.1, 156.8 (q, J 37), and 170.2; m/z
(EI) 414.9906 (13%, M+, C13H13O3NF3I requires 414.9892), 308(5), 266(7), 195(9), 154(3), 139(7), 108(100) and 107(32); [α]22.5D
+ 7.0 (c 1.00 in CHCl3).
General procedure for the coupling reactions of iodides with aryl iodides
Zinc dust (0.236 g, 3.6 mmol, 6.0 eq.) was placed in a dry 25 mL round bottom flask, with sidearm, containing a rugby ball shaped stirrer. The flask was flushed with nitrogen and dry DMF (0.75 mL) and TMSCl (100 µL, 0.8 mmol) were added under nitrogen via syringe. The solution was observed to effervesce and the mixture was vigorously stirred at room temperature for 5 min (the DMF occasionally changes to a yellow colour during this period). The zinc was allowed to settle and the supernatant solution was removed via syringe, followed by drying of the zinc under vacuum by heating with a hot air gun. A solution of the zinc reagent precursor iodide (0.6 mmol, 1.0 eq.) was dissolved in DMF (0.75 mL) under nitrogen and transferred to the zinc via syringe. The solution was stirred at room temperature and the insertion judged to be complete by TLC within 5 min. Pd2(dba)3 (17.9 mg, 0.02 mmol), P(o-tol)3 (23.8 mg, 0.08 mmol) and the aryl iodide (1.3 eq. relative to the iodide) were added to the flask. The flask was covered with aluminium foil and left at room temperature overnight. The reaction was diluted with EtOAc (50 mL), filtered and evaporated under reduced pressure. The residue was warmed at 40 °C under high vacuum to remove the DMF. The crude product was purified by column chromatography.O); 1516 (N–H). Found C, 59.36; H, 4.67; N, 3.52. C19H18F3NO4 requires C, 59.84; H, 4.76; N, 3.67. δH
(400 MHz, CD3OD) 2.58 (1H, dd, J 15.5 and 8.5), 2.64 (1H, dd, J 15.5 and 5.5), 2.69 (1H, dd, J 13.5 and 8), 2.75 (1H, dd, J 14 and 6.5), 4.39–4.49 (1H, m), 5.04 (1H, d, J 12), 5.09 (1H, d, J 12), 6.69 (2H, d, J 8.5) and 6.96 (2H, d, J 8.5). δC
(100 MHz, CD3OD) 39.0, 40.0, 50.5, 67.6, 116.2, 117.4 (q, J 287), 129.2, 129.3, 129.4, 129.5, 131.3, 137.3, 157.3, 158.3 (q, J 37) and 172.1; m/z
(TOF MS ES+) 382.1262 (7%, MH+, C19H19F3NO4 requires 382.1266), 304(10) and 251(100).
O); 1534 (N–H). Found C, 68.69; H, 6.96; N, 3.49. C22H27NO5 requires C, 68.55; H, 7.06; N, 3.63%. δH
(250 MHz, CDCl3) 1.44 (9H, s), 2.59 (2H, br d, J 5), 2.60 (1H, m (signal partially obscured)), 3.01–3.12 (1H, m), 3.83–4.01 (1H, m), 5.16 (2H, s), 5.53 (1H, br d, J 7.5), 6.79 (1H, t, J 7.5), 6.88 (1H, d, J 7.6), 6.96 (1H, d, J 7.5), 7.13 (1H, t, J 7.5), 7.37 (5H, s) and 7.85 (1H, br s). δC
(62.5 MHz, CDCl3) 28.2, 36.1, 36.7, 48.5, 66.6, 80.4, 116.3, 119.8, 123.2, 128.3, 128.4, 128.6, 130.8, 135.4, 155.4, 156.4, and 171.7 (one signal obscured); m/z
(EI+) 385.1875 (3%, M+, C22H27NO5 requires 385.1889), 385(3), 222(19), 178(64) and 91(100); [α]22.1D
+19.6 (c 1.02 in CHCl3).
O); 1691 (CO); 1513 (N–H). Found C, 68.48; H, 7.02; N, 3.51. C22H27NO5 requires C, 68.55; H, 7.06; N, 3.63%. δH
(250 MHz, CDCl3) 1.40 (9H, s), 2.49 (1H, dd, J 16.5 and 6), 2.56 (1H, dd, J 16.5 and 6), 2.73 (1H, dd, J 13 and 7.9), 2.85 (1H, dd, J 13.5 and 6.5), 4.07–4.26 (1H, m), 5.09 (1H, d, J 12), 5.10 (1H, signal partially obscured), 5.17 (1H, d, J 12), 5.85 (1H, s), 6.58–6.78 (3H, m), 7.11 (1H, t, J 8) and 7.36 (5H, s). δC
(62.5 MHz, CDCl3) 28.3, 37.6, 40.2, 48.7, 66.5, 79.7, 113.7, 116.2, 121.2, 128.3, 128.5, 129.5, 135.5, 139.0, 155.4, 156.3 and 171.6 (one signal obscured); m/z
(EI+) 385.1888 (5%, M+, C22H27NO5 requires 385.1889), 278(62), 222(62) and 178(100); [α]22.1D
+6.9 (c 1.02 in CHCl3).
O); 1686 (CO); 1529 (N–H). Found C, 68.32; H, 6.96; N, 3.41. C22H27NO5 requires C, 68.55; H, 7.06; N, 3.63%. δH
(400 MHz, (CD3)2CO) 1.40 (9H, s), 2.58 (2H, d, J 6.5), 2.75 (1H, dd, J 13.5 and 7), 2.83 (1H, dd, J 13.5 and 7), 4.07–4.23 (1H, m), 5.06–5.20 (2H, m), 5.95 (1H, br d, J 7.5), 6.79 (2H, d, J 8), 7.07 (2H, d, J 8), 7.27–7.54 (5H, m) and 8.19 (1H, s). δC
(100 MHz, (CD3)2CO) 28.7, 39.4, 40.4, 50.7, 66.7, 78.9, 116.0, 128.9, 129.0, 129.4, 130.0, 131.3, 137.5, 156.0, 156.9 and 171.9; m/z
(EI+) 385.1887 (3%, M+, C22H27NO5 requires 385.1889), 268(56), 222(42), 178(82) and 91(100); [α]22.1D
+6.9 (c 1.02 in CHCl3).
O); 1683 (CO); 1516 (N–H). Found C, 63.08; H, 8.13; N, 4.22. C17H25NO5 requires C, 63.14; H, 7.79; N, 4.33%. δH
(400 MHz, CD3OD) 1.36 (9H, s), 1.50–1.64 (1H, m), 1.76–1.88 (1H, m), 2.29 (1H, dd, J 16.5 and 7), 2.36 (1H, dd, J 16.5 and 6.5), 3.62 (4H, s (two overlapping signals)), 6.68 (2H, d, J 8.5) and 7.00 (2H, d, J 8.5). δC
(100 MHz, CD3OD) 28.8, 30.6, 31.7, 41.8, 52.1, 53.1, 79.8, 116.0, 130.7, 131.3, 156.8, 158.1 and 175.6; m/z
(EI+) 323.1731 (3%, M+, C17H25NO5 requires 323.1733), 216(32), 160(17), 116(72) and 107(59); [α]22.5D
+1.0 (c 1.02 in MeOH).
O); 1720 (CO); 1690 (CO). Found C, 60.88; H, 7.82; N, 4.42. C15H21NO5 requires C, 61.00; H, 7.17; N, 4.74%; m/z
(EI+) 295.1409 (4%, M+, C22H27NO5 requires 295.1420), 239(25), 178(100), 136(22) and 107(24); [α]22.5D
+38.6 (c 1.04 in CHCl3), literature value [α]25D
+34.2 (c 1.18 in CHCl3).13
O); 1716 (CO); 1689 (CO); 1516 (N–H). Found C, 68.14; H, 7.45; N, 3.51. C22H27NO5 requires C, 68.55; H, 7.06; N, 3.63%. δH
(400 MHz, CDCl3) 1.45 (9H, s), 1.80–1.95 (1H, m), 2.00–2.14 (1H, m), 2.43–2.60 (2H, m), 4.32–4.43 (1H, m), 5.09 (1H, d, J 12.5), 5.18 (1H, d, J 12.5), 5.18 (1H, signal partially obscured), 6.21 (1H, s), 6.72 (2H, d, J 8.5), 6.91 (2H, d, J 8.5) and 7.33 (5H, s). δC
(100 MHz, CDCl3) 28.3, 30.5, 34.4, 53.2, 67.1, 80.3, 115.3, 128.3, 128.4, 128.6, 129.4, 132.2, 135.2, 154.3, 155.5 and 172.6; m/z
(EI+) 385.1880 (6%, M+, C22H27NO5 requires 385.1889), 329(31), 238(24), 209(15), 177(100) and 107(35); [α]22.5D
−18.5 (c 1.08 in MeOH).
O); 1716 (CO); 1688 (CO); 1516 (N–H). Found C, 68.72; H, 7.60; N, 3.42. C23H29NO5 requires C, 69.15; H, 7.32; N, 3.51%. δH
(400 MHz, CDCl3) 1.48 (9H, s), 1.54–1.73 (3H, m), 1.79–1.93 (1H, m), 2.45–2.60 (2H, m), 4.37–4.35 (1H, m), 5.12 (1H, d, J 12.5), 5.20 (1H, signal partially obscured), 5.23 (1H, d, J 12.5), 6.20 (1H, s), 6.77 (2H, d, J 8.5), 6.95 (2H, d, J 8.5) and 7.32–7.43 (5H, m). δC
(100 MHz, CDCl3) 27.6, 28.7, 32.5, 34.7, 53.8, 67.5, 80.7, 115.6, 128.7, 128.9, 129.0, 129.8, 133.7, 135.7, 154.6, 156.0 and 173.3; m/z
(EI+) 399.2055 (1%, M+, C23H29NO5 requires 399.2046), 320(29), 305(100), 208(13), 165(13) and 91(23); [α]22.5D
−21.3 (c 1.04 in MeOH).
O); 1685 (CO); 1530 (N–H). δH
(400 MHz, CDCl3) 1.38 (9H, s), 2.25 (1H, br s), 2.46 (1H, dd, J 16 and 6), 2.54 (1H, dd, J 16 and 6), 2.77 (1H, dd, J 13.5 and 8), 2.90 (1H, dd, J 13.5 and 6.5), 4.09–4.21 (1H, m), 4.60 (2H, s), 5.08 (1H, d, J 12.5), 5.09 (1H, signal partially obscured), 5.14 (1H, d, J 13.5) and 7.01–7.46 (9H, m). δC
(100 MHz, CDCl3) 28.3, 37.6, 40.2, 48.8, 65.0, 66.4, 79.4, 125.2, 127.9, 128.3, 128.5, 128.6, 132.0, 132.9, 135.6, 137.9, 141.2, 155.1 and 171.5; m/z
(EI+) 422.1935 (8%, MNa+, C23H29NO5Na requires 422.1943), 400(9), 344(29), 321(100), 300(33) and 282(28); [α]22.5D
+9.7 (c 1.04 in MeOH).
Acknowledgements
We thank EPSRC for the award of a DTA studentship (IR), and OSI Pharmaceuticals for additional support.References
- P. G. M. Wuts and T. W. Greene, Protective Groups in Organic Synthesis, 3rd ed., John Wiley & Sons, New York, 1999 Search PubMed.
- P. J. Kocienski, Protecting Groups,Georg Thieme Verlag, Stuttgart, 1994 Search PubMed.
- R. F. W. Jackson, N. Wishart, A. Wood, K. James and M. J. Wythes, J. Org. Chem., 1992, 57, 3397 CrossRef CAS.
- R. F. W. Jackson, R. J. Moore, C. S. Dexter, J. Elliot and C. E. Mowbray, J. Org. Chem., 1998, 63, 7875 CrossRef CAS.
- C. S. Dexter, R. F. W. Jackson and J. Elliott, J. Org. Chem., 1999, 64, 7579 CrossRef CAS.
- H. P. Knoess, M. T. Furlong, M. J. Rozema and P. Knochel, J. Org. Chem., 1991, 56, 5974 CrossRef CAS.
- R. F. W. Jackson, I. Rilatt and P. J. Murray, Chem. Commun., 2003, 1242 RSC.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456 CrossRef CAS.
- R. Caputo, E. Cassano, L. Longobardo and G. Palumbo, Tetrahedron, 1995, 51, 12337 CrossRef CAS.
- J. Faust and H. Ripperger, J. Prakt. Chem., 1988, 330, 1033 CAS.
- M. M. Hann, P. G. Sammes, P. D. Kennewell and J. B. Taylor, J. Chem. Soc., Perkin Trans. 1, 1982, 307 RSC.
- M. E. Jung and L. S. Starkey, Tetrahedron, 1997, 53, 8815 CrossRef CAS.
- W. Wang, C. Y. Xiong, J. Y. Zhang and V. J. Hruby, Tetrahedron, 2002, 58, 3101 CrossRef CAS.
- B. Ye and T. R. Burke, J. Org. Chem., 1995, 60, 2640 CrossRef CAS.
- P. W. Sheldrake, L. C. Powling and P. K. Slaich, J. Org. Chem., 1997, 62, 3008 CrossRef CAS.
- S. Kawata, S. Ashizawa and M. Hirama, J. Am. Chem. Soc., 1997, 119, 12012 CrossRef CAS.
Footnote |
| † There is a report14 concerning the coupling of zinc reagent 3a with unprotected 2-iodo-3-hydroxypyridine15 which gave the expected product in 33% yield, but the reaction used 2 equivalents of the organozinc reagent, so the possibility that the free hydroxyl group might have been tolerated in the reaction was not addressed. In another report, which highlights the desirability of avoiding the use of protecting groups, 2-chloro-3-hydroxy-6-iodopyridine was protected as its TBS-ether prior to Pd-catalysed coupling with the zinc reagent 3a, and the protecting group was removed immediately afterwards.16 |
| This journal is © The Royal Society of Chemistry 2004 |