Cyclic PNA-based compound directed against HIV-1 TAR RNA: modelling, liquid-phase synthesis and TAR binding†
Geoffrey
Depecker
a,
Nadia
Patino
a,
Christophe
Di Giorgio
a,
Raphael
Terreux
b,
Daniel
Cabrol-Bass
c,
Christian
Bailly
d,
Anne-Marie
Aubertin
e and
Roger
Condom
*a
aLaboratoire de Chimie Bioorganique, UMR UNSA-CNRS 6001, Faculté des Sciences, Université de Nice-Sophia Antipolis, 06108 Nice Cédex 2, France. E-mail: condom@unice.fr; Fax: (33) 04 92 07 61 51; Tel: (33) 04 92 07 61 52
bInstitut des Sciences Pharmaceutiques et Biologiques, Université Claude Bernard Lyon 1, 8 av Rockefeller, 69008 Lyon Cedex 03, France
cLaboratoire ASI, Faculté des Sciences, Université de Nice Sophia-Antipolis, 06108 Nice Cedex 2, France
dLaboratoire de Pharmacologie Moléculaire Antitumorale, INSERM U124, Place de Verdun, 59045 Lille Cedex, France
eINSERM U74, Institut de Virologie, 3 rue Koberlé, 67000 Strasbourg, France
First published on 27th November 2003
Abstract
A cyclic molecule including a hexameric PNA sequence has been designed and synthesized in order to target the TAR RNA loop of HIV-1 through the formation of a “kissing complex”. For comparison, its linear analogue has also been investigated. The synthesis of the cyclic and linear PNA has been accomplished following a liquid-phase strategy using mixed PNA and fully N-protected (aminoethylglycinamide) fragments. The interactions of this cyclic PNA and its linear analogue with TAR RNA have been studied and the results indicate clearly that no interaction occurs between the cyclic antisense PNA and TAR RNA, whereas a tenuous interaction has been detected with its linear PNA analogue.
Introduction
The TAR RNA element of the HIV-1 genome (Fig. 1) is involved in essential steps of the viral life cycle, including the transcription process,1 reverse transcription and genomic RNA packaging.2 Therefore, this stem loop structure represents a target of considerable interest for inhibiting viral replication. HIV-1 transcriptional elongation is regulated via the formation of a ternary complex, implying the stem-loop TAR structure, the trans-activator viral protein Tat and a Tat-associated kinase, which includes host cellular factor cyclin T1 and kinase CDK9.3 The formation of this complex is crucial for a productive viral replication, as it allows an increased production of full-length transcripts. Thus, inhibition of Tat (or its cofactors) binding on TAR represents a very attractive goal that is intensively studied.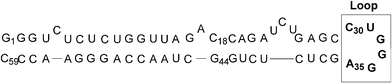 | ||
| Fig. 1 Structure of the TAR RNA of HIV-1. | ||
Among the various approaches investigated, the antisense strategy using synthetic DNA or RNA analogues is the most popular one. Thus, phosphorothioates,4 deoxyphosphoramidate oligomers,5 oligo-2′-O-methylribonucleotides,5,6 oligo-2′-O-methylribonucleoside methylphosphonates,7 locked nucleic acids (LNA),5,6 polyamide nucleic acids (PNA)8 and very recently, oligonucleotides containing 2′-O-methyl G-Clamp ribonucleoside analogues9 were used to target the complex Tat/CyclinT1/CDK9 recognition site on TAR, i.e. the apical stem-loop and the bulge region of TAR. Some of these oligomers (9–16 mers) did bind to TAR RNA very strongly and were able to inhibit the Tat-dependent in vitro transcription. Another interesting approach to target TAR that was described very recently used analogues of mini RNA hairpins10,11 including a six-nucleotide sequence complementary to the TAR loop and capable of forming a “kissing complex” through loop–loop interactions. These ligands were shown to specifically inhibit Tat-dependent transcription in a cell-free assay. However, none of these DNA, RNA or mini RNA hairpin analogues displayed an efficient cellular antiviral activity, likely because of their poor cellular uptake. Nevertheless, it was demonstrated that upon conjugation with a membrane-permeating peptide (transportan), a 16-mer PNA complementary to the bulge and loop regions of TAR retained its affinity for the target sequence and was able to inhibit effectively HIV-1 replication in cell culture.8b
In an earlier paper,12 some of us described the molecular modelling, liquid-phase synthesis and biological activity of a cyclic molecule containing a hexameric PNA moiety, complementary to six of the nine residues of the dimerization initiation site (DIS) loop of HIV-1, a highly conserved stem-loop sequence. This molecule was shown to inhibit the HIV-1 dimerization process in vitro, probably through kissing-loop complex formation. This kind of “loop like” compound, constituted by a low number of PNA units (i.e. 6-mer), should afford advantages over linear analogues such as greater selectivity, or over classical antisense oligomers, such as lower molecular weight.
In this context and on the basis of molecular modelling studies, we designed, synthesized and evaluated the cyclic compound 1 (Fig. 2), which is constituted by i) an antisense 5′-UCCCAG-3′ hexa-PNA complementary to the six residues (C30 to A35) of the TAR loop, and ii) a linker tethering the N- and C-terminal extremities of the oligomer backbone. The length of the linker, which is constituted by two 6-aminocaproic acid residues (14 atoms), was optimized by molecular modelling to allow a kissing complex to occur. Moreover, with the aim of comparing their ability to bind TAR, we synthesized and evaluated the linear antisense 5′-UCCCAG-3′ analogue 2 (Fig. 2).
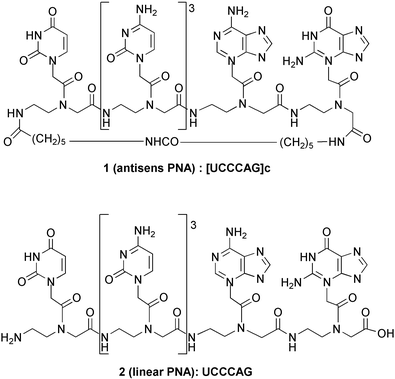 | ||
| Fig. 2 Structure of the target cyclic PNA 1 and of its linear analogue 2. | ||
Herein, we report on the molecular design and the liquid-phase synthesis of the cyclic PNA [UCCCAG]c 1, as well as the synthesis of its linear analogue 2. The ability of these two compounds to bind TAR has been studied via melting temperature measurements, electromobility shift assays and by RNase protection experiments.
Results and discussion
Dynamic molecular modelling studies
On the basis of the 3D structure proposed by Tinoco et al. [i.e. Kissing-loop complex formed between TAR RNA hairpin loop with its complementary hairpin loop sequence (TAR*)14 ], a model of compound 1/TAR RNA complex was built (Fig. 3). For the three first steps, the Insight II (version 97.0) molecular modelling package18 was used with the Hagler (CFF) forcefield.19 Partial charges on the different atoms were computed according to the Gasteiger-Marsili algorithm. Calculations were made using a SGI indigo 2 R10.000 of the LARTIC laboratory.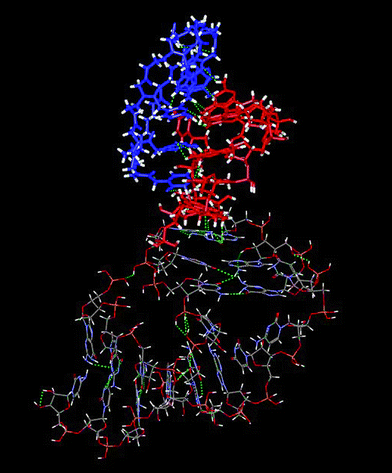 | ||
| Fig. 3 Molecular model of the interaction between compound 1 and TAR RNA of HIV-1. The TAR loop residues are in blue, the cyclic PNA 1 in red. | ||
In a first step, the complementary TAR* RNA sequence has been erased except for the six base residues involved in the kissing complex. The PNA scaffold was then built, resulting in a new complex between the neo-formed linear hexa-PNA with the TAR RNA loop. Additional constraints were set between complementary bases to keep them interacting during the construction time. The resulting structure was then geometrically optimized.
In a second step, short molecular dynamics (200 ps) were carried out to verify the stability of this complex and to relax the whole structure formed after the previous modification.
The third step consisted in adding the linker to close the PNA. Geometric optimization was also applied and the optimal length was found to be 14 atoms. The whole structure was also geometrically optimized.
To validate the design, a 600 ps molecular dynamic simulation with constant temperature and pressure in an aqueous phase were performed. The calculations were carried out on the same SGI workstation using the AMBER 5.0 molecular modelling package.20 The global charge was neutralized with Mg2+ counter ions using the Cornell parameters21 and the different ions were positioned by the Addions algorithm. A parallelepipedal periodic boundary condition (PBC) was set and filled up with the TIP-3P water molecules.22 The parm96 forcefield20 was used. The type and the partial charge of the RNA atoms were set according to the AMBER standard library. For the PNA residues, the partial charge of the different atoms were set using Merz Kollman method.23 The structure of the 6 PNA residues was computed using the Gaussian 98 program with the RHF method24 and a 6-31G** atomic basis set. All bond distances involving a hydrogen atom were constrained by using both algorithms (a 2 fs integration time step was set). The temperature was increased from 100 to 300 K during the first 80 ps of the simulation and then temperature and pressure were kept constant for 600 ps. In the same time, base pair constraints were decreased. Detailed analysis of the trajectories showed good recovery of all interactions. Thus, unpaired bases moved during the approach of system equilibrium to adopt and maintain a stable conformation.
Chemistry
For the development of linear or cyclic PNA based on a liquid-phase approach, some of us have elaborated the fully protected backbone strategy (FPBS) which constitutes an attractive alternative to the classical solid-phase approach.12,13 It consists of building a linear or cyclic fully protected poly(2-aminoethylglycinamide) backbone precursor containing as many different and orthogonal protecting groups on its secondary amines as there are different types of nucleic bases in the targeted PNA. A series of selective deprotection–coupling steps then allows the simultaneous introduction of the required number of identical nucleobase units onto the framework. The challenge of the FPBS lies in the elaboration of a careful synthetic planning in order to achieve the required degree of orthogonality among the protecting groups present in the molecule. It has been successfully applied for the syntheses of two cyclic hexa-PNA containing two (i.e. U, C)13a and three (i.e. U, C, G)12 different nucleobases. However, a limitation of this method lies in the required number of orthogonal acid and amino protecting groups. Thus, the synthesis of these two hexa-PNA has necessitated the introduction of five and six different protecting groups, respectively.In the present work, the targeted PNA 1 and 2 include four different nucleobases (U, C, G and A) and their synthesis, following the FPB strategy, would require a combination of at least eight orthogonal protecting groups. To overcome this difficulty, we elaborated a “mixed” strategy which relies on protected PNA fragments and protected poly(2-aminoethylglycinamide) building blocks.
Synthesis of the cyclic hexa-PNA 1
The retrosynthetic pathway for the synthesis of compound 1 is illustrated in Fig. 4. The key compound is the mixed tri-PNA/tri-Alloc linear hexamer 4 whose N- and C- extremities are both linked to protected amino and acid spacer units, respectively. We have chosen to introduce these spacer units at the beginning of the synthesis rather than on the non-functionalized analogs of 3 and 4, as the structural complexity of these analogs could lead to synthetic problems during their N- and C- functionalizations.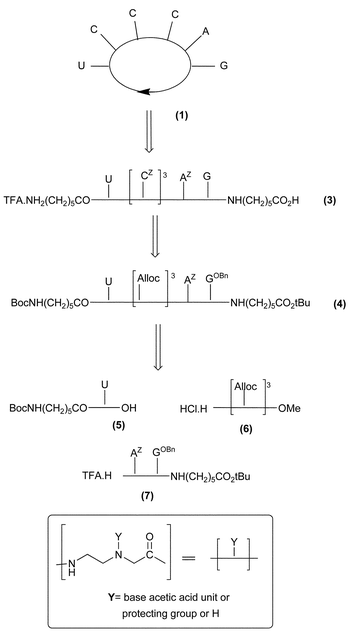 | ||
| Fig. 4 Retrosynthetic pathway for compound 1. | ||
Thus, starting from the N- and C-functionalized mono- and di-PNA fragments 5 and 7, respectively, and from the Alloc protected tris-(2-aminoethylglycinamide) unit 6, a series of condensation and selective deprotection steps allowed obtainment of the mixed tri-PNA/tri-Alloc linear hexamer 4. After Alloc deprotection of 4 and introduction of three N-Z-cytosine acetic acid units, the linear PNA 3 was cyclised to give the desired compound 1. The Alloc protecting group, whose cleavage occurs quickly and cleanly under mild conditions (Pd[P(Ph)3]4 and diethylamine as allyl scavanger), was selected for its orthogonality with the BnO and Z protecting groups of the nucleobases as well as with the Boc and t-Bu protecting groups of the N and C-extremities of the linker.
Concerning the synthesis of the N-acylated uracil mono-PNA 5, this derivative was prepared in three steps (Scheme 1) from 2-aminoethylglycine methyl ester 8 and commercial N-Boc aminocaproic acid 9. These two derivatives were coupled via a DCC–HOSu preactivation to afford compound 10 (87% yield) onto which the uracil acetic acid unit 11 was condensed by means of bromo-tris(dimethylamino)phosphoniumhexafluorophosphate reagent (Brop) (80%). Saponification of the resulting compound 12 with 1 M LiOH gave 5 almost quantitatively.
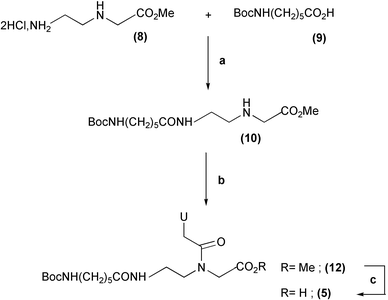 | ||
| Scheme 1 Synthesis of the N-acylated uracil PNA monomer 5. Reagents and conditions: (a) i. 9, DCC–HOSu, DMF, 12 h, rt. ii. −15 °C, 8, NMM, DMF. (b) 11, Brop, TEA, CH2Cl2. (c) 1 M LiOH, dioxane. | ||
The synthesis of the AZGOBn di-PNA 7 is detailed in Scheme 2. It was performed from the two key synthons 15 and 18. These two compounds were both prepared from the N-Mmt protected aminoethyl glycine methyl ester 13. The Z-adenine mono-PNA 15 was obtained by condensation of the Z-adenine acetic acid unit 14 onto the monomer 13 with Brop reagent, followed by saponification of the resulting methyl ester with 1 M LiOH (77% overall yield). The 6-aminocaproic-C-functionalized N-Alloc-protected monomer 18 was prepared in four steps from 13 in 55% overall yield. These steps consisted of the Alloc protection of 13 by reaction with Alloc-Cl, methyl ester hydrolysis with 1 M LiOH, benzotriazol-1-yloxytris(dimethylamino)phosponium hexafluorophosphate (Bop) coupling of the resulting acid 16 with 6-aminocaproic tertiobutyl ester 17, followed by the selective cleavage of the Mmt group with 1% TFA in CH2Cl2. The two 15 and 18 moieties were then linked together using Brop activation, leading to the mixed dimer 19 in 72% yield. Alloc deprotection with Pd(PPh3)4 and diethylamine (80% yield), then conjugation with the OBn-guanine acetic acid unit 20 by means of HATU–HOAt activation afforded 21 in 82% yield. The target 7 was obtained by cleaving the Mmt group on 21 by means of 1% TFA in CH2Cl2 (quantitative).
![Synthesis of the C-functionalized diPNA AZGOBn7. Reagents and conditions:
(a) AdZCH2CO2H 14, DIEA, Brop, DMF. (b) 1 M LiOH, dioxane. (c) Alloc-Cl, DIEA, CH2Cl2. (d) 1 M LiOH, 0.8 M CaCl2, iPrOH–H2O 7 : 3. (e)
16, NH2(CH2)5CO2tBu 17, DIEA, Bop, DMF. (f) 1% TFA–CH2Cl2, TIS. (g) Brop, DIEA, DMF. (h) Pd[P(Phe)3]4, DEA, CH2Cl2. (i) GOBnCH2CO2H 20, DIEA, HOAt, HATU, DMF.](/image/article/2004/OB/b311775h/b311775h-s2.gif) | ||
| Scheme 2 Synthesis of the C-functionalized diPNA AZGOBn7. Reagents and conditions: (a) AdZCH2CO2H 14, DIEA, Brop, DMF. (b) 1 M LiOH, dioxane. (c) Alloc-Cl, DIEA, CH2Cl2. (d) 1 M LiOH, 0.8 M CaCl2, iPrOH–H2O 7 : 3. (e) 16, NH2(CH2)5CO2tBu 17, DIEA, Bop, DMF. (f) 1% TFA–CH2Cl2, TIS. (g) Brop, DIEA, DMF. (h) Pd[P(Phe)3]4, DEA, CH2Cl2. (i) GOBnCH2CO2H 20, DIEA, HOAt, HATU, DMF. | ||
The synthesis of the target cyclic derivative 1 from fragments 5, 6 and 7 is shown in Scheme 3. The tri-Alloc fragment 6, previously described,13b was first condensed with the uracil mono-PNA 5 using Bop activation. This yielded, after saponification, the mixed tetramer 22 (76% overall yield) which was then coupled to the di-PNA 7 with Bop reagent to afford the mixed hexamer backbone 4 (90% yield). After Alloc deprotection of 4 with Pd(PPh3)4, the Z-cytosine acetic acid units 23 were then condensed onto each of the three generated amino functions by means of HATU–HOAt activation giving 24 (90% yield). Treatment of 24 with TFA–CH2Cl2 1 : 1 led to the simultaneous deprotection of the Boc, t-Bu and Bn groups and the linear precursor 3 was isolated in 94% yield. The head-to-tail cyclisation of 3 was accomplished in good yield (60% of 25) using HATU–HOAt activation and semi-high dilution conditions (≤10−2 M) in order to limit oligomerisation. It is worth noting that the presence of the free exocyclic guanine amino function was not a drawback for the issue of this reaction. Finally, the synthesis of the hexa-PNA target 1 was achieved by submitting macrocycle 25 to HBr–AcOH which caused the simultaneous deprotection of the cytosine and adenine nucleobases. Compound 1 was isolated after semi-preparative HPLC (40% yield from 25). Its purity was demonstrated by HPLC analyses and its structure was confirmed by MALDI-TOF experiments.
![Synthesis of compound 1 from synthons 5, 6 and 7. Reagents and conditions:
(a) Bop, DIEA, DMF. (b) 1 M LiOH, THF. (c) Pd[P(Phe)3]4, DEA, CHCl3–DMF. (d) CZCH2CO2H 23, DIEA, HOAt, HATU, DMF. (e) TFA–CH2Cl2 1 : 1. (f) DIEA, HOAt, HATU, DMF. (g) HBr–AcOH.](/image/article/2004/OB/b311775h/b311775h-s3.gif) | ||
| Scheme 3 Synthesis of compound 1 from synthons 5, 6 and 7. Reagents and conditions: (a) Bop, DIEA, DMF. (b) 1 M LiOH, THF. (c) Pd[P(Phe)3]4, DEA, CHCl3–DMF. (d) CZCH2CO2H 23, DIEA, HOAt, HATU, DMF. (e) TFA–CH2Cl2 1 : 1. (f) DIEA, HOAt, HATU, DMF. (g) HBr–AcOH. | ||
Synthesis of linear hexaPNA UCCCAG 2
The synthesis of compound 2 was first envisaged following a similar synthetic pathway as that for compound 1, i.e. starting from the Boc-uracil PNA monomer 26, the triAlloc fragment 6 and the PNA dimer AZGOBn28 (Scheme 4).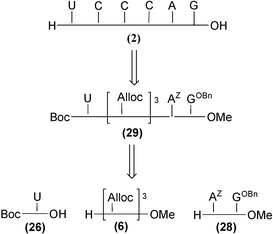 | ||
| Scheme 4 Retrosynthestic scheme for the synthesis of compound 2 starting from PNA fragments 26, 28 and protected trimer 6. | ||
Unfortunately, the key mixed triAlloc–triPNA linear synthon 29 (analogous to compound 4) was found to be unstable in different solvents such as DMF, CHCl3, CH3CN–H2O and MeOH. In particular, when compound 29 was dissolved in MeOH, substitution of the benzyloxy group of the guanine residue by methoxy occured very rapidly (less than two hours).
Furthermore, our attempts to cleanly cleave the three Alloc groups on 29 were unsuccessful as a complex mixture was obtained. These particular events may be attributed to the presence of the uracil moiety in 29. Indeed, it has been reported that this nucleobase is able to catalyse the aminolysis of 6-chloropurine derivatives, via multiple hydrogen-bonding interactions.16 That such behaviors were not evidenced in the case of the analogue 4 could be likely related to the presence of the amino acid linker which may prevent such interactions from occurring.
To overcome these difficulties, an alternative pathway, described in Scheme 5, was thus elaborated. For circumventing side reactions supposed to occur in the presence of the uracil moiety, the uracil monomer 26 was introduced at an advanced stage of the synthesis, i.e. on the already formed PNA pentamer 36 that contains the deprotected G, the three CZ and the AZ nucleobases.
![Synthesis of linear hexaPNA 2 from PNA fragments 26 and 28, and protected fragment 33. Reagents and conditions:
(a)
13, GOBnCH2CO2H 20, Bop, DIEA, DMF. (b) 1% TFA–CH2Cl2, TIS. (c)
13, AdZCH2CO2H 14, Brop, DIEA, DMF. (d) 1 M LiOH, dioxane. (e) Brop, DIEA, DMF. (f) 1 M LiOH, 0.8 M CaCl2, iPrOH–H2O 7 : 3. (g) Bop, DIEA, DMF. (h) i.: Pd[P(Phe)3]4, DEA, CH2Cl2. ii. CZCH2CO2H 23, DIEA, HOAt, HATU, DMF. (i) TFA–CH2Cl2 1 : 1. (j) UCH2CO2H 38, Bop, 2.6-lutidine, DMF. (k) HBr–AcOH, H2O (few drops).](/image/article/2004/OB/b311775h/b311775h-s5.gif) | ||
| Scheme 5 Synthesis of linear hexaPNA 2 from PNA fragments 26 and 28, and protected fragment 33. Reagents and conditions: (a) 13, GOBnCH2CO2H 20, Bop, DIEA, DMF. (b) 1% TFA–CH2Cl2, TIS. (c) 13, AdZCH2CO2H 14, Brop, DIEA, DMF. (d) 1 M LiOH, dioxane. (e) Brop, DIEA, DMF. (f) 1 M LiOH, 0.8 M CaCl2, iPrOH–H2O 7 : 3. (g) Bop, DIEA, DMF. (h) i.: Pd[P(Phe)3]4, DEA, CH2Cl2. ii. CZCH2CO2H 23, DIEA, HOAt, HATU, DMF. (i) TFA–CH2Cl2 1 : 1. (j) UCH2CO2H 38, Bop, 2.6-lutidine, DMF. (k) HBr–AcOH, H2O (few drops). | ||
Thus, the synthesis of the linear hexa-PNA started with condensation between the key AZGOBn dimer 28 and triAlloc fragment 33. This condensation was performed by means of the Bop reagent and afforded the mixed pentamer 34 in 85% yield. After quantitative removal of the three Alloc groups on 34 by Pd[PPh3]4, three Z-cytosine acetic acid units 23 were then inserted with HATU–HOAt as coupling reagents, to give the N-Boc penta-PNA ester 35. Treatment with a 1 : 1 TFA–CH2Cl2 solution led to cleavage of the Boc and Bn groups, and the TFA salt of penta-PNA 36 was isolated in quantitative yield. This derivative was then conjugated to the uracil monomer 26 by means of Bop reagent leading to the hexa-PNA 38 (75%). The one-step cleavage of all the protecting groups in 38 was realized with a HBr–AcOH solution containing a few drops of water. The targeted linear hexaPNA 2 was isolated in 50% yield after semi-preparative HPLC. Its purity was demonstrated by HPLC analyses and its structure was confirmed by MALDI-TOF experiments.
Concerning key synthons 28, 33 and 26, the first one was prepared by condensing the acid Z-adenine monomer 15 with the guanine monomer 30 using Brop activation, followed by the selective and smooth TFA-mediated cleavage of the Mmt group in the resulting 31 (86% yield for two steps). The BnO-guanine monomer 30 was obtained in two steps from 13 (65% overall yield), which consisted of the condensation of the BnO-guanine acetic acid 20 onto 13 with the Bop reagent and the TFA-mediated Mmt-deprotection.
The key synthon 33 was obtained by the Ca2+-catalyzed saponification of the triAlloc fragment 3213b (quantitative).
The N-Boc uracil PNA 26, was prepared in two steps (82% yield) from the N-Boc protected aminoethylglycine ester backbone 37, by condensing the uracil acetic acid unit 38 onto 37 with the Bop reagent, then by saponification of the resulting PNA monomer with 1 M LiOH.
TAR binding assays.
The interactions of the cyclic and linear PNA 1–2 with the TAR RNA were investigated by melting temperature (Tm) measurements, electromobility shift assays and RNase footprinting experiments.25 All these experiments indicated clearly that no hybridisation (hence no interaction) occurred between the cyclic antisense PNA 1 and TAR RNA whereas an interaction, though tenuous, was detected with its linear PNA analogue 2 (Fig. 5). Indeed, the single transition at 49 °C of a 27-mer oligoribonucleotide (which corresponds to the 18–44 of the 59-bases of the TAR RNA sequence and contains the pyrimidine bulge and the CUGGGA apical loop of TAR, see Fig. 1) remained unchanged even for a PNA 1 : TAR molar ratio > 1. By contrast, a slight Tm increase (from 49 °C to 51.4 °C) was detected with the linear PNA 2 at a PNA 2 : TAR molar ratio of 1. The ΔTm never exceeded 3 °C even for a PNA 2 : TAR molar ratio > 1. Concerning the electromobility shift assays, we used the full length 59-mer TAR RNA which was prepared by in vitro transcription, radiolabeled then incubated with the different PNA 1–2. No retarded band corresponding to PNA–RNA complexes could be detected. Similarly, RNase footprinting experiments using endonuclease A failed to detect recognition of the apical loop by the PNA. Different experimental conditions were tested (low salt buffer, higher ionic strength, heating of the PNA to unfold it, various incubation periods) but in all cases, formation of a specific complex was never observed.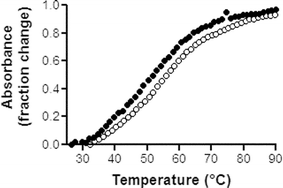 | ||
| Fig. 5 Representative Tm plots for the 27-mer RNA oligonucleotide corresponding to the 18–44 portion of the TAR RNA, (●) in the absence and (○) presence of PNA 2, at a PNA : RNA ratio of 1. Tm measurements were performed in triplicate, in BPE buffer pH 7.0 (6 mM Na2HPO4, 2 mM NaH2PO4, 1 mM EDTA), at 260 nm with a heating rate of 1 °C min−1.25 The Tm values were obtained from first-derivative plots. | ||
The inability of compound 1 to bind to TAR could be explained in the light of the results obtained by Toulmé and coworkers.15,16 This group has identified, by in vitro selection, mini RNA hairpin aptamers which recognize the HIV-1 TAR-RNA through very stable kissing complexes. These mini RNA hairpins contain the octameric loop sequence 5′-GTCCCAGA-3′, which includes the six-nucleotide sequence complementary to the TAR loop flanked by a G and A residue. This GA pair was found to be crucial for the formation of a stable kissing complex between TAR and these aptamers. It should be emphasized that this GA pair was also shown to be essential in the case of N3′ P5′ deoxyphosphoramidate10 and 2′-OMe analogues.10b,11 The absence of such a GA pair flanking the cyclic hexa-PNA 1 could account for its incapacity to bind to TAR.
Concerning the linear antisense hexa-PNA 2, only a slight affinity for TAR was detected, whereas the linear 2′-O-methyl octamer GUCCCAGA has been reported to be unable to form any complex with TAR.10b Although it has been previously reported that the linear RNA hexamer TCCCAG binds to TAR,17 such linear oligomers targeting only the six residues of the TAR loop do not appear as valuable candidates for inhibiting the Tat–TAR interaction and thus HIV replication.
Conclusion
A cyclic compound, containing a hexa-PNA sequence complementary to the TAR RNA loop of HIV-1, and its linear analogue have been successfully synthesized, following a liquid-phase strategy using mixed PNA and fully N-protected (aminoethylglycinamide) fragments. This procedure offers the advantage, over the FPB strategy, of requiring less orthogonal protecting groups and enables the preparations of PNA containing the four nucleobases A, C, G and U. However, the presence of the uracil nucleobase is suspected of inducing unpredictable side reactions, emphasizing the interest of the FPB strategy in which the troublesome base acetic acid units are introduced at advanced stages of the synthesis.TAR binding assays have shown that cyclic hexa-PNA 1 is unable to form a stable kissing complex with the loop of TAR, whereas only a slight binding to TAR has been detected for its linear analogue 2. In the light of the results of Toulmé and coworkers, the molecular modelling, synthesis and TAR binding capability of an octameric cyclic PNA-based compound [GTCCCAGA]c, including the six-nucleotide sequence complementary of the TAR loop flanked by a G and A residue, are currently under progress.
Acknowledgments
We thank the ‘Centre National de la Recherche Scientifique’ and Sidaction for their grants.References
- For a review, see: (a) K. A. Roebuck and M. Saifuddin, Gene Expr., 1999, 8, 67–84 Search PubMed; (b) K. A. Jones and B. M. Peterlin, Annu. Rev. Biochem., 1994, 63, 717–743 CrossRef CAS; (c) B. Berkhout and K. T. Jeang, J. Virol., 1989, 63, 5501–5504 CAS.
- (a) D. Harrich, C. W. Hooker and E. Parry, J. Virol., 2000, 74, 5639–5646 CrossRef CAS; (b) D. Harrich and B. Hooker, Rev. Med. Virol., 2002, 12, 31–45 CrossRef CAS; (c) J. L. Clever, D. Mirandar and T. G. Parslow, J. Virol., 2002, 76, 12381–12387 CrossRef CAS.
- (a) P. Wei, M. E. Garber, S. M. Fang, W. H. Fischer and K. A. Jones, Cell, 1998, 92, 451–62 CAS; (b) A. Fraldi, P. Licciardo, B. Majello, A. Giordano and L. Lania, J. Cell Biochem., 2001, 36, 247–53 CrossRef; (c) S. Richter, Y. H. Ping and T. M. Rana, Proc. Natl Acad. Sci. USA., 2002, 99, 7928–7933 CrossRef CAS; (d) S. Richter, H. Cao and T. M. Rana, Biochemistry, 2002, 41, 6391–6397 CrossRef CAS.
- T. Vickers, B. F. Baker, P. D. Cook, M. Zounes, R. W. Buckheit, J. Germany and D. J. Ecker, Nucleic Acids Res., 1991, 19, 3359–3368 CAS.
- F. Boulmé, F. Freund, S. Moreau, P. E. Nielsen, S. Gryaznov, J. J. Toulmé and S. Litvak, Nucleic Acids Res., 1998, 26, 5492–5500 CrossRef CAS.
- (a) A. Arzumanov, A. P. Walsh, V. K. Rajwanshi, J. Wengel and M. J. Gait, Biochemistry, 2001, 40, 14645–14654 CrossRef CAS; (b) A. Arzumanov, A. P. Walsh, X. Liu, V. K. Rajwanshi, J. Wengel and M. J. Gait, Nucleosides Nucleotides & Nucleic Acids, 2001, 20, 471–480 Search PubMed; (c) B. Mestre, A. Arzumanov, M. Sing, F. Boulmé, S. Litvak and M. J. Gait, Biochim. Biophys. Acta, 1999, 1445, 86–98 CrossRef CAS.
- (a) T. Hamma and P. S. Miller, Antisense Nucleic Acid Drug Dev., 2003, 13, 19–30 CrossRef CAS; (b) T. Hamma, A. Saleh, I. Huq, T. M. Rana and P. S. Miller, Bioorg. Med. Chem. Lett., 2003, 13, 1845–1848 CrossRef CAS; (c) P. S. Miller, R. A. Cassidy, T. Hamma and N. S. Kondo, Pharmacol. Ther., 2000, 85, 159–163 CrossRef CAS; (d) T. Hamma and P. S. Miller, Biochemistry, 1999, 38, 15333–15342 CrossRef CAS.
- (a) N. Kaushik, A. Basu and V. N. Pandey, Antiviral Res., 2002, 56, 13–27 CrossRef CAS; (b) N. Kaushik, A. Basu, P. Palumbo, R. L. Myers and V. N. Pandey, J. Virol., 2002, 76, 3881–3891 CrossRef CAS; (c) T. Mayhood, N. Kaushik, P. K. Pandey, F. Kashanchi, L. Deng and V. N. Pandey, Biochemistry, 2000, 39, 11532–11539 CrossRef CAS.
- S. C. Holmes, A. A. Arzumanov and M. J. Gait, Nucleic Acids Res., 2003, 31, 2759–2768 CrossRef CAS.
- (a) F. Darfeuille, A. Arzumanov, S. Gryaznov, M. J. Gait, C. Di Primo and J. J. Toulmé, Proc. Natl Acad. Sci. USA., 2002, 99, 9709–9714 CrossRef CAS; (b) F. Darfeuille, A. Arzumanov, M. J. Gait, C. Di Primo and J. J. Toulmé, Biochemistry, 2002, 41, 12186–12192 CrossRef CAS; (c) F. Darfeuille, C. Cazenave, S. Gryaznov, F. Duconge, C. Di Primo and J. J. Toulmé, Nucleosides Nucleotides & Nucleic Acids, 2001, 20, 441–449 Search PubMed.
- (a) L. Zapata, K. Bathany, J. M. Schmitter and S. Moreau, Eur. J. Org. Chem., 2003, 1022–1028 CrossRef CAS; (b) J. Michel, K. Bathany, J. M. Schmitter, J. P. Monti and S. Moreau, Tetrahedron, 2002, 58, 7975–7982 CrossRef CAS.
- C. Schwergold, G. Depecker, C. Di Giorgio, N. Patino, F. Jossinet, B. Ehresmann, R. Terreux, D. Cabrol-Bass and R. Condom, Tetrahedron, 2002, 58, 5675–5687 CrossRef CAS.
- (a) G. Depecker, C. Di Giorgio, N. Patino and R. Condom, Tetrahedron Lett., 2001, 42, 8303–8306 CrossRef CAS; (b) C. Di Giorgio, S. Pairot, C. Schwergold, N. Patino, R. Condom, A. Farese-Di Giorgio and R. Guedj, Tetrahedron, 1999, 55, 1937–1958 CrossRef CAS.
- (a) L. R. Comolli, J. G. Pelton and I. Tinoco, Nucleic Acids Res., 1998, 26, 4688–4695 CrossRef CAS; (b) K. Y. Chang and I. Tinoco, J. Mol. Biol., 1997, 269, 52–66 CrossRef CAS; (c) K. Y. Chang and I. Tinoco, Proc. Natl Acad. Sci. USA, 1994, 91, 8705–8709 CAS.
- (a) F. Darfeuille, C. Cazenave, S. Gryaznov, F. Ducongé, C. Di Primo and J. J. Toulmé, Nucleosides Nucleotides & Nucleic Acids, 2001, 20, 441–449 Search PubMed; (b) F. Ducongé, C. Di Primo and J. J. Toulmé, J Biol Chem., 2000, 275, 21287–21294 CrossRef CAS; (c) F. Ducongé and J. J. Toulmé, RNA, 1999, 5, 1605–14 CrossRef CAS; (d) C. Boiziau, E. Dausse, L. Yurchenko and J. J. Toulmé, J. Biol. Chem., 1999, 274, 12730–12737 CrossRef CAS.
- F. Beaurain, C. Di Primo, J. J. Toulmé and M. Laguerre, Nucleic Acids Res., 2003, 31, 4275–4284 CrossRef CAS.
- C. H. Tung, J. Wang, M. J. Leibowitz and S. Stein, Bioconjugate Chem., 1995, 6, 292–295 CrossRef CAS.
- Insight II release 97.0, Accelerys Inc., San Diego, USA.
- J. R. Maple, M.-J. Hwang, K. J. Jalkanen, T. P. Stockfisch and A. T. Hagler, J. Comp. Chem., 1998, 19, 430–458 Search PubMed.
- D. A. Case, D. A. Pearlman, J. W. Caldwell, T. E. Cheatham III, W. S. Ross, C. L. Simmerling, T. A. Darden, K. M. Merz, R. V. Stanton, A. L. Cheng, J. J. Vincent, M. Crowley, D. M. Ferguson, R. J. Radmer, G. L. Seibel, U. C. Singh, P. K. Weiner, and P. A. Kollman, AMBER 5, University of California, San Francisco, 1997 Search PubMed.
- W. D. Cornell, P. Cieplack, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Imprey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- B. H. Besler, K. M. Merz and P. A. Kollman, J. Comp. Chem., 1990, 11, 431–439 Search PubMed.
- D. R. Hartree, Proc. Cambridge Philos. Soc., 1928, 24, 89–110 Search PubMed.
- V. Peytou, R. Condom, N. Patino, R. Guedj, A. M. Aubertin, N. Gelus, C. Bailly, R. Terreux and D. Cabrol-Bass, J. Med. Chem., 1999, 42, 4042–4043 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: experimental details. See http://www.rsc.org/suppdata/ob/b3/b311775h/ |
| This journal is © The Royal Society of Chemistry 2004 |